Mechanisms Underlying the Skin-Gut Cross Talk in the Development of IgE-Mediated Food Allergy

 ,
,  , , and
, , and 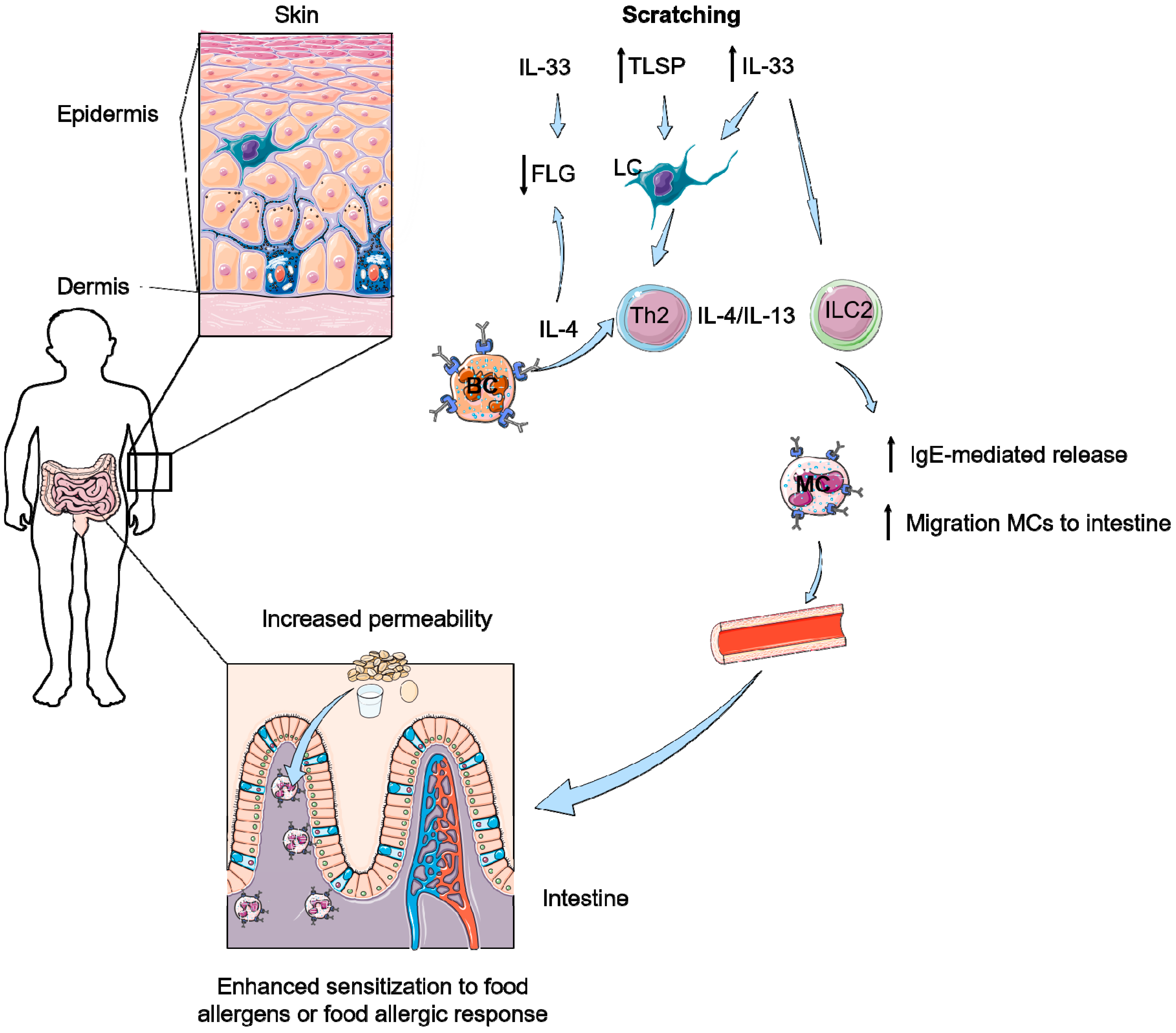{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Skin Barrier and Skin Sensitization
- Skin is an important permeability and immune barrier.
- Disrupted skin barrier leads to increased sensitization to food allergens in the skin.
3. Environmental Factors Induce Sensitization to Food Allergens via the Skin
- Detergents and environmental allergens, like house dust mite or Alternaria alternata allergens, can disrupt skin barrier.
- Cutaneous exposure of allergens prior to ingestion leads to increased sensitization.
- Tolerance is induced if allergens are ingested prior to cutaneous exposure.
4. TSLP-Mediated Type 2 Inflammation in the Skin
- Disrupted skin barrier leads to increased sensitization to food allergens in the skin. This process is mediated by TSLP-induced DC and basophils, producing IL-4 and resulting in enhanced type 2 responses.
5. Major Role for Type 2 Innate Lymphoid Cells (ILC2) and Epithelial Cytokines in the Development of Food Allergy
- Skin damage results in the release of IL-33, TSLP.
- Specifically, IL-33 cells activate DCs and ILC2 cells.
- Through activating ILC2 cells and DCs, epithelial cytokines, e.g., TSLP and IL-25, can mediate a type 2 inflammation reaction in an antigen independent manner.
6. How Can Pruritus Lead to Food-Induced Anaphylaxis?
- Scratching induces enhanced IL-33 levels in the skin and in serum.
- IL-33 together with IL-4 and Th2 cells are able to induce accumulation of mast cells and IL-9 producing mucosal mast cells (MMC9) in the intestine.
- IL-33 results in more IgE-mediated degranulation of these MCs and MMC9 cells, leading to food allergy.
