Sweet but Bitter: Focus on Fructose Impact on Brain Function in Rodent Models




{kind=link}
{kind=link}
Abstract
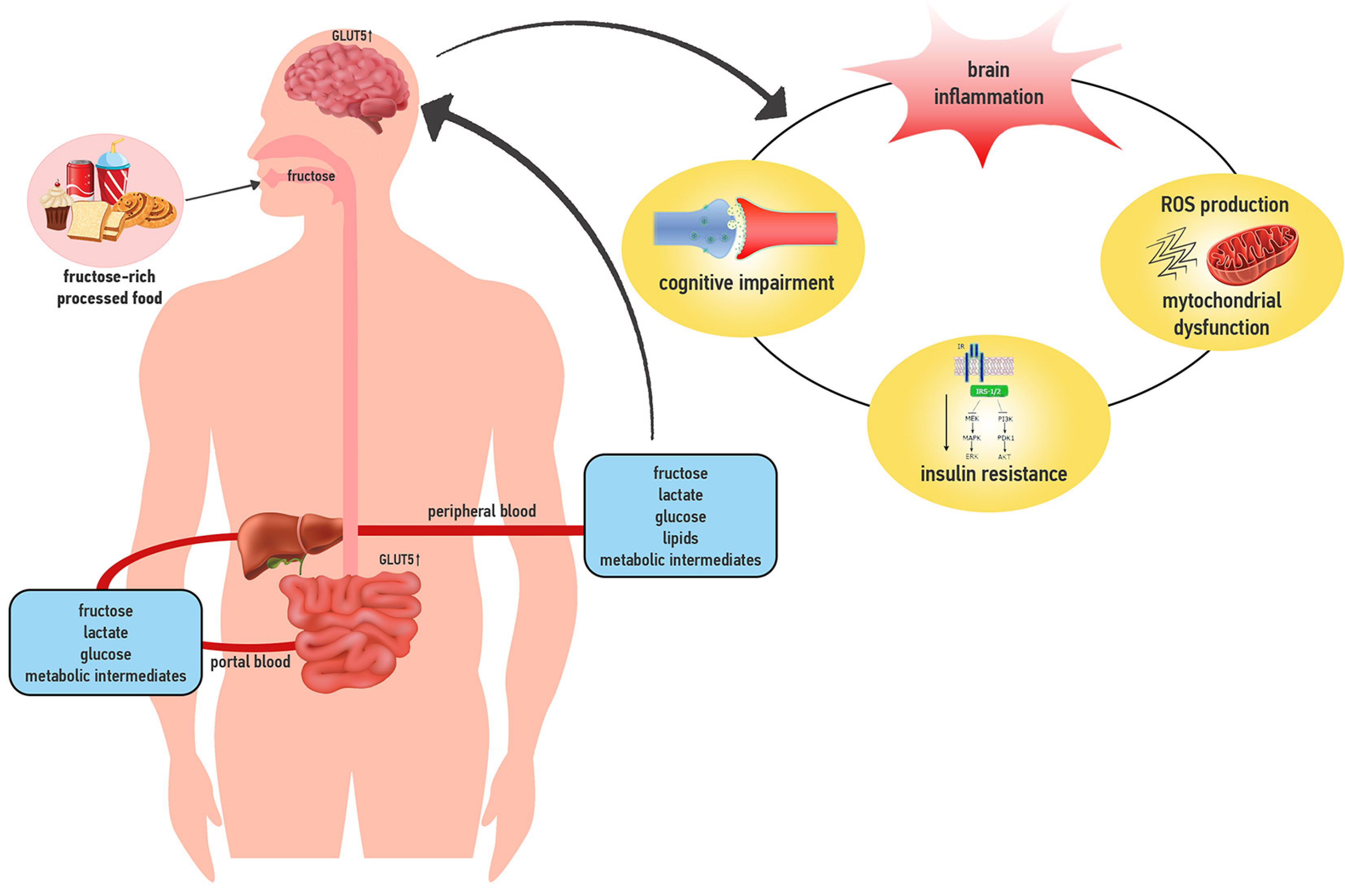
:1. Introduction
2. Fructose Effects on the Regulation of Food Intake
3. Fructose and Neuroinflammation
4. Fructose, Brain Mitochondrial Function and Oxidative Stress
5. Fructose and Insulin Signaling
6. Fructose, Cognitive Function, Aging and Neurodegenerative Diseases
7. Direct Effects of Fructose on the Brain
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Soft drink consumption and obesity: It is all about fructose. Curr. Opin. Lipidol. 2010, 21, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L. Role of fructose-containing sugars in the epidemics of obesity and metabolic syndrome. Annu. Rev. Med. 2012, 63, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.B.; Manson, J.E.; Ludwig, D.S.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 2004, 292, 927–934. [Google Scholar] [CrossRef]
- Livesey, G.; Taylor, R. Fructose consumption and consequences for glycation, plasma triacylglycerol, and body weight: Meta-analyses and meta-regression models of intervention studies. Am. J. Clin. Nutr. 2008, 88, 1419–1437. [Google Scholar] [CrossRef]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collino, M.; Rogazzo, M.; Medana, C.; Nigro, D.; Boccuzzi, G.; Aragno, M. Advanced glycation end products promote hepatosteatosis by interfering with SCAP-SREBP pathway in fructose-drinking mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G398–G407. [Google Scholar] [CrossRef] [Green Version]
- Bray, G.A. Fructose: Should we worry? Int. J. Obes. 2008, 32 (Suppl. 7), S127–S131. [Google Scholar] [CrossRef] [Green Version]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Campos, V.C.; Tappy, L. Physiological handling of dietary fructose-containing sugars: Implications for health. Int. J. Obes. 2016, 40 (Suppl. 1), S6–S11. [Google Scholar] [CrossRef]
- Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.M.; Jiao, R.Q.; Kong, L.D. High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients 2017, 9, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappy, L. Fructose-containing caloric sweeteners as a cause of obesity and metabolic disorders. J. Exp. Biol. 2018, 221 (Suppl. 1), jeb164202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappy, L. Fructose metabolism and noncommunicable diseases: Recent findings and new research perspectives. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.C.; Martin, R.J.; Whitney, M.L.; Edwards, G.L. Intracerebroventricular injection of fructose stimulates feeding in rats. Nutr. Neurosci. 2002, 5, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.H.; Wolfgang, M.; Tokutake, Y.; Chohnan, S.; Lane, M.D. Differential effects of central fructose and glucose on hypothalamic malonyl-coa and food intake. Proc. Natl. Acad. Sci. USA 2008, 105, 16871–16875. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, M.; Malbert, C.H.; Lallès, J.P.; Bobillier, E.; Val-Laillet, D. Effects of chronic intake of starch-, glucose- and fructose-containing diets on eating behaviour in adult minipigs. Appl. Anim. Behav. Sci. 2014, 157, 61–71. [Google Scholar] [CrossRef]
- Lane, M.D.; Cha, S.H. Effect of glucose and fructose on food intake via malonyl-CoA signaling in the brain. Biochem. Biophys. Res. Commun. 2009, 382, 1–5. [Google Scholar] [CrossRef]
- Shapiro, A.; Mu, W.; Roncal, C.; Cheng, K.Y.; Johnson, R.J.; Scarpace, P.J. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1370–R1375. [Google Scholar] [CrossRef] [Green Version]
