
Hypocaloric Dieting Unsettles the Neuroenergetic Homeostasis in Humans

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Participants and Methods
2.1. Participants
2.2. Experimental Design
2.3. Standardized Buffet Testing
2.4. 31 Phosphor Magnetic Resonance Spectroscopy
2.5. Analyses of Blood Samples
2.6. Statistical Analyses
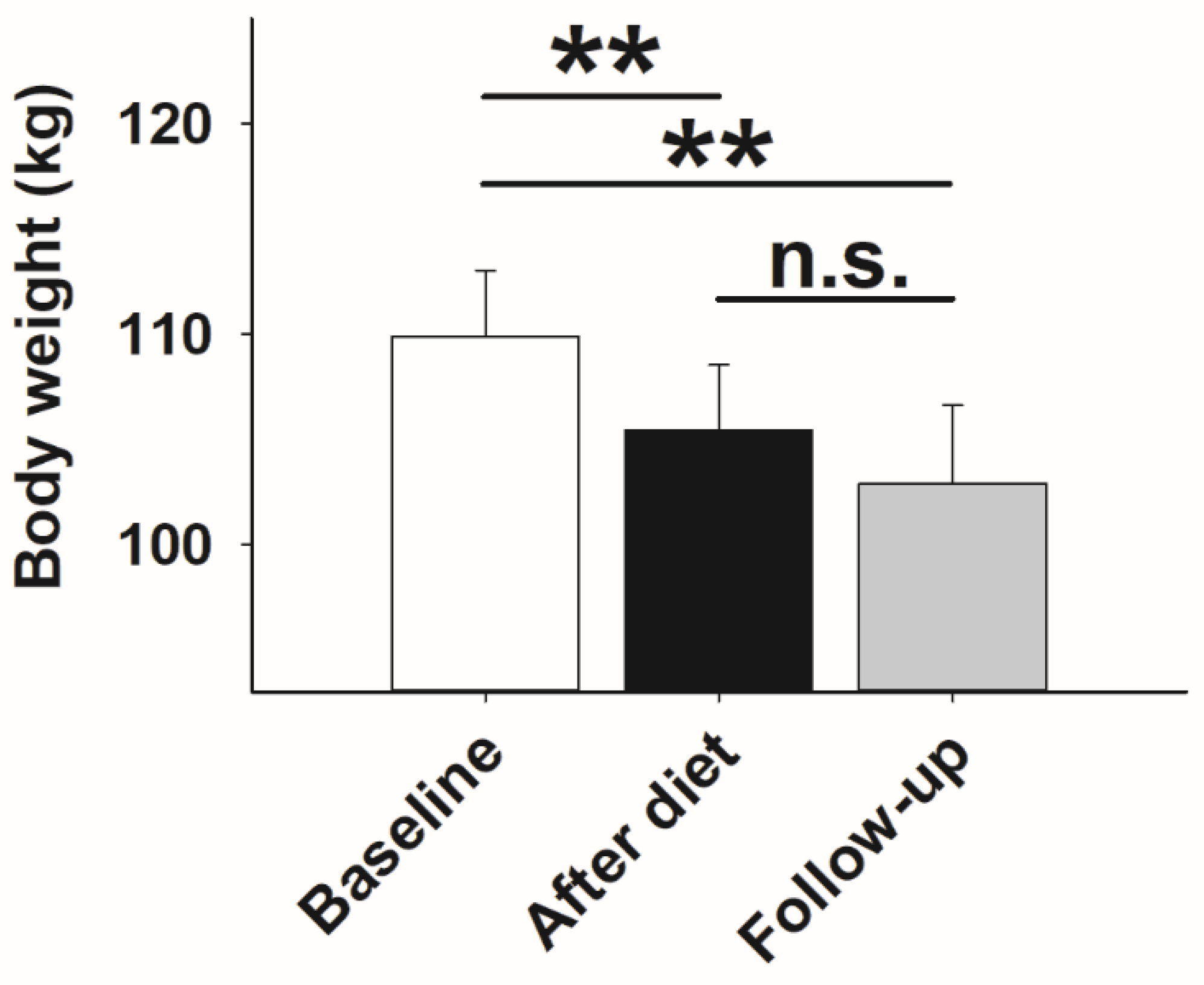
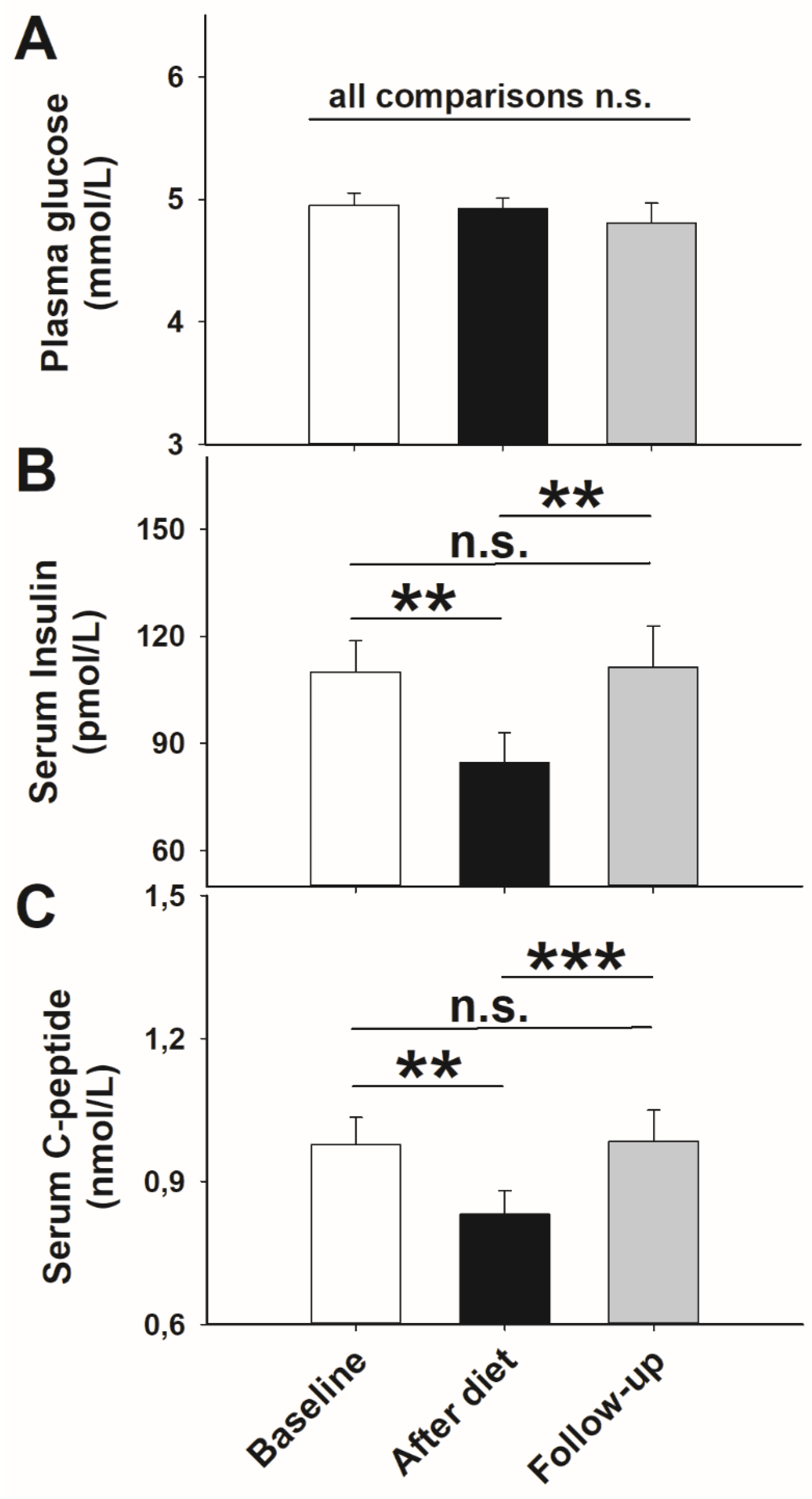
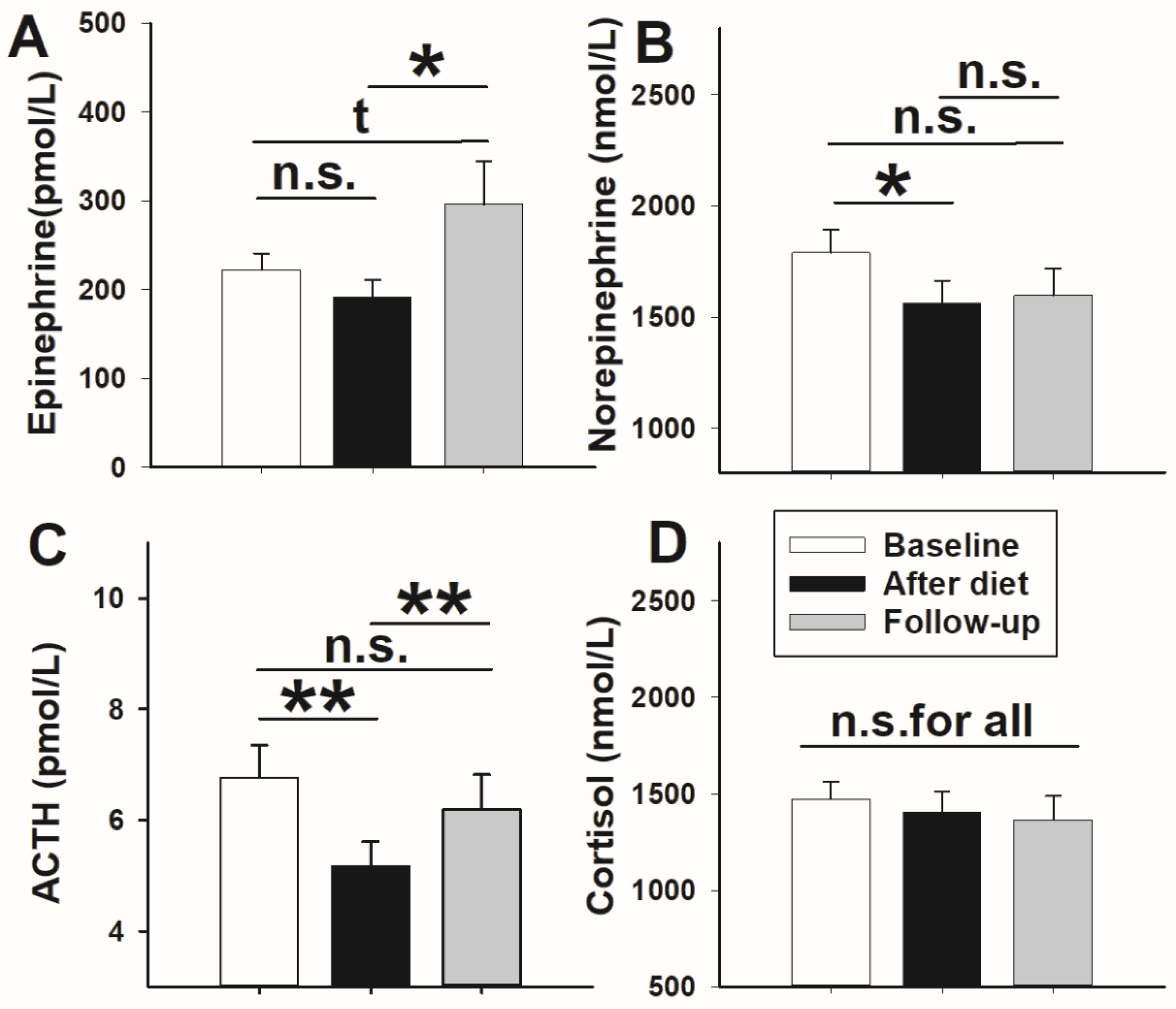
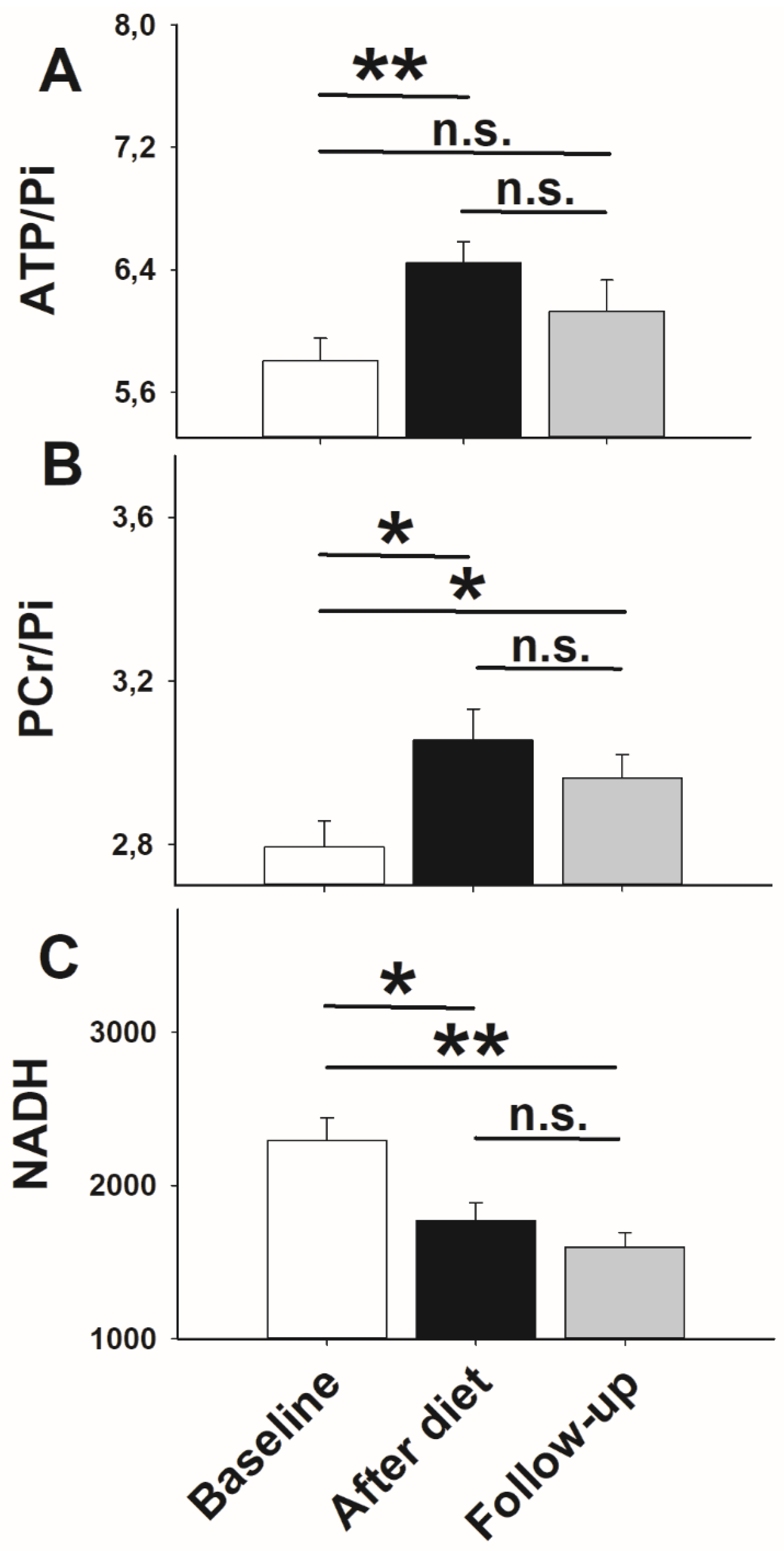
3. Results
3.1. Glucose Metabolism and Stress Axis Activity
3.2. High-Energy Phosphate Measurements
3.3. Standardized Buffet Tests
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Curtin, L.R. Prevalence and Trends in Obesity among US Adults, 1999–2008. JAMA-J. Am. Med. Assoc. 2010, 303, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Peralta, M.; Ramos, M.; Lipert, A.; Martins, J.; Marques, A. Prevalence and trends of overweight and obesity in older adults from 10 European countries from 2005 to 2013. Scand. J. Public Health 2018, 46, 522–529. [Google Scholar] [CrossRef] [PubMed]
- OECD. Obesity and the Economics of Prevention; OECD Publishing: Paris, France, 2014. [Google Scholar]
- Wing, R.R.; Phelan, S. Long-term weight loss maintenance. Am. J. Clin. Nutr. 2005, 82 (Suppl. 1), 222S–225S. [Google Scholar] [CrossRef] [PubMed]
- Jauch-Chara, K.; Oltmanns, K.M. Obesity--a neuropsychological disease? Systematic review and neuropsychological model. Prog. Neurobiol. 2014, 114, 84–101. [Google Scholar] [CrossRef]