- Scratching increases numbers of intestinal mast cells and increased permeability of the intestines resulting in the development of food allergy.
- A skin-to-gut axis is inevitable as food allergy symptoms in the intestine apparently can be induced by increased IL-33 levels in serum, which is induced by a damaged skin barrier due to scratching or AD.
7. The Role of Skin Microbiota in the Development of Food Allergy
- Staphylococcus aureus colonization is related to reduce microbial diversity in the skin and increased prevalence of atopic dermatitis and food allergy.
8. The Role of Intestinal Microbiota on the Development of Atopy and Atopic Dermatitis
- There is no conclusive evidence that specific microbial species are responsible for the development of allergy or atopic dermatitis.
- Short chain fatty acids produced by intestinal microbiota are linked to reduced allergic inflammation.
9. Future Human Research Priorities
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Glossary of Terms
| ATP | Adenosine triphosphate |
| AD | atopic dermatitis |
| DC | dendritic cell |
| FLG | Filaggrin |
| HDM | House dust mite |
| Ig | Immune globuline |
| IL | interleukin |
| ILC | innate lymphoid cells |
| LPS | lipopolysaccharide |
| MC | mast cells |
| MMC9 | IL-9-producing mucosal mast cell |
| OVA | ovalbumin |
| SCFA | short chain fatty acids |
| SDS | sodium dodecyl sulfate |
| S. aureus | Staphylococcus aureus |
| TEWL | transepidermal water loss |
| Th2 | T helper cell 2 |
| TSLP | thymic stromal lymphopoietin |
| Treg | regulatory T cell |
References
- Han, H.; Thelen, T.D.; Comeau, M.R.; Ziegler, S.F. Thymic stromal lymphopoietin-mediated epicutaneous inflammation promotes acute diarrhea and anaphylaxis. J. Clin. Investig. 2014, 124, 5442–5452. [Google Scholar] [CrossRef] [Green Version]
- Noti, M.; Wojno, E.D.T.; Kim, B.S.; Siracusa, M.C.; Giacomin, P.R.; Nair, M.G.; Benitez, A.J.; Ruymann, K.R.; Muir, A.B.; Hill, D.A.; et al. Thymic stromal lymphopoietin-elicited basophil responses promote eosinophilic esophagitis. Nat. Med. 2013, 19, 1005–1013. [Google Scholar] [CrossRef]
- Noti, M.; Kim, B.S.; Siracusa, M.C.; Rak, G.D.; Kubo, M.; Moghaddam, A.E.; Sattentau, Q.A.; Comeau, M.R.; Spergel, J.M.; Artis, D. Exposure to food allergens through inflamed skin promotes intestinal food allergy through the thymic stromal lymphopoietin-basophil axis. J. Allergy Clin. Immunol. 2014, 133, 1390–1399.e6. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Borcard, L.; Walsh, K.P.; Pena Rodriguez, M.; Mueller, C.; Kim, B.S.; Kubo, M.; Artis, D.; Noti, M. Basophil-derived IL-4 promotes epicutaneous antigen sensitization concomitant with the development of food allergy. J. Allergy Clin. Immunol. 2018, 141, 223–234.e5. [Google Scholar] [CrossRef] [Green Version]
- Lack, G. Early exposure hypothesis: Where are we now? Clin. Transl. Allergy 2011, 1, S71. [Google Scholar] [CrossRef] [Green Version]
- Brough, H.A.; Nadeau, K.C.; Sindher, S.B.; Alkotob, S.S.; Chan, S.; Bahnson, H.; Leung, D.Y.M.; Lack, G. Epicutaneous sensitization in the development of food allergy: What is the evidence and how can this be prevented? Allergy 2020, 75, 2185–2205. [Google Scholar] [CrossRef]
- Martin, P.E.; Eckert, J.K.; Koplin, J.J.; Lowe, A.J.; Gurrin, L.C.; Dharmage, S.C.; Vuillermin, P.; Tang, M.L.K.K.; Ponsonby, A.-L.L.; Matheson, M.; et al. Which infants with eczema are at risk of food allergy? Results from a population-based cohort. Clin. Exp. Allergy 2015, 45, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Xu, W.; Headley, M.B.; Jessup, H.K.; Lee, K.S.; Omori, M.; Comeau, M.R.; Marshak-Rothstein, A.; Ziegler, S.F. Thymic stromal lymphopoietin (TSLP)-mediated dermal inflammation aggravates experimental asthma. Mucosal Immunol. 2012, 5, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Leyva-Castillo, J.M.; Hener, P.; Jiang, H.; Li, M. TSLP Produced by Keratinocytes Promotes Allergen Sensitization through Skin and Thereby Triggers Atopic March in Mice. J. Investig. Dermatol. 2013, 133, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Hener, P.; Frossard, N.; Kato, S.; Metzger, D.; Li, M.; Chambon, P. Thymic stromal lymphopoietin overproduced by keratinocytes in mouse skin aggravates experimental asthma. Proc. Natl. Acad. Sci. USA 2009, 106, 1536–1541. [Google Scholar] [CrossRef] [Green Version]
- Brough, H.A.; Liu, A.H.; Sicherer, S.; Makinson, K.; Douiri, A.; Brown, S.J.; Stephens, A.C.; Irwin McLean, W.H.; Turcanu, V.; Wood, R.A.; et al. Atopic dermatitis increases the effect of exposure to peanut antigen in dust on peanut sensitization and likely peanut allergy. J. Allergy Clin. Immunol. 2015, 135, 164–170.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Toit, G.; Roberts, G.; Sayre, P.H.; Bahnson, H.T.; Radulovic, S.; Santos, A.F.; Brough, H.A.; Phippard, D.; Basting, M.; Feeney, M.; et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N. Engl. J. Med. 2015, 372, 803–813. [Google Scholar] [CrossRef] [Green Version]