- Teff, K.L.; Elliott, S.S.; Tschop, M.; Kieffer, T.J.; Rader, D.; Heiman, M.; Townsend, R.R.; Keim, N.L.; D’Alessio, D.; Havel, P.J. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Havel, P.J. Endocrine and metabolic effects of consuming beverages sweetened with fructose, glucose, sucrose, or high-fructose corn syrup. Am. J. Clin. Nutr. 2008, 88, 1733S–1737S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, K.A.; Chan, O.; Arora, J.; Belfort-DeAguiar, R.; Dzuira, J.; Roehmholdt, B.; Cline, G.W.; Naik, S.; Sinha, R.; Constable, R.T.; et al. Effects of fructose vs glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA 2013, 309, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindqvist, A.; Baelemans, A.; Erlanson-Albertsson, C. Effects of sucrose, glucose and fructose on peripheral and central appetite signals. Regul. Pept. 2008, 150, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bursać, B.N.; Vasiljević, A.D.; Nestorović, N.M.; Veličković, N.A.; Vojnović Milutinović, D.D.; Matić, G.M.; Djordjevic, A.D. High-fructose diet leads to visceral adiposity and hypothalamic leptin resistance in male rats-Do glucocorticoids play a role? J. Nutr. Biochem. 2014, 25, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Duan, W.; Guo, Z. Meal size and frequency affect neuronal plasticity and vulnerability to disease: Cellular and molecular mechanisms. J. Neurochem. 2003, 84, 417–431. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Dong, F.; Ren, J.; Driscoll, M.J.; Culver, B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp. Neurol. 2005, 191, 318–325. [Google Scholar] [CrossRef]
- Souza, C.G.; Moreira, J.D.; Siqueira, I.R.; Pereira, A.G.; Rieger, D.K.; Souza, D.O.; Souza, T.M.; Portela, L.V.; Perry, M.L. Highly palatable diet consumption increases protein oxidation in rat frontal cortex and anxiety-like behavior. Life Sci. 2007, 81, 198–203. [Google Scholar] [CrossRef]
- Crescenzo, R.; Spagnuolo, M.S.; Cancelliere, R.; Iannotta, L.; Mazzoli, A.; Gatto, C.; Iossa, S.; Cigliano, L. Effect of Initial Aging and High-Fat/High-Fructose Diet on Mitochondrial Bioenergetics and Oxidative Status in Rat Brain. Mol. Neurobiol. 2019, 56, 7651–7663. [Google Scholar] [CrossRef]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.R.; Haley-Zitlin, V.; Rosenberger, D.S.; Granholm, A.C. Damaging effects of a high-fat diet to the brain and cognition: A review of proposed mechanisms. Nutr. Neurosci. 2014, 17, 241–251. [Google Scholar] [CrossRef]
- Estrada, J.A.; Contreras, I. Nutritional Modulation of Immune and Central Nervous System Homeostasis: The Role of Diet in Development of Neuroinflammation and Neurological Disease. Nutrients 2019, 11, 1076. [Google Scholar] [CrossRef] [Green Version]
- Mazzoli, A.; Spagnuolo, M.S.; Gatto, C.; Nazzaro, M.; Cancelliere, R.; Crescenzo, R.; Iossa, S.; Cigliano, L. Adipose Tissue and Brain Metabolic Responses to Western Diet-Is There a Similarity between the Two? Int. J. Mol. Sci. 2020, 21, 786. [Google Scholar] [CrossRef] [Green Version]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Liverini, G.; Iossa, S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur. J. Nutr. 2013, 52, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Luccia, B.; Crescenzo, R.; Mazzoli, A.; Cigliano, L.; Venditti, P.; Walser, J.C.; Widmer, A.; Baccigalupi, L.; Ricca, E.; Iossa, S. Rescue of Fructose-Induced Metabolic Syndrome by Antibiotics or Faecal Transplantation in a Rat Model of Obesity. PLoS ONE 2015, 10, e0134893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.B.; Meng, Y.H.; Chang, S.; Zhang, R.Y.; Shi, C. High fructose causes cardiac hypertrophy via mitochondrial signaling pathway. Am. J. Transl. Res. 2016, 8, 4869–4880. [Google Scholar] [PubMed]