- Jauch-Chara, K.; Binkofski, F.; Loebig, M.; Reetz, K.; Jahn, G.; Melchert, U.H.; Schweiger, U.; Oltmanns, K.M. Blunted brain energy consumption relates to insula atrophy and impaired glucose tolerance in obesity. Diabetes 2015, 64, 2082–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmoller, A.; Hass, T.; Strugovshchikova, O.; Melchert, U.H.; Scholand-Engler, H.G.; Peters, A.; Schweiger, U.; Hohagen, F.; Oltmanns, K.M. Evidence for a relationship between body mass and energy metabolism in the human brain. J. Cereb. Blood Flow Metab. 2010, 30, 1403–1410. [Google Scholar] [CrossRef]
- Wardzinski, E.K.; Kistenmacher, A.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Impaired brain energy gain upon a glucose load in obesity. Metabolism 2018, 85, 90–96. [Google Scholar] [CrossRef]
- Jauch-Chara, K.; Friedrich, A.; Rezmer, M.; Melchert, U.H.; Scholand-Engler, G.; Hallschmid, M.; Oltmanns, K.M. Intranasal insulin suppresses food intake via enhancement of brain energy levels in humans. Diabetes 2012, 61, 2261–2268. [Google Scholar] [CrossRef] [Green Version]
- Jauch-Chara, K.; Kistenmacher, A.; Herzog, N.; Schwarz, M.; Schweiger, U.; Oltmanns, K.M. Repetitive electric brain stimulation reduces food intake in humans. Am. J. Clin. Nutr. 2014, 100, 1003–1009. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Conley, K.E.; Ali, A.S.; Flores, B.; Jubrias, S.A.; Shankland, E.G. Mitochondrial NAD(P)H In vivo: Identifying Natural Indicators of Oxidative Phosphorylation in the (31)P Magnetic Resonance Spectrum. Front. Physiol. 2016, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Bachert-Baumann, P.; Ermark, F.; Zabel, H.J.; Sauter, R.; Semmler, W.; Lorenz, W.J. In vivo nuclear Overhauser effect in 31P-(1H) double-resonance experiments in a 1.5-T whole-body MR system. Magn. Reson. Med. 1990, 15, 165–172. [Google Scholar] [CrossRef]