- Du Toit, G.; Roberts, G.; Sayre, P.H.; Plaut, M.; Bahnson, H.T.; Mitchell, H.; Radulovic, S.; Chan, S.; Fox, A.; Turcanu, V.; et al. Identifying infants at high risk of peanut allergy: The Learning Early about Peanut Allergy (LEAP) screening study. J. Allergy Clin. Immunol. 2013, 131, 135–143.e12. [Google Scholar] [CrossRef]
- Natsume, O.; Kabashima, S.; Nakazato, J.; Yamamoto-Hanada, K.; Narita, M.; Kondo, M.; Saito, M.; Kishino, A.; Takimoto, T.; Inoue, E.; et al. Two-step egg introduction for prevention of egg allergy in high-risk infants with eczema (PETIT): A randomised, double-blind, placebo-controlled trial. Lancet 2017, 389, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Peavy, R.D.; Metcalfe, D.D. Understanding the mechanisms of anaphylaxis. Curr. Opin. Allergy Clin. Immunol. 2008, 8, 310–315. [Google Scholar] [CrossRef]
- Schleimer, R.P.; Berdnikovs, S. Etiology of epithelial barrier dysfunction in type 2 inflammatory disease. J. Allergy Clin. Immunol. 2017, 139, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egawa, G.; Kabashima, K. Multifactorial skin barrier deficiency and atopic dermatitis: Essential topics to prevent the atopic march. J. Allergy Clin. Immunol. 2016, 138, 350–358.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, D.J.; Hosking, C.S.; De Benedictis, F.M.; Oranje, A.P.; Diepgen, T.L.; Bauchau, V.; Warner, J.O.; Naspitz, C.K.; Simons, F.E.R.; Diepgen, T.L.; et al. Confirmation of the association between high levels of immunoglobulin E food sensitization and eczema in infancy: An international study. Clin. Exp. Allergy 2008, 38, 161–168. [Google Scholar] [CrossRef]
- Irvine, A.D.; McLean, W.H.I.; Leung, D.Y.M. Filaggrin mutations associated with skin and allergic diseases. N. Engl. J. Med. 2011, 365, 1315–1327. [Google Scholar] [CrossRef] [Green Version]
- Van Smeden, J.; Janssens, M.; Kaye, E.C.J.; Caspers, P.J.; Lavrijsen, A.P.; Vreeken, R.J.; Bouwstra, J.A. The importance of free fatty acid chain length for the skin barrier function in atopic eczema patients. Exp. Dermatol. 2014, 23, 45–52. [Google Scholar] [CrossRef]
- Kelleher, M.M.; Dunn-Galvin, A.; Hourihane, J.O.O.B.; Murray, D.; Campbell, L.E.; McLean, W.H.I.I.; Irvine, A.D. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J. Allergy Clin. Immunol. 2015, 135, 930–935.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelleher, M.M.; Dunn-Galvin, A.; Gray, C.; Murray, D.M.; Kiely, M.; Kenny, L.; McLean, W.H.I.; Irvine, A.D.; Hourihane, J.O. Skin barrier impairment at birth predicts food allergy at 2 years of age. J. Allergy Clin. Immunol. 2016, 137, 1111–1116.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.J.; Asai, Y.; Cordell, H.J.; Campbell, L.E.; Zhao, Y.; Liao, H.; Northstone, K.; Henderson, J.; Alizadehfar, R.; Ben-Shoshan, M.; et al. Loss-of-function variants in the filaggrin gene are a significant risk factor for peanut allergy. J. Allergy Clin. Immunol. 2011, 127, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusunoki, T.; Okafuji, I.; Yoshioka, T.; Saito, M.; Nishikomori, R.; Heike, T.; Sugai, M.; Shimizu, A.; Nakahata, T. SPINK5 polymorphism is associated with disease severity and food allergy in children with atopic dermatitis. J. Allergy Clin. Immunol. 2005, 115, 636–638. [Google Scholar] [CrossRef]
- Brough, H.A.; Simpson, A.; Makinson, K.; Hankinson, J.; Brown, S.; Douiri, A.; Belgrave, D.C.M.; Penagos, M.; Stephens, A.C.; McLean, W.H.I.; et al. Peanut allergy: Effect of environmental peanut exposure in children with filaggrin loss-of-function mutations. J. Allergy Clin. Immunol. 2014, 134, 867–875.e1. [Google Scholar] [CrossRef]
- Walker, M.T.; Green, J.E.; Ferrie, R.P.; Queener, A.M.; Kaplan, M.H.; Cook-Mills, J.M. Mechanism for initiation of food allergy: Dependence on skin barrier mutations and environmental allergen costimulation. J. Allergy Clin. Immunol. 2018, 141, 1711–1725.e9. [Google Scholar] [CrossRef] [Green Version]
- Akdis, C.A.; Arkwright, P.D.; Brüggen, M.C.; Busse, W.; Gadina, M.; Guttman-Yassky, E.; Kabashima, K.; Mitamura, Y.; Vian, L.; Wu, J.; et al. Type 2 immunity in the skin and lungs. Allergy 2020, 74, 1582–1605. [Google Scholar] [CrossRef]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet-Schiltz, O.; Gonzalez-De-Peredo, A.; Girard, J.P. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef]
- Landheer, J.; Giovannone, B.; Mattson, J.D.; Tjabringa, S.; Bruijnzeel-Koomen, C.A.F.M.; McClanahan, T.; De Waal Malefyt, R.; Knol, E.; Hijnen, D. Epicutaneous application of house dust mite induces thymic stromal lymphopoietin in nonlesional skin of patients with atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 1252–1254. [Google Scholar] [CrossRef]
