Dietary Management, Clinical Status and Outcome of Patients with Citrin Deficiency in the UK

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Subjects
2.2. Inclusion Criteria
2.3. Exclusion Criteria
2.4. Data Collection
2.5. Dietary Intake
2.6. Growth
2.7. Ethical Statement
2.8. Statisticsa
3. Results
3.1. Subjects
3.2. Signs and Symptoms at Presentation
3.3. Diagnosis and Biochemistry
3.4. Dietary Treatment
3.5. Diet Prescriptions
3.6. Vitamin and Mineral Supplements and Tube Feeding
3.7. Dietary Intake
3.8. Food Preferences
3.9. Emergency Regimens
3.10. Hospital Admissions
3.11. Drug Therapy
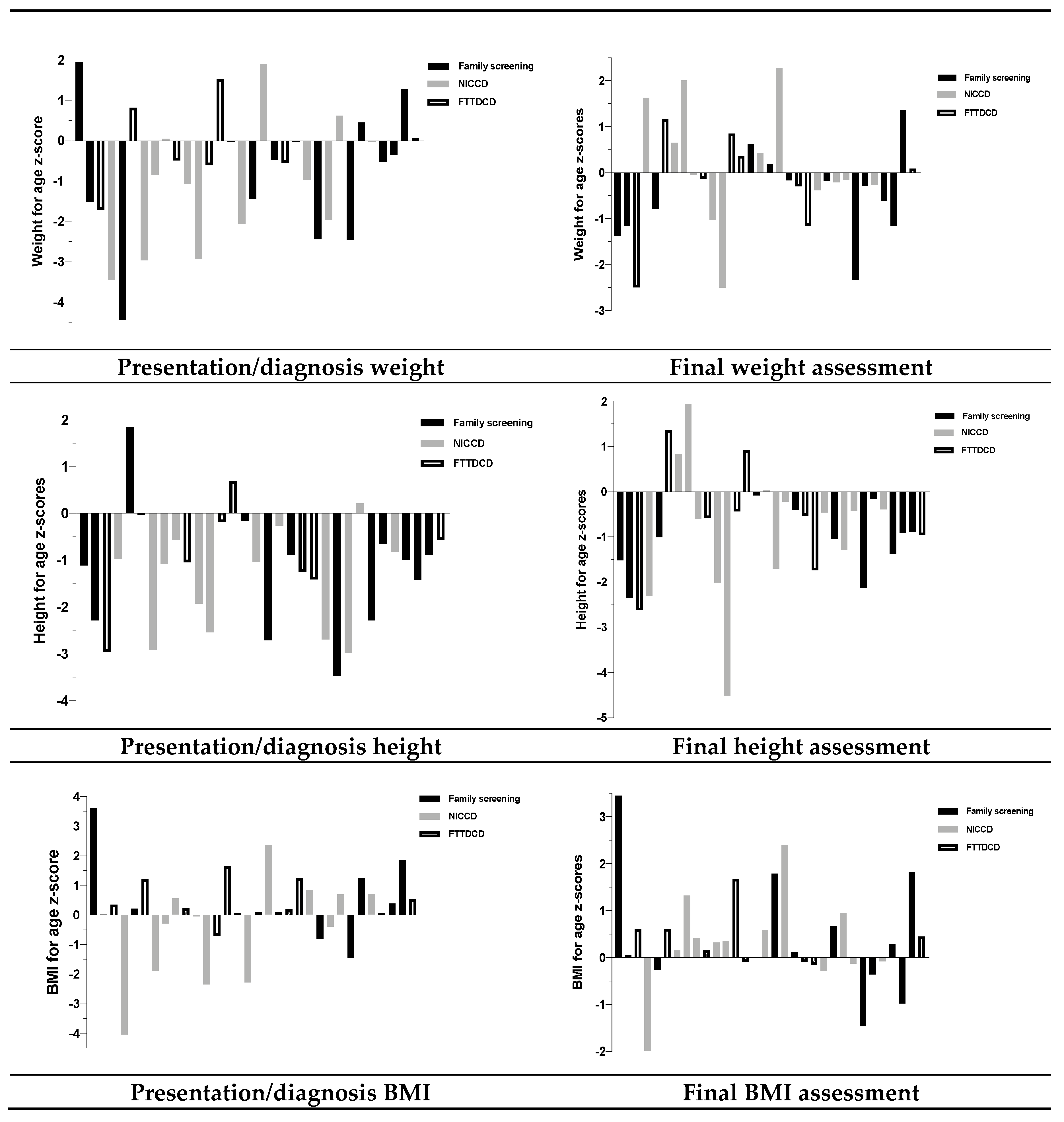
3.12. Growth
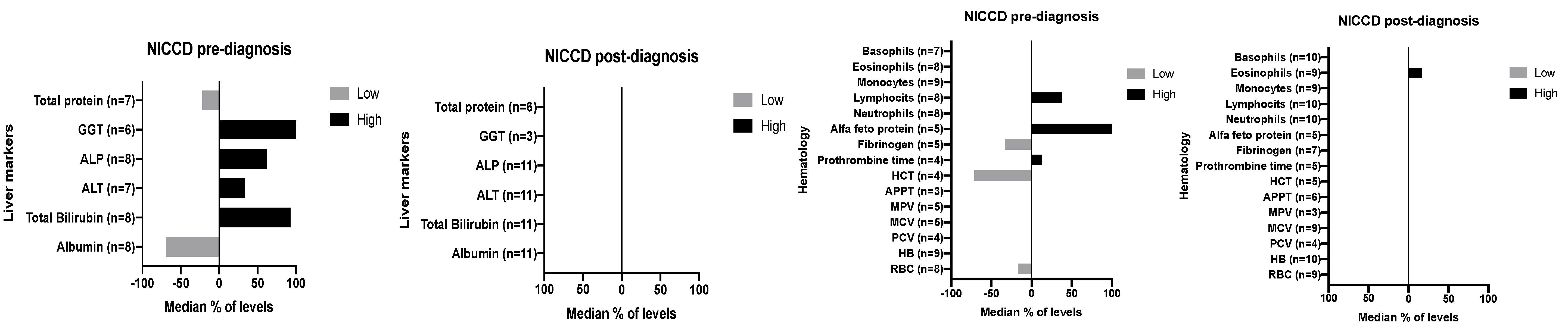
3.13. Biochemical Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kobayashi, K.; Sinasac, D.S.; Iijima, M.; Boright, A.P.; Begum, L.; Lee, J.R.; Yasuda, T.; Ikeda, S.; Hirano, R.; Terazono, H.; et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat. Genet. 1999, 22, 159–163. [Google Scholar] [CrossRef]
- Dimmock, D.; Kobayashi, K.; Iijima, M.; Tabata, A.; Wong, L.J.; Saheki, T.; Lee, B.; Scaglia, F. Citrin deficiency: A novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics 2007, 119, e773–e777. [Google Scholar] [CrossRef]
- Okano, Y.; Ohura, T.; Sakamoto, O.; Inui, A. Current treatment for citrin deficiency during NICCD and adaptation/compensation stages: Strategy to prevent CTLN2. Mol. Genet. Metab. 2019, 127, 175–183. [Google Scholar] [CrossRef]