- Barker, P.B.; Golay, X.; Artemov, D.; Ouwerkerk, R.; Smith, M.A.; Shaka, A.J. Broadband proton decoupling for in vivo brain spectroscopy in humans. Magn. Reson. Med 2001, 45, 226–232. [Google Scholar] [CrossRef]
- Naressi, A.; Couturier, C.; Castang, I.; de Beer, R.; Graveron-Demilly, D. Java-based graphical user interface for MRUI, a software package for quantitation of in vivo/medical magnetic resonance spectroscopy signals. Comput. Biol. Med. 2001, 31, 269–286. [Google Scholar] [CrossRef]
- Vanhamme, L.; van den Boogaart, A.; Van, H.S. Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J. Magn. Reson. 1997, 129, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Iosifescu, D.V.; Renshaw, P.E. 31P-magnetic resonance spectroscopy and thyroid hormones in major depressive disorder: Toward a bioenergetic mechanism in depression? Harv. Rev. Psychiatry 2003, 11, 51–63. [Google Scholar]
- Rango, M.; Bozzali, M.; Prelle, A.; Scarlato, G.; Bresolin, N. Brain activation in normal subjects and in patients affected by mitochondrial disease without clinical central nervous system involvement: A phosphorus magnetic resonance spectroscopy study. J. Cereb. Blood Flow Metab. 2001, 21, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viondury, J.; Meyerhoff, D.J.; Cozzone, P.J.; Weiner, M.W. What Might be the Impact on Neurology of the Analysis of Brain Metabolism by In-Vivo Magnetic-Resonance Spectroscopy. J. Neurol. 1994, 241, 354–371. [Google Scholar] [CrossRef]
- Smith, C.D.; Ain, K.B. Brain metabolism in hypothyroidism studied with 31P magnetic-resonance spectroscopy. Lancet 1995, 345, 619–620. [Google Scholar] [CrossRef]
- Raichle, M.E.; Mintun, M.A. Brain work and brain imaging. Annu. Rev. Neurosci. 2006, 29, 449–476. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Neuroscience. Let there be (NADH) light. Science 2004, 305, 50–52. [Google Scholar] [CrossRef]