- Strid, J.; Hourihane, J.; Kimber, I.; Callard, R.; Strobel, S. Epicutaneous exposure to peanut protein prevents oral tolerance and enhances allergic sensitization. Clin. Exp. Allergy 2005, 35, 757–766. [Google Scholar] [CrossRef]
- Leung, D.Y.M.; Calatroni, A.; Zaramela, L.S.; LeBeau, P.K.; Dyjack, N.; Brar, K.; David, G.; Johnson, K.; Leung, S.; Ramirez-Gama, M.; et al. The nonlesional skin surface distinguishes atopic dermatitis with food allergy as a unique endotype. Sci. Transl. Med. 2019, 11, eaav2685. [Google Scholar] [CrossRef]
- Pellerin, L.; Henry, J.; Hsu, C.-Y.; Balica, S.; Jean-Decoster, C.; Méchin, M.-C.; Hansmann, B.; Rodriguez, E.; Weindinger, S.; Schmitt, A.-M.; et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013, 131, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Reche, P.A.; Soumelis, V.; Gorman, D.M.; Clifford, T.; Liu, M.; Travis, M.; Zurawski, S.M.; Johnston, J.; Liu, Y.-J.; Spits, H.; et al. Human Thymic Stromal Lymphopoietin Preferentially Stimulates Myeloid Cells. J. Immunol. 2001, 167, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell–mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Oyoshi, M.K.; Larson, R.P.; Ziegler, S.F.; Geha, R.S. Mechanical injury polarizes skin dendritic cells to elicit a T(H)2 response by inducing cutaneous thymic stromal lymphopoietin expression. J. Allergy Clin. Immunol. 2010, 126, 976–984.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, B.E.; Lee, J.; Han, Y.; Jun, H.Y.; Kim, H.; Choi, J.; Leung, D.Y.M.; Ahn, K. Epidermal thymic stromal lymphopoietin predicts the development of atopic dermatitis during infancy. J. Allergy Clin. Immunol. 2016, 137, 1282–1285.e4. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.-H.; de Waal Malefyt, R.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An Epithelial Cell Cytokine that Regulates T Cell Differentiation by Conditioning Dendritic Cell Maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef]
- Ito, T.; Wang, Y.-H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.-F.; Yao, Z.; Cao, W.; Liu, Y.-J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Leyva-Castillo, J.M.; Hener, P.; Michea, P.; Karasuyama, H.; Chan, S.; Soumelis, V.; Li, M. Skin thymic stromal lymphopoietin initiates Th2 responses through an orchestrated immune cascade. Nat. Commun. 2013, 4, 2847. [Google Scholar] [CrossRef]
- Hsieh, K.-Y.; Tsai, C.-C.; Wu, C.H.H.; Lin, R.-H. Epicutaneous exposure to protein antigen and food allergy. Clin. Exp. Allergy 2003, 33, 1067–1075. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity 2015, 43, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Moltke, J.; Liang, H.-E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-B.; Chen, C.-Y.; Liu, B.; Mugge, L.; Angkasekwinai, P.; Facchinetti, V.; Dong, C.; Liu, Y.-J.; Rothenberg, M.E.; Hogan, S.P.; et al. IL-25 and CD4+Th2 cells enhance ILC2-derived IL-13 production that promotes IgE-mediated experimental food allergy. J. Allergy Clin. Immunol. 2016, 137, 1216–1225.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camelo, A.; Rosignoli, G.; Ohne, Y.; Stewart, R.A.; Overed-sayer, C.; Sleeman, M.A.; May, R.D. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv. 2017, 1, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Burton, O.T.; Wise, P.; Charbonnier, L.M.; Georgiev, P.; Oettgen, H.C.; Rachid, R.; Chatila, T.A. Regulatory T cell reprogramming toward a Th2-Cell-like lineage impairs oral tolerance and promotes food allergy. Immunity 2015, 42, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Burton, O.T.; Medina Tamayo, J.; Stranks, A.J.; Miller, S.; Koleoglou, K.J.; Weinberg, E.O.; Oettgen, H.C. IgE promotes type 2 innate lymphoid cells in murine food allergy. Clin. Exp. Allergy 2018, 48, 288–296. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Burton, O.T.; Oettgen, H.C.; Chatila, T. IL-4 production by group 2 innate lymphoid cells promotes food allergy by blocking regulatory T-cell function. J. Allergy Clin. Immunol. 2016, 138, 801–811.e9. [Google Scholar] [CrossRef] [Green Version]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimäki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in Atopic Dermatitis: Expression Profiles and Modulation by Triggering Factors. J. Investig. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.J.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa-Mineoka, R.; Okuzawa, Y.; Masuda, K.; Katoh, N. Increased serum levels of interleukin 33 in patients with atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Seltmann, J.; Roesner, L.M.; Von Hesler, F.W.; Wittmann, M.; Werfel, T. IL-33 impacts on the skin barrier by downregulating the expression of filaggrin. J. Allergy Clin. Immunol. 2015, 135, 1659–1661.e4. [Google Scholar] [CrossRef]