- Numakura, C.; Tamiya, G.; Ueki, M.; Okada, T.; Maisawa, S.I.; Kojima-Ishii, K.; Murakami, J.; Horikawa, R.; Tokuhara, D.; Ito, K.; et al. Growth impairment in individuals with citrin deficiency. J. Inherit. Metab. Dis. 2019, 42, 501–508. [Google Scholar] [CrossRef]
- Saheki, T.; Song, Y.Z. Citrin Deficiency. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hayasaka, K.; Numakura, C.; Toyota, K.; Kimura, T. Treatment with lactose (galactose)-restricted and medium-chain triglyceride-supplemented formula for neonatal intrahepatic cholestasis caused by citrin deficiency. JIMD Rep. 2012, 2, 37–44. [Google Scholar] [CrossRef]
- Hayasaka, K. Metabolic basis and treatment of citrin deficiency. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef]
- Saheki, T.; Inoue, K.; Tushima, A.; Mutoh, K.; Kobayashi, K. Citrin deficiency and current treatment concepts. Mol. Genet. Metab. 2010, 100 (Suppl. 1), S59–S64. [Google Scholar] [CrossRef]
- Hayasaka, K.; Numakura, C.; Yamakawa, M.; Mitsui, T.; Watanabe, H.; Haga, H.; Yazaki, M.; Ohira, H.; Ochiai, Y.; Tahara, T.; et al. Medium-chain triglycerides supplement therapy with a low-carbohydrate formula can supply energy and enhance ammonia detoxification in the hepatocytes of patients with adult-onset type II citrullinemia. J. Inherit. Metab. Dis. 2018, 41, 777–784. [Google Scholar] [CrossRef]
- Nutritics, R. Edition (v5. 09) [Computer Software]. Available online: https://www.nutritics.com/p/home (accessed on 8 October 2019).