- Wang, W.; Ding, X.Q.; Gu, T.T.; Song, L.; Li, J.M.; Xue, Q.C.; Kong, L.D. Pterostilbene and allopurinol reduce fructose-induced podocyte oxidative stress and inflammation via microrna-377. Free Radic. Biol. Med. 2015, 83, 214–226. [Google Scholar] [CrossRef]
- Crescenzo, R.; Mazzoli, A.; Di Luccia, B.; Bianco, F.; Cancelliere, R.; Cigliano, L.; Liverini, G.; Baccigalupi, L.; Iossa, S. Dietary fructose causes defective insulin signalling and ceramide accumulation in the liver that can be reversed by gut microbiota modulation. Food Nutr. Res. 2017, 61, 1331657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crescenzo, R.; Cigliano, L.; Mazzoli, A.; Cancelliere, R.; Carotenuto, R.; Tussellino, M.; Liverini, G.; Iossa, S. Early Effects of a Low Fat, Fructose-Rich Diet on Liver Metabolism, Insulin Signaling, and Oxidative Stress in Young and Adult Rats. Front. Physiol. 2018, 9, 411. [Google Scholar] [CrossRef] [Green Version]
- Cigliano, L.; Spagnuolo, M.S.; Crescenzo, R.; Cancelliere, R.; Iannotta, L.; Mazzoli, A.; Liverini, G.; Iossa, S. Short-Term Fructose Feeding Induces Inflammation and Oxidative Stress in the Hippocampus of Young and Adult Rats. Mol. Neurobiol. 2018, 55, 2869–2883. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Pallottini, V.; Mazzoli, A.; Iannotta, L.; Tonini, C.; Morone, B.; Ståhlman, M.; Crescenzo, R.; Strazzullo, M.; Iossa, S.; et al. A Short-Term Western Diet Impairs Cholesterol Homeostasis and Key Players of Beta Amyloid Metabolism in Brain of Middle Aged Rats. Mol. Nutr. Food Res. 2020, e2000541. [Google Scholar] [CrossRef]
- De Sousa Rodrigues, M.E.; Bekhbat, M.; Houser, M.C.; Chang, J.; Walker, D.I.; Jones, D.P.; Oller do Nascimento, C.M.; Barnum, C.J.; Tansey, M.G. Chronic psychological stress and high-fat high-fructose diet disrupt metabolic and inflammatory gene networks in the brain, liver, and gut and promote behavioral deficits in mice. Brain Behav. Immun. 2017, 59, 158–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.M.; Ge, C.X.; Xu, M.X.; Wang, W.; Yu, R.; Fan, C.Y.; Kong, L.D. Betaine recovers hypothalamic neural injury by inhibiting astrogliosis and inflammation in fructose-fed rats. Mol. Nutr. Food Res. 2015, 59, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 2015, 25, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.X.; Yu, R.; Shao, L.F.; Zhang, Y.X.; Ge, C.X.; Liu, X.M.; Wu, W.Y.; Li, J.M.; Kong, L.D. Up-regulated fractalkine (FKN) and its receptor CX3CR1 are involved in fructose-induced neuroinflammation: Suppression by curcumin. Brain Behav. Immun. 2016, 58, 69–81. [Google Scholar] [CrossRef]
- Yin, Q.; Ma, Y.; Hong, Y.; Hou, X.; Chen, J.; Shen, C.; Sun, M.; Shang, Y.; Dong, S.; Zeng, Z.; et al. Lycopene attenuates insulin signaling deficits, oxidative stress, neuroinflammation, and cognitive impairment in fructose-drinking insulin resistant rats. Neuropharmacology 2014, 86, 389–396. [Google Scholar] [CrossRef]
- Djordjevic, A.; Bursać, B.; Veličković, N.; Vasiljević, A.; Matić, G. The impact of different fructose loads on insulin sensitivity, inflammation, and PSA-NCAM-mediated plasticity in the hippocampus of fructose-fed male rats. Nutr. Neurosci. 2015, 18, 66–75. [Google Scholar] [CrossRef]