- Han, H.; Roan, F.; Johnston, L.K.; Smith, D.E.; Bryce, P.J.; Ziegler, S.F. IL-33 promotes gastrointestinal allergy in a TSLP-independent manner. Mucosal Immunol. 2018, 11, 394–403. [Google Scholar] [CrossRef]
- Galand, C.; Leyva-Castillo, J.M.; Yoon, J.; Han, A.; Lee, M.S.; McKenzie, A.N.J.; Stassen, M.; Oyoshi, M.K.; Finkelman, F.D.; Geha, R.S. IL-33 promotes food anaphylaxis in epicutaneously sensitized mice by targeting mast cells. J. Allergy Clin. Immunol. 2016, 138, 1356–1366. [Google Scholar] [CrossRef] [Green Version]
- Parveen, S.; Saravanan, D.B.; Saluja, R.; Elden, B.T. IL-33 mediated amplification of allergic response in human mast cells. J. Recept. Signal Transduct. 2019, 39, 359–367. [Google Scholar] [CrossRef]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef] [Green Version]
- Tordesillas, L.; Goswami, R.; Benedé, S.; Grishina, G.; Dunkin, D.; Järvinen, K.M.; Maleki, S.J.; Sampson, H.A.; Berin, M.C. Skin exposure promotes a Th2-dependent sensitization to peanut allergens. J. Clin. Investig. 2014, 124, 4965–4975. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegle, C.G.K.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung tissue homeostasis following acute influenza virus infection. Nat. Immunol. 2012, 12, 1045–1054. [Google Scholar] [CrossRef]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misery, L.; Brenaut, E.; Le Garrec, R.; Abasq, C.; Genestet, S.; Marcorelles, P.; Zagnoli, F. Neuropathic pruritus. Nat. Rev. Neurol. 2014, 10, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Seegräber, M.; Srour, J.; Walter, A.; Knop, M.; Wollenberg, A. Dupilumab for treatment of atopic dermatitis. Expert Rev. Clin. Pharmacol. 2018, 11, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, J.L.; Fluhr, J.W.; Fowler, A.J.; Bruckner, T.; Diepgen, T.L.; Williams, M.L. The Objective Severity Assessment of Atopic Dermatitis Score. Arch. Dermatol. 2003, 139, 1417–1422. [Google Scholar] [CrossRef] [Green Version]
- Leyva-Castillo, J.M.; Galand, C.; Kam, C.; Burton, O.; Gurish, M.; Musser, M.A.; Goldsmith, J.D.; Hait, E.; Nurko, S.; Brombacher, F.; et al. Mechanical Skin Injury Promotes Food Anaphylaxis by Driving Intestinal Mast Cell Expansion. Immunity 2019, 50, 1262–1275.e4. [Google Scholar] [CrossRef]
- Fleischer, D.M.; Bock, S.A.; Spears, G.C.; Wilson, C.G.; Miyazawa, N.K.; Gleason, M.C.; Gyorkos, E.A.; Murphy, J.R.; Atkins, D.; Leung, D.Y.M. Oral Food Challenges in Children with a Diagnosis of Food Allergy. J. Pediatr. 2011, 158, 578–583.e1. [Google Scholar] [CrossRef]
- Ahrens, R.; Osterfeld, H.; Wu, D.; Chen, C.-Y.; Arumugam, M.; Groschwitz, K.; Strait, R.; Wang, Y.-H.; Finkelman, F.D.; Hogan, S.P. Intestinal mast cell levels control severity of oral antigen-induced anaphylaxis in mice. Am. J. Pathol. 2012, 180, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Bartnikas, L.M.; Gurish, M.F.; Burton, O.T.; Leisten, S.; Janssen, E.; Oettgen, H.C.; Beaupré, J.; Lewis, C.N.; Austen, K.F.; Schulte, S.; et al. Epicutaneous sensitization results in IgE-dependent intestinal mast cell expansion and food-induced anaphylaxis. J. Allergy Clin. Immunol. 2013, 131, 451–460.e6. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Park, J.H.; Park, D.I.; Kim, H.J.; Cho, Y.K.; Sohn, C.I.; Jeon, W.K.; Kim, B.I.; Chae, S.W. Mucosal mast cell count is associated with intestinal permeability in patients with diarrhea predominant irritable bowel syndrome. J. Neurogastroenterol. Motil. 2013, 19, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Sampson, H.A.; O’Mahony, L.; Burks, A.W.; Plaut, M.; Lack, G.; Akdis, C.A. Mechanisms of food allergy. J. Allergy Clin. Immunol. 2018, 141, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodoun, M.V.; Tomar, S.; Tocker, J.E.; Wang, Y.H.; Finkelman, F.D. Prevention of food allergy development and suppression of established food allergy by neutralization of thymic stromal lymphopoietin, IL-25, and IL-33. J. Allergy Clin. Immunol. 2018, 141, 171–179.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurashima, Y.; Kiyono, H. New era for mucosal mast cells: Their roles in inflammation, allergic immune responses and adjuvant development. Exp. Mol. Med. 2014, 46, e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Lee, J.B.; Liu, B.; Ohta, S.; Wang, P.Y.; Kartashov, A.V.; Mugge, L.; Abonia, J.P.; Barski, A.; Izuhara, K.; et al. Induction of Interleukin-9-Producing Mucosal Mast Cells Promotes Susceptibility to IgE-Mediated Experimental Food Allergy. Immunity 2015, 43, 788–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, E.E.; Groschwitz, K.; Abonia, J.P.; Brandt, E.B.; Cohen, E.; Blanchard, C.; Ahrens, R.; Seidu, L.; McKenzie, A.; Strait, R.; et al. IL-9- and mast cell-mediated intestinal permeability predisposes to oral antigen hypersensitivity. J. Exp. Med. 2008, 205, 897–913. [Google Scholar] [CrossRef]