- WHO/UK Growth Charts. Available online: https://www.rcpch.ac.uk/resources/growth-charts (accessed on 8 October 2019).
- Stevens, G.A.; Finucane, M.M.; Paciorek, C.J.; Flaxman, S.R.; White, R.A.; Donner, A.J.; Ezzati, M. Trends in mild, moderate, and severe stunting and underweight, and progress towards MDG 1 in 141 developing countries: A systematic analysis of population representative data. Lancet 2012, 380, 824–834. [Google Scholar] [CrossRef]
- WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr. 2006, 450, 76–85. [CrossRef]
- De Onis, M.; Onyango, A.W.; Borghi, E.; Siyam, A.; Nishida, C.; Siekmann, J. Development of a WHO growth reference for school-aged children and adolescents. Bull. World Health Organ. 2007, 85, 660–667. [Google Scholar] [CrossRef]
- SACO Nutrition. Dietary Reference Values for Energy. 2011. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/339317/SACN_Dietary_Reference_Values_for_Energy.pdf (accessed on 27 November 2019).
- Guidelines, B. Emergency Management Guidance. Available online: http://www.bimdg.org.uk/store/guidelines/ER-AandE_Citrin_2017_135427_18012017.pdf (accessed on 27 November 2019).
- Saheki, T.; Kobayashi, K.; Terashi, M.; Ohura, T.; Yanagawa, Y.; Okano, Y.; Hattori, T.; Fujimoto, H.; Mutoh, K.; Kizaki, Z.; et al. Reduced carbohydrate intake in citrin-deficient subjects. J. Inherit. Metab. Dis. 2008, 31, 386–394. [Google Scholar] [CrossRef]
- Imamura, Y.; Kobayashi, K.; Shibatou, T.; Aburada, S.; Tahara, K.; Kubozono, O.; Saheki, T. Effectiveness of carbohydrate-restricted diet and arginine granules therapy for adult-onset type II citrullinemia: A case report of siblings showing homozygous SLC25A13 mutation with and without the disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2003, 26, 68–72. [Google Scholar] [CrossRef]
- Yazaki, M.; Takei, Y.; Kobayashi, K.; Saheki, T.; Ikeda, S. Risk of worsened encephalopathy after intravenous glycerol therapy in patients with adult-onset type II citrullinemia (CTLN2). Intern. Med. 2005, 44, 188–195. [Google Scholar] [CrossRef][Green Version]
- Otsuka, H.; Sasai, H.; Abdelkreem, E.; Kawamoto, N.; Kawamoto, M.; Kamiya, T.; Tanimoto, Y.; Kikuchi, A.; Kure, S.; Numakura, C.; et al. Effectiveness of Medium-Chain Triglyceride Oil Therapy in Two Japanese Citrin-Deficient Siblings: Evaluation Using Oral Glucose Tolerance Tests. Tohoku J. Exp. Med. 2016, 240, 323–328. [Google Scholar] [CrossRef]
- Unita, S.; Hirashima, N.; Shimada, M.; Tsunekawa, T.; Tanaka, D.; Kondo, T.; Urata, N.; Kondo, H.; Saito, M.; Iwase, H.; et al. Successful treatment of adult-onset type II citrullinemia with a low-carbohydrate diet and L-arginine after DNA analysis produced a definitive diagnosis. Clin. J. Gastroenterol. 2020. [Google Scholar] [CrossRef]
- Awrich, A.E.; Stackhouse, W.J.; Cantrell, J.E.; Patterson, J.H.; Rudman, D. Hyperdibasicaminoaciduria, hyperammonemia, and growth retardation: Treatment with arginine, lysine, and citrulline. J. Pediatrics 1975, 87, 731–738. [Google Scholar] [CrossRef]
- Li, M.X.; Nakajima, T.; Fukushige, T.; Kobayashi, K.; Seiler, N.; Saheki, T. Aberrations of ammonia metabolism in ornithine carbamoyltransferase-deficient spf-ash mice and their prevention by treatment with urea cycle intermediate amino acids and an ornithine aminotransferase inactivator. Biochim. Biophys. Acta 1999, 1455, 1–11. [Google Scholar] [CrossRef][Green Version]