- Foster, G.D.; Wadden, T.A.; Kendall, P.C.; Stunkard, A.J.; Vogt, R.A. Psychological effects of weight loss and regain: A prospective evaluation. J. Consult. Clin. Psychol. 1996, 64, 752–757. [Google Scholar] [CrossRef]
- Sarlio-Lahteenkorva, S.; Rissanen, A.; Kaprio, J. A descriptive study of weight loss maintenance: 6 and 15 year follow-up of initially overweight adults. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, T.; Bruun, J.M.; Madsen, E.L.; Richelsen, B. Weight loss maintenance in severely obese adults after an intensive lifestyle intervention: 2- to 4-year follow-up. Obes. Silver. Spring 2007, 15, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Mingrone, G. Effects of weight loss and calorie restriction on carbohydrate metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 431–439. [Google Scholar] [CrossRef]
- Brekke, H.K.; Bertz, F.; Rasmussen, K.M.; Bosaeus, I.; Ellegard, L.; Winkvist, A. Diet and exercise interventions among overweight and obese lactating women: Randomized trial of effects on cardiovascular risk factors. PLoS ONE 2014, 9, e88250. [Google Scholar] [CrossRef]
- Holter, M.M.; Dutia, R.; Stano, S.M.; Prigeon, R.L.; Homel, P.; McGinty, J.J., Jr.; Belsley, S.J.; Ren, C.J.; Rosen, D.; Laferrere, B. Glucose Metabolism After Gastric Banding and Gastric Bypass in Individuals With Type 2 Diabetes: Weight Loss Effect. Diabetes Care 2017, 40, 7–15. [Google Scholar] [CrossRef] [Green Version]
- de Luis, D.A.; Aller, R.; Izaola, O.; Gonzalez Sagrado, M.; Conde, R. Effect of two different hypocaloric diets in transaminases and insulin resistance in nonalcoholic fatty liver disease and obese patients. Nutr. Hosp. 2010, 25, 730–735. [Google Scholar]