- Tomar, S.; Ganesan, V.; Sharma, A.; Zeng, C.; Waggoner, L.; Smith, A.; Kim, C.H.; Licona-Limón, P.; Reinhardt, R.L.; Flavell, R.A.; et al. IL-4-BATF signaling directly modulates IL-9 producing mucosal mast cell (MMC9) function in experimental food allergy. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Sehra, S.; Yao, W.; Nguyen, E.T.; Glosson-Byers, N.L.; Akhtar, N.; Zhou, B.; Kaplan, M.H. TH9 cells are required for tissue mast cell accumulation during allergic inflammation. J. Allergy Clin. Immunol. 2015, 136, 433–440.e1. [Google Scholar] [CrossRef] [Green Version]
- Chamlin, S.L.; Kao, J.; Frieden, I.J.; Sheu, M.Y.; Fowler, A.J.; Fluhr, J.W.; Williams, M.L.; Elias, P.M. Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: Changes in barrier function provide a sensitive indicator of disease activity. J. Am. Acad. Dermatol. 2002, 47, 198–208. [Google Scholar] [CrossRef]
- Leung, D.Y.M. Atopic dermatitis: New insights and opportunities for therapeutic intervention. J. Allergy Clin. Immunol. 2000, 105, 860–876. [Google Scholar] [CrossRef]
- Elias, P.M.; Feingold, K.R. Does the tail wag the dog? Role of the barrier in the pathogenesis of inflammatory dermatoses and therapeutic implications. Arch. Dermatol. 2001, 137, 1079–1081. [Google Scholar]
- Kennedy, E.A.; Connolly, J.; Hourihane, J.O.; Fallon, P.G.; McLean, W.H.I.; Murray, D.; Jo, J.-H.; Segre, J.A.; Kong, H.H.; Irvine, A.D. Skin microbiome before development of atopic dermatitis: Early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J. Allergy Clin. Immunol. 2017, 139, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; Mullikin, J.; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudikoff, D.; Lebwohl, M. Atopic dermatitis. Lancet 1998, 351, 1715–1721. [Google Scholar] [CrossRef]
- Bunikowski, R.; Mielke, M.E.A.; Skarabis, H.; Worm, M.; Anagnostopoulos, I.; Kolde, G.; Wahn, U.; Renz, H. Evidence for a disease-promoting effect of Staphylococcus aureus-derived exotoxins in atopic dermatitis. J. Allergy Clin. Immunol. 2000, 105, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Tsilochristou, O.; du Toit, G.; Sayre, P.H.; Roberts, G.; Lawson, K.; Sever, M.L.; Bahnson, H.T.; Radulovic, S.; Basting, M.; Plaut, M.; et al. Association of Staphylococcus aureus colonization with food allergy occurs independently of eczema severity. J. Allergy Clin. Immunol. 2019, 144, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.C.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.-C.; Xing, Z.; Berin, C.M.; Soderholm, J.D.; Feng, B.-S.; Wu, L.; Yeh, C. TIM-4 expressed by mucosal dendritic cells plays a critical role in food antigen-specific Th2 differentiation and intestinal allergy. Gastroenterology 2007, 133, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Salem, I.; Ramser, A.; Isham, N.; Ghannoum, M.A. The gut microbiome as a major regulator of the gut-skin axis. Front. Microbiol. 2018, 9, 1459. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-Y.; Lee, E.; Park, Y.M.; Hong, S.J. Microbiome in the gut-skin axis in atopic dermatitis. Allergy Asthma Immunol. Res. 2018, 10, 354–362. [Google Scholar] [CrossRef]
- O’Neill, C.A.; Monteleone, G.; McLaughlin, J.T.; Paus, R. The gut-skin axis in health and disease: A paradigm with therapeutic implications. BioEssays 2016, 38, 1167–1176. [Google Scholar] [CrossRef]
- Esposito, S.; Isidori, C.; Pacitto, A.; Salvatori, C.; Sensi, L.; Frati, F.; Di Cara, G.; Marcucci, F. Epicutaneous immunotherapy in rhino-conjunctivitis and food allergies: A review of the literature. J. Transl. Med. 2018, 16, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdijk, O.; Marsland, B.J. The microbiome: Toward preventing allergies and asthma by nutritional intervention. Curr. Opin. Immunol. 2019, 60, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Thijs, C.; van den Brandt, P.A.; Kummeling, I.; Snijders, B.; Stelma, F.; Adams, H.; van Ree, R.; Stobberingh, E.E. Gut microbiota composition and development of atopic manifestations in infancy: The KOALA Birth Cohort Study. Gut 2007, 56, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Björkstén, B.; Naaber, P.; Sepp, E.; Mikelsaar, M. The intestinal microflora in allergic Estonian and Swedish 2-year-old children. Clin. Exp. Allergy 1999, 29, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Björkstén, B.; Sepp, E.; Julge, K.; Voor, T.; Mikelsaar, M. Allergy development and the intestinal microflora during the first year of life. J. Allergy Clin. Immunol. 2001, 108, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Yap, G.C.; Loo, E.X.L.; Aw, M.; Lu, Q.; Shek, L.P.C.; Lee, B.W. Molecular analysis of infant fecal microbiota in an Asian at-risk cohort-correlates with infant and childhood eczema. BMC Res. Notes 2014, 7, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepp, E.; Julge, K.; Mikelsaar, M.; Björkstén, B. Intestinal microbiota and immunoglobulin E responses in 5-year-old Estonian children. Clin. Exp. Allergy 2005, 35, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Adlerberth, I.; Strachan, D.P.; Matricardi, P.M.; Ahrné, S.; Orfei, L.; Åberg, N.; Perkin, M.R.; Tripodi, S.; Hesselmar, B.; Saalman, R.; et al. Gut microbiota and development of atopic eczema in 3 European birth cohorts. J. Allergy Clin. Immunol. 2007, 120, 343–350. [Google Scholar] [CrossRef]