- Saheki, T.; Moriyama, M.; Kuroda, E.; Funahashi, A.; Yasuda, I.; Setogawa, Y.; Gao, Q.; Ushikai, M.; Furuie, S.; Yamamura, K.-I.; et al. Pivotal role of inter-organ aspartate metabolism for treatment of mitochondrial aspartate-glutamate carrier 2 (citrin) deficiency, based on the mouse model. Sci. Rep. 2019, 9, 4179. [Google Scholar] [CrossRef]
- Fukushima, K.; Yazaki, M.; Nakamura, M.; Tanaka, N.; Kobayashi, K.; Saheki, T.; Takei, H.; Ikeda, S. Conventional diet therapy for hyperammonemia is risky in the treatment of hepatic encephalopathy associated with citrin deficiency. Intern. Med. 2010, 49, 243–247. [Google Scholar] [CrossRef]
- Okano, Y.; Kobayashi, K.; Ihara, K.; Ito, T.; Yoshino, M.; Watanabe, Y.; Kaji, S.; Ohura, T.; Nagao, M.; Noguchi, A.; et al. Fatigue and quality of life in citrin deficiency during adaptation and compensation stage. Mol. Genet. Metab. 2013, 109, 9–13. [Google Scholar] [CrossRef]
- Sargent, J.D.; Stukel, T.A.; Dalton, M.A.; Freeman, J.L.; Brown, M.J. Iron deficiency in Massachusetts communities: Socioeconomic and demographic risk factors among children. Am. J. Public Health 1996, 86, 544–550. [Google Scholar] [CrossRef]
- Tamamori, A.; Okano, Y.; Ozaki, H.; Fujimoto, A.; Kajiwara, M.; Fukuda, K.; Kobayashi, K.; Saheki, T.; Tagami, Y.; Yamano, T. Neonatal intrahepatic cholestasis caused by citrin deficiency: Severe hepatic dysfunction in an infant requiring liver transplantation. Eur. J. Pediatrics 2002, 161, 609–613. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Weight-for-Age z-Score | Height-for-Age z-Score | BMI-for-Age z-Score(5–19 Years) | |
|---|---|---|---|
| Definitions | Marginally underweight: <−1 Moderately underweight: <−2 Severely underweight: <−3 [12] | Marginally stunted: <−1 Moderately stunted: <−2 Severely stunted: <−3 [12] | Overweight: >+1 (or BMI ≥ 25 *) Obesity: >+2 (or BMI ≥ 30 *) [13,14] |
| Subject | Ethnicity | Consanguinity | Gender | Mutations | Co-existing Conditions | Full Term | Breast/Bottle Feeding on Presentation | Presentation Age | Diagnostic Age | Current Age | Biochemistry at Presentation/Diagnosis | Symptoms Post-Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | Asian | Yes | Female | c.1763G>A (p.Arg588Gln) | Coeliac disease | NA | Regular infant formula | 1 month | 2 months | 11 years | Neonatal cholestasis, abnormal lactate. | Jaundice, abdominal pain due to constipation. Headache, lethargic morning leg pain. |
| 7 | White | Yes | Male | 1465T>C (C489R) | - | Yes | Breastfeeding | 2 months | 1 year | 14 years | Neonatal cholestasis. High citrulline, threonine and methionine. | Severe abdominal pain. |
| 8 | White | Yes | Male | c.1173T>G (P.Tyr391X) | Asthma | Yes | Regular infant formula | 1 month | 6 months | 6 years | Neonatal intrahepatic cholestasis and conjugated hyperbilirubinemias. | Jaundice, pale stools. |
| 9 | Asian | No | Female | c.852_855delTATG p.(Met285Profs*2) | - | Yes | Soya infant formula | 4 months | 5 months | 5 years | Cholestasis, high citrulline, galactosuria. | Hepatitis, jaundice, hypoglycaemia. |