- Harrell, C.S.; Zainaldin, C.; McFarlane, D.; Hyer, M.M.; Stein, D.; Sayeed, I.; Neigh, G.N. High-fructose diet during adolescent development increases neuroinflammation and depressive-like behavior without exacerbating outcomes after stroke. Brain Behav. Immun. 2018, 73, 340–351. [Google Scholar] [CrossRef]
- Jiménez-Maldonado, A.; Ying, Z.; Byun, H.R.; Gomez-Pinilla, F. Short-term fructose ingestion affects the brain independently from establishment of metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 24–33. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Bergamo, P.; Crescenzo, R.; Iannotta, L.; Treppiccione, L.; Iossa, S.; Cigliano, L. Brain Nrf2 Pathway, Autophagy, and Synaptic Function Proteins Are Modulated by a Short-Term Fructose Feeding in Young and Adult Rats. Nutr. Neurosci. 2020, 23, 309–320. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodeg. 2020, 15, 30. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, R.; Noble, E.; Vergnes, L.; Ying, Z.; Reue, K.; Gomez-Pinilla, F. Dietary fructose aggravates the pathobiology of traumatic brain injury by influencing energy homeostasis and plasticity. J. Cereb. Blood Flow Metab. 2016, 36, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Nigro, D.; Cento, A.S.; Chiazza, F.; Collino, M.; Aragno, M. High-fructose intake as risk factor for neurodegeneration: Key role for carboxy methyllysine accumulation in mice hippocampal neurons. Neurobiol. Dis. 2016, 89, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Macongonde, E.A.; Costa, N.L.; Ferreira, B.K.; Biella, M.S.; Frederico, M.J.; Oliveira, M.R.; Ávila Júnior, S.; Silva, F.R.; Ferreira, G.C.; Streck, E.L.; et al. Neutrotoxic effects of fructose administration in rat brain: Implications for fructosemia. An. Acad. Bras. Cienc. 2015, 87 (Suppl. 2), 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, O.H.; Larsen, L.H.; Ørstrup, L.K.; Hansen, L.H.; Grunnet, N.; Quistorff, B. Developmental programming by high fructose decreases phosphorylation efficiency in aging offspring brain mitochondria, correlating with enhanced UCP5 expression. J. Cereb. Blood Flow Metab. 2014, 34, 1205–1211. [Google Scholar] [CrossRef]
- Wu, C.W.; Hung, C.Y.; Hirase, H.; Tain, Y.L.; Lee, W.C.; Chan, J.Y.H.; Fu, M.H.; Chen, L.W.; Liu, W.C.; Liang, C.K.; et al. Pioglitazone reversed the fructose-programmed astrocytic glycolysis and oxidative phosphorylation of female rat offspring. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E622–E634. [Google Scholar] [CrossRef]
- Yamada, H.; Munetsuna, E.; Yamazaki, M.; Mizuno, G.; Sadamoto, N.; Ando, Y.; Fujii, R.; Shiogama, K.; Ishikawa, H.; Suzuki, K.; et al. Maternal fructose-induced oxidative stress occurs via Tfam and Ucp5 epigenetic regulation in offspring hippocampi. FASEB J. 2019, 33, 11431–11442. [Google Scholar] [CrossRef]
- Kloster, A.; Hyer, M.M.; Dyer, S.; Salome-Sanchez, C.; Neigh, G.N. High Fructose Diet Induces Sex-specific Modifications in Synaptic Respiration and Affective-like Behaviors in Rats. Neuroscience 2020. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Cigliano, L.; Liverini, G.; Iossa, S. Increased skeletal muscle mitochondrial efficiency in rats with fructose-induced alteration in glucose tolerance. Br. J. Nutr. 2013, 110, 1996–2003. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.; Vilela, T.C.; Taschetto, L.; Vuolo, F.; Petronilho, F.; Dal-Pizzol, F.; Streck, E.L.; Ferreira, G.C.; Schuck, P.F. Evaluation of the effects of fructose on oxidative stress and inflammatory parameters in rat brain. Mol. Neurobiol. 2014, 50, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Sangüesa, G.; Cascales, M.; Griñán, C.; Sánchez, R.M.; Roglans, N.; Pallàs, M.; Laguna, J.C.; Alegret, M. Impairment of Novel Object Recognition Memory and Brain Insulin Signaling in Fructose- but Not Glucose-Drinking Female Rats. Mol. Neurobiol. 2018, 55, 6984–6999. [Google Scholar] [CrossRef] [PubMed]