- de Luis, D.A.; Izaola, O.; Aller, R.; de la Fuente, B.; Bachiller, R.; Romero, E. Effects of a high-protein/low carbohydrate versus a standard hypocaloric diet on adipocytokine levels and insulin resistance in obese patients along 9 months. J. Diabetes Complicat. 2015, 29, 950–954. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Goldsmith, R.; Bloomfield, D.; Magnano, A.; Weimer, L.; Heymsfield, S.; Gallagher, D.; Mayer, L.; Murphy, E.; Leibel, R.L. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J. Clin. Investig. 2005, 115, 3579–3586. [Google Scholar] [CrossRef] [PubMed]
- Straznicky, N.E.; Grima, M.T.; Sari, C.I.; Lambert, E.A.; Phillips, S.E.; Eikelis, N.; Mariani, J.A.; Kobayashi, D.; Hering, D.; Dixon, J.B.; et al. Comparable Attenuation of Sympathetic Nervous System Activity in Obese Subjects with Normal Glucose Tolerance, Impaired Glucose Tolerance, and Treatment Naive Type 2 Diabetes following Equivalent Weight Loss. Front. Physiol. 2016, 7, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortier, M.; Castellano, C.A.; Croteau, E.; Langlois, F.; Bocti, C.; St-Pierre, V.; Vandenberghe, C.; Bernier, M.; Roy, M.; Descoteaux, M.; et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement 2019, 15, 625–634. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Gupta, L.; Khandelwal, D.; Kalra, S.; Gupta, P.; Dutta, D.; Aggarwal, S. Ketogenic diet in endocrine disorders: Current perspectives. J. Postgrad. Med. 2017, 63, 242–251. [Google Scholar]