| 11 | Asian | Yes | Male | - | - | Yes | Casein hydrolysate with medium chain triglyceride (MCT) infant formula | 1 month | 1 month | 1 years | Increased phenylalanine and tyrosine on newborn screening. | Asymptomatic. |
| 12 | Asian | No | Male | c.1763 G>A P. (Arg 588GIn) | - | Yes | Casein hydrolysate with MCT infant formula | 2 weeks | 8 months | 1 years | NA | Prolonged jaundice. |
| 16 | Asian | Yes | Male | c.1763 G>A (p.Arg588Gln) | - | Yes | Casein hydrolysate with MCT infant formula | 1 month | 8 months | 6 years | Deranged liver function tests, clotting associated with positive reducing substances in urine and raised plasma citrulline. | Asymptomatic. |
| 18 | Asian | No | Male | c.1763G>A (p.Arg588Gln) | Hypoplastic left heart syndrome | NA | NA | 1 month | 8 years | 20 years | Cholestasis. | Acute liver failure, hypoglycaemia (resolved with age). Abdominal pain, sweating on morning waking. |
| 22 | Asian | Yes | Female | c.1763G>A (p.Arg588Gln) | - | NA | Casein hydrolysate with MCT infant formula | 1 week | 7 months | 10 years | Raised phenylalanine and tyrosine, neonatal jaundice. | Recurrent abdominal pain and headaches. |
| 24 | Asian | No | Male | c.852_855delTATG p.(Met285Profs*2) | - | NA | Breastfeeding | 1 day | 2 months | 4 years | Neonatal cholestasis. | Asymptomatic |
| 25 | Asian | No | Male | c.848 G>T p. (Gly283 Val) | - | Yes | Breastfeeding | 2 months | 5 months | 3 years | Increased citrulline. | Conjugated jaundice. |
| 28 | Asian | No | Male | c.1763G>A (p.Arg588Gln) | - | Yes | Casein hydrolysate with MCT infant formula | 1 month | 1 month | 8 years | Neonatal cholestasis, elevated phenylalanine and tyrosine on newborn screening. | Conjugated jaundice, hypoglycaemia. |
| Subject | Ethnicity | Consanguinity | Gender | Mutations | Co-existing Disorders | Presentation Age | Diagnostic Age | Current Age | Biochemistry at Presentation/Diagnosis | Symptoms Post-Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | Asian | Yes | Female | c.1763G>A (p.Arg588Gln) | Coeliac disease | 1 year | 6 years | 8 years | NA | Abdominal pain and hypoketotic hypoglycaemia (sweating when eating sugar). |
| 6 | Asian | Yes | Male | 1610-1612 delTA Gins AT | GSD type IX | 2 years | 2 years | 8 years | NA | Night sweats. |
| 10 | Asian | No | Male | c.550C>T p. (Arg 184*) | - | 11 years | 12 years | 15 years | Neonatal hepatitis, carnitine deficiency. | Conjugated jaundice, hypoglycaemia, febrile seizure. |
| 13 | Asian | Yes | Female | c.1766 C>T (p.Ser589 Phe) | - | 1 year | 8 years | 15 years | NA | Prolonged jaundice, abdominal pain. |
| 14 | Asian | No | Female | c. 1763G> A (p.Arg588Gln) | - | 3 years | 3 years | 14 years | NA | Abdominal pain. |
| 20 | Asian | No | Female | c.1781G>A (p.Gly594As) | Hypothyroidism | 7 years | 8 years | 11 years | Decompensated cirrhosis and thyroid antibodies, elevated plasma citrulline. | Asymptomatic. |
| 21 | Asian | No | Male | R588Q | - | 12 years | 12 years | 12 years | Neonatal hepatitis syndrome, raised citrulline, mildly elevated ammonia. | Asymptomatic. |
| 32 | Asian | No | Female | c.1763G> A (p.Arg588Gln) | - | 1 year | 14 years | 22 years | Cirrhosis and steatosis. Failure to thrive and hypertonia with generalised aminoaciduria, galactosemia and echogenic enlarged liver. | Floppiness and rickets. Abdominal pain when fasting for Ramadan (unknown cause). Failure to thrive. |