- Rivera, D.S.; Lindsay, C.B.; Codocedo, J.F.; Carreño, L.E.; Cabrera, D.; Arrese, M.A.; Vio, C.P.; Bozinovic, F.; Inestrosa, N.C. Long-Term, Fructose-Induced Metabolic Syndrome-Like Condition Is Associated with Higher Metabolism, Reduced Synaptic Plasticity and Cognitive Impairment in Octodon degus. Mol. Neurobiol. 2018, 55, 9169–9187. [Google Scholar] [CrossRef]
- Bermejo-Millo, J.C.; Guimaraes, M.; De Luxan-Delgado, B.; Potes, Y.; Pérez-Martínez, Z.; Díaz-Luis, A.; Caballero, B.; Solano, J.J.; Vega-Naredo, I.; Coto-Montes, A. High-fructose consumption impairs the redox system and protein quality control in the brain of Syrian hamsters: Therapeutic effects of melatonin. Mol. Neurobiol. 2018, 55, 7973–7986. [Google Scholar] [CrossRef]
- Levi, B.; Werman, M.J. Long-term fructose consumption accelerates glycation and several age-related variables in male rats. J. Nutr. 1998, 128, 1442–1449. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.; Van Hinsbergh, V.W. Fructose-mediated nonenzymatic glycation: Sweet coupling or bad modification. Diabetes Metab. Res. Rev. 2004, 20, 369–382. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 973–978. [Google Scholar] [CrossRef]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE Axis in Neuroinflammation, Neurodegeneration and Its Emerging Role in the Pathogenesis of Amyotrophic Lateral Sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Frimat, M.; Daroux, M.; Litke, R.; Nevière, R.; Tessier, F.J.; Boulanger, E. Kidney, heart and brain: Three organs targeted by ageing and glycation. Clin. Sci. (Lond.) 2017, 131, 1069–1092. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: Relevance to Alzheimer’s disease. J. Alzheimers Dis. 2005, 7, 45–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; De la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; De la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Vagelatos, N.T.; Eslick, G.D. Type 2 Diabetes as a Risk Factor for Alzheimer’s Disease: The Confounders, Interactions, and Neuropathology Associated With This Relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, C.; Kiely, A.P.; Coakley, M.F.; Manning, S.; Long-Smith, C.M. Insulin and IGF-1 signalling: Longevity, protein homoeostasis and Alzheimer’s disease. Biochem. Soc. Trans. 2012, 40, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin, cognition, dementia. Eur. J. Pharmacol. 2013, 719, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdile, G.; Fuller, S.J.; Martins, R.N. The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 2015, 84, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Pan, Y.; Wang, R.; Kang, L.L.; Xue, Q.C.; Wang, X.N.; Kong, L.D. Quercetin inhibits AMPK/TXNIP activation and reduces inflammatory lesions to improve insulin signaling defect in the hypothalamus of high fructose-fed rats. J. Nutr. Biochem. 2014, 25, 420–428. [Google Scholar] [CrossRef]
- Wu, H.W.; Ren, L.F.; Zhou, X.; Han, D.W. A high-fructose diet induces hippocampal insulin resistance and exacerbates memory deficits in male Sprague-Dawley rats. Nutr. Neurosci. 2015, 18, 323–328. [Google Scholar] [CrossRef]