- Kalliomäki, M.; Kirjavainen, P.; Eerola, E.; Kero, P.; Salminen, S.; Isolauri, E. Distinct patterns of neonatal gut microflora in infants in whom atopy was and was not developing. J. Allergy Clin. Immunol. 2001, 107, 129–134. [Google Scholar] [CrossRef]
- Watanabe, S.; Narisawa, Y.; Arase, S.; Okamatsu, H.; Ikenaga, T.; Tajiri, Y.; Kumemura, M. Differences in fecal microflora between patients with atopic dermatitis and healthy control subjects. J. Allergy Clin. Immunol. 2003, 111, 587–591. [Google Scholar] [CrossRef]
- Johansson, M.A.; Sjögren, Y.M.; Persson, J.O.; Nilsson, C.; Sverremark-Ekström, E. Early colonization with a group of Lactobacilli decreases the risk for allergy at five years of age despite allergic heredity. PLoS ONE 2011, 6, e23031. [Google Scholar] [CrossRef] [PubMed]
- Kendler, M.; Uter, W.; Rueffer, A.; Shimshoni, R.; Jecht, E. Comparison of fecal microflora in children with atopic eczema/dermatitis syndrome according to IgE sensitization to food. Pediatr. Allergy Immunol. 2006, 17, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Gore, C.; Munro, K.; Lay, C.; Bibiloni, R.; Morris, J.; Woodcock, A.; Custovic, A.; Tannock, G.W. Bifidobacterium pseudocatenulatum is associated with atopic eczema: A nested case-control study investigating the fecal microbiota of infants. J. Allergy Clin. Immunol. 2008, 121, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.K.; Skov, T.; Paludan-Müller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011, 128, 646–652. [Google Scholar] [CrossRef]
- Storrø, O.; Øien, T.; Langsrud, O.; Rudi, K.; Dotterud, C.; Johnsen, R. Temporal variations in early gut microbial colonization are associated with allergen-specific immunoglobulin E but not atopic eczema at 2 years of age. Clin. Exp. Allergy 2011, 41, 1545–1554. [Google Scholar] [CrossRef]
- Wang, M.; Karlsson, C.; Olsson, C.; Adlerberth, I.; Wold, A.E.; Strachan, D.P.; Martricardi, P.M.; Åberg, N.; Perkin, M.R.; Tripodi, S.; et al. Reduced diversity in the early fecal microbiota of infants with atopic eczema. J. Allergy Clin. Immunol. 2008, 121, 129–134. [Google Scholar] [CrossRef]
- Ismail, I.H.; Oppedisano, F.; Joseph, S.J.; Boyle, R.J.; Licciardi, P.V.; Robins-Browne, R.M.; Tang, M.L.K. Reduced gut microbial diversity in early life is associated with later development of eczema but not atopy in high-risk infants. Pediatr. Allergy Immunol. 2012, 23, 674–681. [Google Scholar] [CrossRef]
- Kalliomäki, M.; Salminen, S.; Arvilommi, H.; Kero, P.; Koskinen, P.; Isolauri, E. Probiotics in primary prevention of atopic disease: A randomised placebo-controlled trial. Lancet 2001, 357, 1076–1079. [Google Scholar] [CrossRef]
- Kalliomäki, M.; Salminen, S.; Poussa, T.; Arvilommi, H.; Isolauri, E. Probiotics and prevention of atopic disease: 4-year follow-up of a randomised placebo-controlled trial. Lancet 2003, 361, 1869–1871. [Google Scholar] [CrossRef]
- Rautava, S.; Kainonen, E.; Salminen, S.; Isolauri, E. Maternal probiotic supplementation during pregnancy and breast-feeding reduces the risk of eczema in the infant. J. Allergy Clin. Immunol. 2012, 130, 1355–1360. [Google Scholar] [CrossRef]
- Enomoto, T.; Sowa, M.; Nishimori, K.; Shimazu, S.; Yoshida, A.; Yamada, K.; Furukawa, F.; Nakagawa, T.; Yanagisawa, N.; Iwabuchi, N.; et al. Effects of bifidobacterial supplementation to pregnant women and infants in the prevention of allergy development in infants and on fecal microbiota. Allergol. Int. 2014, 63, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.; Gerhold, K.; Zimmermann, K.; Ockeloen, C.W.; Rossberg, S.; Wagner, P.; Sulser, C.; Bunikowski, R.; Witt, I.; Wauer, J.; et al. Oral application of bacterial lysate in infancy decreases the risk of atopic dermatitis in children with 1 atopic parent in a randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 2012, 129, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kwon, J.H.; Ahn, S.H.; Lee, S.I.; Han, Y.S.; Choi, Y.O.; Lee, S.Y.; Ahn, K.M.; Ji, G.E. Effect of probiotic mix (Bifidobacterium bifidum, Bifidobacterium lactis, Lactobacillus acidophilus) in the primary prevention of eczema: A double-blind, randomized, placebo-controlled trial. Pediatr. Allergy Immunol. 2010, 21, e386–e393. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, R.J.; Brantsæter, A.L.; Magnus, M.C.; Haugen, M.; Myhre, R.; Jacobsson, B.; Longnecker, M.P.; Meltzer, H.M.; London, S.J. Probiotic milk consumption in pregnancy and infancy and subsequent childhood allergic diseases. J. Allergy Clin. Immunol. 2014, 133, 165–171.e8. [Google Scholar] [CrossRef] [Green Version]