| Subject | Ethnicity | Consanguinity | Gender | Mutations | Co-existing Disorders | Diagnosis Age by Family Screening | Current Age | Biochemistry at Presentation/Diagnosis | Symptoms Post-Diagnosis |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Asian | Yes | Male | c.1763G>A(p.Arg588Gln) | Coeliac disease | 6 years | 6 years | NA | Asymptomatic. |
| 2 | Asian | Yes | Female | c.1763G>A(p.Arg588Gln) | - | 8 years | 10 years | NA | Abdominal pain. |
| 5 | Asian | Yes | Female | c.1763G>A(p.Arg588Gln) | - | 5 years | 6 years | NA | Asymptomatic |
| 15 | Asian | No | Female | c.1763G>A(p.Arg588Gln) | - | 8 years | 19 years | NA | Long history of severe abdominal pain. |
| 17 | Asian | Yes | Female | c.1763G>A(p.Arg588Gln) | - | 4 years | 4 years | Elevated citrulline and threonine. | Hypoglycaemia, abdominal pain with reduced appetite. |
| 19 | Asian | No | Male | c.1781G>A(p.Gly594Asp) | Hypothyroidism | 1 year | 4 years | Cholestasis, raised ammonia and lactate, galactosuria, high citrulline. | Asymptomatic. |
| 23 | Asian | Yes | Male | c.1763G>A(p.Arg588Gln) | - | 2 years | 2 years | Raised phenylalanine and tyrosine. | Asymptomatic. |
| 26 | Asian | No | Female | c.1763G>A(p.Arg588Gln) | - | 35 years | 44 years | NA | Abdominal pain, feels nauseous with certain foods, abdominal cramps and frequent bowel movements. |
| 27 | Asian | Yes | Male | c.1763G>A(p.Arg588Gln) | - | 5 years | 13 years | NA | Asymptomatic. |
| 29 | Asian | Yes | Female | c.1763G>A(p.Arg588Gln) | - | 9 years | 19 years | NA | Abdominal pain. |
| 30 | Asian | Yes | Female | c.1763G>A(p.Arg588Gln) | - | 15 years | 22 years | NA | Abdominal pain. |
| 31 | Asian | Yes | Female | c.1763G>A(p.Arg588Gln) | - | 12 years | 20 years | Unconjugated jaundice. | Abdominal pain. |
| Subject | Presentation | Growth at Presentation (Height-for-Age z-Scores) | Last Growth (Height-for-Age z-Scores) | Food Aversions/Preferences | Current Prescribed Dietary Management | Median Intake from Dietary Assessments | Oral Emergency Feed for Illness Management |
|---|---|---|---|---|---|---|---|
| 1 (Family 1) | Family screening | −1.11 | −1.52 | Likes custard. | Normal diet. | CHO: 42% Protein: 18% Fat: 40% | Not prescribed. |
| 2 (Family 1) | Family screening | −2.29 | −2.35 | Avoids: sweets, puddings, fruit juice, cakes, biscuits. Prefers: meat, fish, cheese, eggs, soya, nuts. | Low sugar, high protein, high fat diet. | CHO: 28% Protein: 20% Fat: 53% | Low sugar soya milk. |
| 3 (Family 1) | FTTDCD | −2.96 | −2.62 | Avoids: sweets, puddings, fruit juice, cakes and biscuits. Prefers: meat, fish, cheese, eggs, soya, nuts. | Low sugar, high protein, high fat diet + MCT oil. One oral feed in the middle of the night. | CHO: 30% Protein: 19% Fat: 51% | Low sugar soya milk + MCT emulsion + soya protein powder. |
| 4 | NICCD | −0.98 | −2.31 | Initial self- restriction of sweets but now eats them (with symptoms). Prefers high protein foods. | Normal diet (but feeds 4 hourly day and night). | CHO: 44% Protein: 18% Fat: 43% | Not prescribed. |
| 5 (Family 2) | Family screening | 1.85 | −1.01 | Dislikes sweets. | Normal diet (but feeds 4 hourly day and night). | NA | Not prescribed. |