- Liu, W.C.; Wu, C.W.; Tain, Y.L.; Fu, M.H.; Hung, C.Y.; Chen, I.C.; Chen, L.W.; Wu, K.L.H. Oral Pioglitazone Ameliorates Fructose-Induced Peripheral Insulin Resistance and Hippocampal Gliosis but Not Restores Inhibited Hippocampal Adult Neurogenesis. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 274–285. [Google Scholar] [CrossRef]
- Kovačević, S.; Nestorov, J.; Matić, G.; Elaković, I. Chronic Stress Combined with a Fructose Diet Reduces Hypothalamic Insulin Signaling and Antioxidative Defense in Female Rats. Neuroendocrinology 2019, 108, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Norman, E.D.; Lee, K.; Cutler, R.G.; Telljohann, R.S.; Egan, J.M.; Mattson, M.P. Diet-induced Insulin Resistance Impairs Hippocampal Synaptic Plasticity and Cognition in Middle-Aged Rats. Hippocampus 2008, 18, 1085–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, C.S.; Burgado, J.; Kelly, S.D.; Johnson, Z.P.; Neigh, G.N. High-fructose Diet During Periadolescent Development Increases Depressive-Like Behavior and Remodels the Hypothalamic Transcriptome in Male Rats. Psychoneuroendocrinology 2015, 62, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Flaherty, B.; Neigh, G.N.; Rainnie, D. High-fructose diet initiated during adolescence does not affect basolateral amygdala excitability or affective-like behavior in Sprague Dawley rats. Behav. Brain. Res. 2019, 365, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Franco-Pérez, J.; Manjarrez-Marmolejo, J.; Ballesteros-Zebadúa, P.; Neri-Santos, A.; Montes, S.; Suarez-Rivera, N.; Hernández-Cerón, M.; Pérez-Koldenkova, V. Chronic Consumption of Fructose Induces Behavioral Alterations by Increasing Orexin and Dopamine Levels in the Rat Brain. Nutrients 2018, 10, 1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ergorul, C.; Eichenbaum, H. The hippocampus and memory for “what,” “where,” and “when”. Learn. Mem. 2004, 11, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.R. Cognitive effects of insulin in the central nervous system. Neurosci. Biobehav. Rev. 2001, 25, 311–323. [Google Scholar] [CrossRef]
- Ross, A.P.; Bartness, T.J.; Mielke, J.G.; Parent, M.B. A high fructose diet impairs spatial memory in male rats. Neurobiol. Learn. Mem. 2009, 92, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Van der Borght, K.; Köhnke, R.; Göransson, N.; Deierborg, T.; Brundin, P.; Erlanson-Albertsson, C.; Lindqvist, A. Reduced Neurogenesis in the Rat Hippocampus Following High Fructose Consumption. Regul. Pept. 2011, 167, 26–30. [Google Scholar] [CrossRef]
- Farr, S.A.; Yamada, K.A.; Butterfield, D.A.; Abdul, H.M.; Xu, L.; Miller, N.E.; Banks, W.A.; Morley, J.E. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology 2008, 149, 2628–2636. [Google Scholar] [CrossRef]
- Cisternas, P.; Salazar, P.; Serrano, F.G.; Montecinos-Oliva, C.; Arredondo, S.B.; Varela-Nallar, L.; Barja, S.; Vio, C.P.; Gomez-Pinilla, F.; Inestrosa, N.C. Fructose consumption reduces hippocampal synaptic plasticity underlying cognitive performance. Biochim. Biophys. Acta 2015, 1852, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, C.; Masnik, A.M.; Mun, J.G.; Du, K.; Clark, D.; Dilger, R.N.; Dilger, A.C.; Rhodes, J.S. Fructose Decreases Physical Activity and Increases Body Fat Without Affecting Hippocampal Neurogenesis and Learning Relative to an Isocaloric Glucose Diet. Sci. Rep. 2015, 5, 9589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.L.; Wu, C.W.; Tain, Y.L.; Huang, L.T.; Chao, Y.M.; Hung, C.Y.; Wu, J.C.; Chen, S.R.; Tsai, P.C.; Chan, J.Y. Environmental