- Oltmanns, K.M.; Melchert, U.H.; Scholand-Engler, H.G.; Howitz, M.C.; Schultes, B.; Schweiger, U.; Hohagen, F.; Born, J.; Peters, A.; Pellerin, L. Differential energetic response of brain vs. skeletal muscle upon glycemic variations in healthy humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R12–R16. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Bakshi, V.; Lin, A.L. Early Shifts of Brain Metabolism by Caloric Restriction Preserve White Matter Integrity and Long-Term Memory in Aging Mice. Front. Aging Neurosci. 2015, 7, 213. [Google Scholar] [CrossRef] [Green Version]
- Duelli, R.; Kuschinsky, W. Brain glucose transporters: Relationship to local energy demand. News Physiol. Sci. 2001, 16, 71–76. [Google Scholar] [CrossRef]
- Liu, D.; Pitta, M.; Mattson, M.P. Preventing NAD(+) depletion protects neurons against excitotoxicity: Bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann. N. Y. Acad. Sci. 2008, 1147, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol. Neurobiol. 2016, 54, 1046–1077. [Google Scholar] [CrossRef]
- Duelli, R.; Maurer, M.H.; Staudt, R.; Heiland, S.; Duembgen, L.; Kuschinsky, W. Increased cerebral glucose utilization and decreased glucose transporter Glut1 during chronic hyperglycemia in rat brain. Brain Res. 2000, 858, 338–347. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Triguero, D.; Farrell, C.R. Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes 1990, 39, 1040–1044. [Google Scholar] [CrossRef]
- Vincent, A.M.; McLean, L.L.; Backus, C.; Feldman, E.L. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 2005, 19, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharm. 2014, 2, 125. [Google Scholar]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistenmacher, A.; Goetsch, J.; Ullmann, D.; Wardzinski, E.K.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Psychosocial stress promotes food intake and enhances the neuroenergetic level in men. Stress 2018, 21, 1–10. [Google Scholar] [CrossRef]