| 6 (Family 2) | FTTDCD | −0.03 | 1.36 | Dislikes sweets. Likes meat. | Low sugar, high protein, high fat diet (MCT). | NA | Liquid chocolate yoghurt. |
| 7 | NICCD | −2.91 | 0.84 | Dislikes CHO foods, e.g., bread, rice, potatoes, sweets. Prefers high protein foods, e.g., bacon, eggs, nuts. | Low sugar, high protein high fat diet (LCT/MCT). CHO limited to 30g/day. | CHO: 18% Protein: 27% Fat: 55% | Soya protein powder + LCT/MCT emulsions + water. |
| 8 | NICCD | −1.08 | 1.94 | None described. | Normal diet. | CHO: 47% Protein: 19% Fat: 33% | Not prescribed. |
| 9 | NICCD | −0.57 | −0.60 | Avoids sugar and likes cheese, fish, potatoes. | Normal diet. | CHO: 42% Protein: 23% Fat: 35% | Soya milk. |
| 10 | FTTDCD | −1.05 | −0.58 | Avoids sugar. | Normal diet. | CHO: 45% Protein: 19% Fat: 36% | Not prescribed. |
| 11 | NICCD | −0.43 | −2.01 | Eats sugary foods. Dislikes cheese. | Low sugar, high protein, high fat diet. Casein hydrolysed formula with MCT. | CHO: 40% Protein: 11% Fat: 49% | Hydrolysed formula (55% MCT fat). |
| 12 | NICCD | −2.54 | −4.51 | Eats sweet puddings. | Casein hydrolysed formula with MCT. | CHO: 43% Protein: 12% Fat: 45% | Hydrolysed formula (55% MCT fat). |
| 13 | FTTDCD | −0.19 | −0.44 | Does not eat cake or biscuits; rarely eats rice, pasta and bread. Prefers high protein foods (yoghurt, milk, meat, eggs). | Low sugar, high protein/ high fat diet. | CHO: 47% Protein: 19% Fat: 38% | Not prescribed. |
| 14 (Family 3) | FTTDCD | 0.69 | 0.91 | Dislikes sweets. Avoids CHO and prefers high protein foods (meat/cheese/milk). | Normal diet. | CHO: 28% Protein: 26% Fat: 46% | Not prescribed. |
| 15 (Family 3) | Family screening | −0.16 | −0.08 | Dislikes sweets. Avoids vegetables and fruits. Craves high protein foods, particularly yoghurt. Likes cream. | Low sugar, high protein, high fat diet. | CHO: 32% Protein: 20% Fat: 48% | Full fat milk. |
| 16 | NICCD | −1.04 | 0.02 | Dislikes sweets. Avoids high CHO containing foods. Prefers high protein foods (egg, meat, cheese). | Normal diet. | CHO: 43% Protein: 17% Fat: 37% | Full fat milk. |
| 17 | Family screening | −2.71 | −1.70 | Avoids sugary foods. Likes yoghurt. | Lactose free diet (soya milk). | CHO: 41% Protein: 15% Fat: 47% | Soya milk. |
| 18 | NICCD | −0.26 | −0.22 | Self-selects a high protein and high fat diet. Dislikes sweets and fizzy drinks. | Low sugar, high protein, high fat diet. | CHO: 42% Protein: 19% Fat: 38% | Full fat milk, nuts. |
| 19 (Family 4) | Family screening | −0.89 | −0.40 | Likes CHO containing foods and sweets. | Normal diet. | CHO: 44% Protein: 16% Fat: 38% | Not prescribed. |
| 20 (Family 4) | FTTDCD | −1.25 | −0.53 | Dislikes CHO containing foods and fruit. Likes meat. | Normal diet. | CHO: 42% Protein: 15% Fat: 43% | Not prescribed. |
| 21 | FTTDCD | −1.41 | −1.74 | Dislikes fruit and vegetables. Prefers chicken. | Normal diet. | CHO: 46% Protein: 17% Fat: 35% | Not prescribed. |
| 22 | NICCD | −2.69 | −0.46 | Dislikes vegetables. Prefers cheese. | Low sugar, high protein, high fat diet. | CHO: 40% Protein: 17% Fat: 41% | Not prescribed. |
| 23 | Family screening | −3.47 | −1.04 | Likes high protein foods. | Low sugar, high protein, high fat diet. | CHO: 43% Protein: 14% Fat: 43% | Not prescribed. |