stimulation rescues maternal high fructose intake-impaired learning and memory in female offspring: Its correlation with redistribution of histone deacetylase 4. Neurobiol. Learn. Mem. 2016, 130, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.H.F.; Clark, O.E.; Williamson, L.L. Maternal High Fructose Diet and Neonatal Immune Challenge Alter Offspring Anxiety-Like Behavior and Inflammation Across the Lifespan. Life Sci. 2018, 197, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Yamada, H.; Munetsuna, E.; Ishikawa, H.; Mizuno, G.; Mukuda, T.; Mouri, A.; Nabeshima, T.; Saito, K.; Suzuki, K.; et al. Excess maternal fructose consumption impairs hippocampal function in offspring via epigenetic modification of BDNF promoter. FASEB J. 2018, 32, 2549–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Luo, D.; Zheng, M.; Hao, Y.; Hou, L.; Zhang, S. Effect of pioglitazone on insulin resistance in fructose-drinking rats correlates with AGEs/RAGE inhibition and block of NADPH oxidase and NF kappa B activation. Eur. J. Pharmacol. 2010, 629, 153–158. [Google Scholar] [CrossRef]
- Luo, D.; Hou, X.; Hou, L.; Wang, M.; Xu, S.; Dong, C.; Liu, X. Effect of pioglitazone on altered expression of Aβ metabolism-associated molecules in the brain of fructose-drinking rats, a rodent model of insulin resistance. Eur. J. Pharmacol. 2011, 664, 14–19. [Google Scholar] [CrossRef]
- Evans, D.B.; Rank, K.B.; Bhattacharya, K.; Thomsen, D.R.; Gurney, M.E.; Sharma, S.K. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau’s ability to promote microtubule assembly. J. Biol. Chem. 2000, 275, 24977–24983. [Google Scholar] [CrossRef] [Green Version]
- Rankin, C.A.; Sun, Q.; Gamblin, T.C. Pre-assembled tau filaments phosphorylated by GSK-3b form large tangle-like structures. Neurobiol. Dis. 2008, 31, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T.; Yanagihara, Y.T.; Ohyagi, Y.; Nakamura, N.; Iinuma, K.M.; Yamasaki, R.; Asai, H.; Maeda, M.; Murakami, K.; Irie, K.; et al. Insulin deficiency promotes formation of toxic amyloid-β42 conformer co-aggregating with hyper-phosphorylated tau oligomer in an Alzheimer’s disease model. Neurobiol. Dis. 2020, 137, 104739. [Google Scholar] [CrossRef]
- Jones, H.F.; Butler, R.N.; Brooks, D.A. Intestinal fructose transport and malabsorption in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G202–G206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, C.D.; Dawson, A.M. The absorption of monosaccharides in man. Clin. Sci. 1964, 27, 371–379. [Google Scholar] [PubMed]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58 (Suppl. 5), 754S–765S. [Google Scholar] [CrossRef] [PubMed]
- Mantych, G.J.; James, D.E.; Devaskar, S.U. Jejunal/kidney glucose transporter isoform (GLUT-5) is expressed in the human blood-brain barrier. Endocrinology 1993, 132, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Maher, F.; Vannucci, S.J.; Simpson, I.A. Glucose transporter proteins in brain. FASEB J. 1994, 8, 1003–1011. [Google Scholar] [CrossRef]
- Ueno, M.; Nishi, N.; Nakagawa, T.; Chiba, Y.; Tsukamoto, I.; Kusaka, T.; Miki, T.; Sakamoto, H.; Yamaguchi, F.; Tokuda, M. Immunoreactivity of glucose transporter 5 is located in epithelial cells of the choroid plexus and ependymal cells. Neuroscience 2014, 260, 149–157. [Google Scholar] [CrossRef]
- Douard, V.; Ferraris, R.P. The role of fructose transporters in diseases linked to excessive fructose intake. J. Physiol. 2013, 591, 401–414. [Google Scholar] [CrossRef]