- Jeong, K.; Kim, M.; Jeon, S.A.; Kim, Y.H.; Lee, S. A randomized trial of Lactobacillus rhamnosus IDCC 3201 tyndallizate (RHT3201) for treating atopic dermatitis. Pediatr. Allergy Immunol. 2020, 31, 783–792. [Google Scholar] [CrossRef]
- Abrahamsson, T.R.; Jakobsson, T.; Böttcher, M.F.; Fredrikson, M.; Jenmalm, M.C.; Björkstén, B.; Oldaeus, G. Probiotics in prevention of IgE-associated eczema: A double-blind, randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 2007, 119, 1174–1180. [Google Scholar] [CrossRef]
- Taylor, A.L.; Dunstan, J.A.; Prescott, S.L. Probiotic supplementation for the first 6 months of life fails to reduce the risk of atopic dermatitis and increases the risk of allergen sensitization in high-risk children: A randomized controlled trial. J. Allergy Clin. Immunol. 2007, 119, 184–191. [Google Scholar] [CrossRef]
- Chapat, L.; Chemin, K.; Dubois, B.; Bourdet-Sicard, R.; Kaiserlian, D. Lactobacillus casei reduces CD8+ T cell-mediated skin inflammation. Eur. J. Immunol. 2004, 34, 2520–2528. [Google Scholar] [CrossRef]
- Oyoshi, M.K.; Elkhal, A.; Scott, J.E.; Wurbel, M.; Hornick, J.L.; Campbell, J.J.; Geha, R.S. Epicutaneous challenge of orally immunized mice redirects antigen-specific gut-homing T cells to the skin. J. Clin. Investig. 2011, 121, 2210–2220. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Turcanu, V.; Stephens, A.C.; Fox, A.T.; Grieve, A.P.; Lack, G. Cutaneous lymphocyte antigen and α4β7 T-lymphocyte responses are associated with peanut allergy and tolerance in children. Allergy Eur. J. Allergy Clin. Immunol. 2012, 67, 336–342. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A.N.; McKenzie, C.I.; Shen, S.; Stanley, D.; MacIa, L.; Mason, L.J.; Roberts, L.K.; Wong, C.H.Y.; Shim, R.; Robert, R.; et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat. Commun. 2015, 6, 7320. [Google Scholar] [CrossRef] [PubMed]
- Thio, C.L.P.; Chi, P.Y.; Lai, A.C.Y.; Chang, Y.J. Regulation of type 2 innate lymphoid cell–dependent airway hyperreactivity by butyrate. J. Allergy Clin. Immunol. 2018, 142, 1867–1883.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, J.A.; Zhang, L.J.; Williams, M.R.; Gangoiti, J.A.; Huang, C.M.; Gallo, R.L. Inhibition of HDAC8 and HDAC9 by microbial short-chain fatty acids breaks immune tolerance of the epidermis to TLR ligands. Sci. Immunol. 2016, 1, eaah4609. [Google Scholar] [CrossRef]
- Schwarz, A.; Bruhs, A.; Schwarz, T. The Short-Chain Fatty Acid Sodium Butyrate Functions as a Regulator of the Skin Immune System. J. Investig. Dermatol. 2017, 137, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Roduit, C.; Frei, R.; Ferstl, R.; Loeliger, S.; Westermann, P.; Rhyner, C.; Schiavi, E.; Barcik, W.; Rodriguez-Perez, N.; Wawrzyniak, M.; et al. High levels of butyrate and propionate in early life are associated with protection against atopy. Allergy Eur. J. Allergy Clin. Immunol. 2019, 74, 799–809. [Google Scholar] [CrossRef]
- Folkerts, J.; Redegeld, F.; Folkerts, G.; Blokhuis, B.; van den Berg, M.P.M.; de Bruijn, M.J.W.; van IJcken, W.F.J.; Junt, T.; Tam, S.; Galli, S.J.; et al. Butyrate inhibits human mast cell activation via epigenetic regulation of FcεRI-mediated signaling. Allergy 2020, 75, 1966–1978. [Google Scholar] [CrossRef]
- Xian, M.; Wawrzyniak, P.; Rückert, B.; Duan, S.; Meng, Y.; Sokolowska, M.; Globinska, A.; Zhang, L.; Akdis, M.; Akdis, C.A. Anionic surfactants and commercial detergents decrease tight junction barrier integrity in human keratinocytes. J. Allergy Clin. Immunol. 2016, 138, 890–893.e9. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Splunter, M.; Liu, L.; van Neerven, R.J.J.; Wichers, H.J.; Hettinga, K.A.; de Jong, N.W. Mechanisms Underlying the Skin-Gut Cross Talk in the Development of IgE-Mediated Food Allergy. Nutrients 2020, 12, 3830. https://doi.org/10.3390/nu12123830
van Splunter M, Liu L, van Neerven RJJ, Wichers HJ, Hettinga KA, de Jong NW. Mechanisms Underlying the Skin-Gut Cross Talk in the Development of IgE-Mediated Food Allergy. Nutrients. 2020; 12(12):3830. https://doi.org/10.3390/nu12123830
Chicago/Turabian Stylevan Splunter, Marloes, Liu Liu, R.J. Joost van Neerven, Harry J. Wichers, Kasper A. Hettinga, and Nicolette W. de Jong. 2020. "Mechanisms Underlying the Skin-Gut Cross Talk in the Development of IgE-Mediated Food Allergy" Nutrients 12, no. 12: 3830. https://doi.org/10.3390/nu12123830
APA Stylevan Splunter, M., Liu, L., van Neerven, R. J. J., Wichers, H. J., Hettinga, K. A., & de Jong, N. W. (2020). Mechanisms Underlying the Skin-Gut Cross Talk in the Development of IgE-Mediated Food Allergy. Nutrients, 12(12), 3830. https://doi.org/10.3390/nu12123830