| 24 | NICCD | −2.97 | −1.29 | Dislikes rice, potatoes and pasta but likes chicken and fish. | Low sugar, high protein, high fat diet. | NA | Full fat milk. |
| 25 | NICCD | 0.22 | −0.43 | Likes meat. | Low sugar, high protein, high fat diet. | NA | Full fat milk. |
| 26 (Family 5) | Family screening | −2.29 | −2.12 | Avoids many CHO containing food, fruit and vegetables. Prefers high protein foods. | Normal diet. | CHO: 41% Protein: 22% Fat: 36% | Full fat milk. |
| 27 (Family 5) | Family screening | −0.64 | −0.15 | No food aversions. Likes dairy products. | Normal diet. | CHO: 44% Protein: 20% Fat: 36% | Full fat milk. |
| 28 (Family 5) | NICCD | −0.82 | −0.39 | No food aversions. Likes chicken. | Normal diet. | NA | Full fat milk. |
| 29 (Family 5) | Family screening | −0.99 | −1.38 | Likes chicken and cheese. Does not like dairy products. | Normal diet. | CHO: 49% Protein: 13% Fat: 38% | Full fat milk. |
| 30 (Family 5) | Family screening | −1.43 | −0.91 | No preferences. | Normal diet. | CHO: 48% Protein: 20% Fat: 32% | Full fat milk. |
| 31 (Family 5) | Family screening | −0.89 | −0.89 | Dislikes sweet foods. Poor tolerance to dairy foods. | Normal diet. | CHO: 31% Protein: 23% Fat: 42% | Full fat milk. |
| 32 | FTTDCD | −0.57 | −0.96 | Dislikes sugary foods. Prefers savoury foods. | Normal diet. | CHO: 37% Protein: 23% Fat: 38% | Soya milk, nuts. |
| Carbohydrates Median % [Range] | Protein Median % [Range] | Fat Median % [Range] | |
|---|---|---|---|
| Total (n = 27) | 42% [18–49] | 19% [11–27] | 40% [32–55] |
| NICCD (n = 9) | 42% [18–47] | 18% [11–27] | 41% [33–55] |
| FTTDCD (n = 7) | 42% [28–47] | 19% [15–26] | 38% [35–51] |
| Family screening (n = 11) | 42% [28–49] | 20% [13–23] | 40% [32–53] |
| Carbohydrate | Protein | Fat | |
|---|---|---|---|
| Normal diet (n = 16) | 43% [28–49] | 19% [13–26] | 38% [32–51] |
| Prescribed low-carbohydrate, high-protein, high fat diet (n = 11) | 40% [18–47] | 19% [11–27] | 45% [38–55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.; Ashmore, C.; Batzios, S.; Daly, A.; Dawson, C.; Dixon, M.; Evans, S.; Green, D.; Gribben, J.; Hunjan, I.; et al. Dietary Management, Clinical Status and Outcome of Patients with Citrin Deficiency in the UK. Nutrients 2020, 12, 3313. https://doi.org/10.3390/nu12113313
Pinto A, Ashmore C, Batzios S, Daly A, Dawson C, Dixon M, Evans S, Green D, Gribben J, Hunjan I, et al. Dietary Management, Clinical Status and Outcome of Patients with Citrin Deficiency in the UK. Nutrients. 2020; 12(11):3313. https://doi.org/10.3390/nu12113313
Chicago/Turabian StylePinto, Alex, Catherine Ashmore, Spyros Batzios, Anne Daly, Charlotte Dawson, Marjorie Dixon, Sharon Evans, Diane Green, Joanna Gribben, Inderdip Hunjan, and et al. 2020. "Dietary Management, Clinical Status and Outcome of Patients with Citrin Deficiency in the UK" Nutrients 12, no. 11: 3313. https://doi.org/10.3390/nu12113313
APA StylePinto, A., Ashmore, C., Batzios, S., Daly, A., Dawson, C., Dixon, M., Evans, S., Green, D., Gribben, J., Hunjan, I., Jameson, E., Newby, C., Pierre, G., Rajwal, S., Robertson, L., Santra, S., Sharrard, M., Vara, R., White, L., ... MacDonald, A. (2020). Dietary Management, Clinical Status and Outcome of Patients with Citrin Deficiency in the UK. Nutrients, 12(11), 3313. https://doi.org/10.3390/nu12113313