- Patel, C.; Sugimoto, K.; Douard, V.; Shah, A.; Inui, H.; Yamanouchi, T.; Ferraris, R.P. Effect of Dietary Fructose on Portal and Systemic Serum Fructose Levels in Rats and in KHK-/- And GLUT5-/- Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G779–G790. [Google Scholar] [CrossRef] [Green Version]
- Funari, V.A.; Voevodski, K.; Leyfer, D.; Yerkes, K.; Cramer, D.; Tolan, D.R. Quantitative gene-expression profiles in real time from expressed sequence tag database. Gene Expr 2010, 14, 321–336. [Google Scholar] [CrossRef]
- Chain, E.B.; Rose, S.P.; Masi, I.; Pocchiari, F. Metabolism of hexoses in rat cerebral cortex slices. J. Neurochem. 1969, 16, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.H.; Cohen, J. The measurement by a microtitration technique of carbon dioxide production in rat brain slices. Studies with glucose, fructose, and xylitol. Anal. Biochem. 1976, 71, 444–451. [Google Scholar] [CrossRef]
- Wada, H.; Okada, Y.; Uzuo, T.; Nakamura, H. The effects of glucose, mannose, fructose and lactate on the preservation of neural activity in the hippocampal slices from the guinea pig. Brain Res. 1998, 788, 144–150. [Google Scholar] [CrossRef]
- Izumi, Y.; Zorumski, C.F. Glial-neuronal interactions underlying fructose utilization in rat hippocampal slices. Neuroscience 2009, 161, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppelt, S.A.; Zhang, W.; Tolan, D.R. Specific regions of the brain are capable of fructose metabolism. Brain Res. 2017, 1657, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Roncal-Jimenez, C.A.; Lanaspa-Garcia, M.A.; Oppelt, S.A.; Kuwabara, M.; Jensen, T.; Milagres, T.; Andres-Hernando, A.; Ishimoto, T.; Garcia, G.E.; et al. Role of fructose and and fructokinase in acute dehydration-induced vasopressin gene expression and secretion in mice. J. Neurophysiol. 2017, 117, 646–654. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Fowler, M.J.; Rathbone, A.J.; Allen, L.M.; Ransom, B.R.; Ray, D.E.; Brown, A.M. Fructose metabolism in the adult mouse optic nerve, a central white matter tract. J. Cereb. Blood Flow Metab. 2007, 27, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.L.; Hung, C.Y.; Chan, J.Y.; Wu, C.W. An increase in adenosine-50-triphosphate (ATP) content in rostral ventrolateral medulla is engaged in the high fructose diet-induced hypertension. J. Biomed. Sci. 2014, 21, 8. [Google Scholar] [CrossRef] [Green Version]
- Hassel, B.; Elsais, A.; Froland, A.S.; Tauboll, E.; Gjerstad, L.; Quan, Y.; Dingledine, R.; Rise, F. Uptake and metabolism, of fructose by rat neocortical cells in vivo and by isolated nerve terminals in vitro. J. Neurochem. 2015, 133, 572–581. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spagnuolo, M.S.; Iossa, S.; Cigliano, L. Sweet but Bitter: Focus on Fructose Impact on Brain Function in Rodent Models. Nutrients 2021, 13, 1. https://doi.org/10.3390/nu13010001
Spagnuolo MS, Iossa S, Cigliano L. Sweet but Bitter: Focus on Fructose Impact on Brain Function in Rodent Models. Nutrients. 2021; 13(1):1. https://doi.org/10.3390/nu13010001
Chicago/Turabian StyleSpagnuolo, Maria Stefania, Susanna Iossa, and Luisa Cigliano. 2021. "Sweet but Bitter: Focus on Fructose Impact on Brain Function in Rodent Models" Nutrients 13, no. 1: 1. https://doi.org/10.3390/nu13010001
APA StyleSpagnuolo, M. S., Iossa, S., & Cigliano, L. (2021). Sweet but Bitter: Focus on Fructose Impact on Brain Function in Rodent Models. Nutrients, 13(1), 1. https://doi.org/10.3390/nu13010001