- Brownell, K.D.; Greenwood, M.R.; Stellar, E.; Shrager, E.E. The effects of repeated cycles of weight loss and regain in rats. Physiol. Behav. 1986, 38, 459–464. [Google Scholar] [CrossRef]
- Cunarro, J.; Casado, S.; Lugilde, J.; Tovar, S. Hypothalamic Mitochondrial Dysfunction as a Target in Obesity and Metabolic Disease. Front. Endocrinol. Lausanne 2018, 9, 283. [Google Scholar] [CrossRef]
- Jin, S.; Diano, S. Mitochondrial Dynamics and Hypothalamic Regulation of Metabolism. Endocrinology 2018, 159, 3596–3604. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.; Rabe, D.; Duysen, K.; Melchert, U.H.; Oltmanns, K.M. Lactate infusion increases brain energy content during euglycemia but not hypoglycemia in healthy men. NMR Biomed. 2019, 32, e4167. [Google Scholar] [CrossRef]
- Lu, M.; Zhu, X.H.; Chen, W. In vivo (31) P MRS assessment of intracellular NAD metabolites and NAD(+) /NADH redox state in human brain at 4 T. NMR Biomed. 2016, 29, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wardzinski, E.K.; Hyzy, C.; Duysen, K.U.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Hypocaloric Dieting Unsettles the Neuroenergetic Homeostasis in Humans. Nutrients 2021, 13, 3433. https://doi.org/10.3390/nu13103433
Wardzinski EK, Hyzy C, Duysen KU, Melchert UH, Jauch-Chara K, Oltmanns KM. Hypocaloric Dieting Unsettles the Neuroenergetic Homeostasis in Humans. Nutrients. 2021; 13(10):3433. https://doi.org/10.3390/nu13103433
Chicago/Turabian StyleWardzinski, Ewelina K., Carolin Hyzy, Kai Uwe Duysen, Uwe H. Melchert, Kamila Jauch-Chara, and Kerstin M. Oltmanns. 2021. "Hypocaloric Dieting Unsettles the Neuroenergetic Homeostasis in Humans" Nutrients 13, no. 10: 3433. https://doi.org/10.3390/nu13103433