
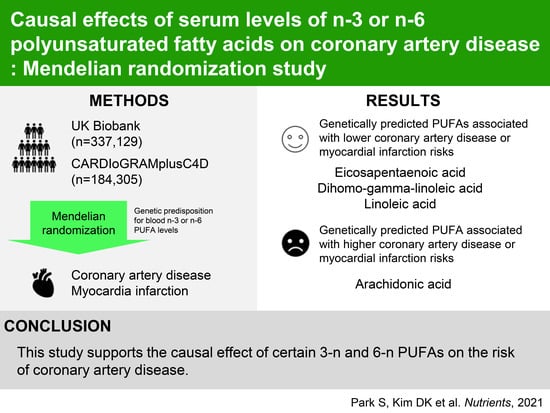
Causal Effects of Serum Levels of n-3 or n-6 Polyunsaturated Fatty Acids on Coronary Artery Disease: Mendelian Randomization Study

 ,
,  , , ,
, , ,
Abstract
1. Introduction
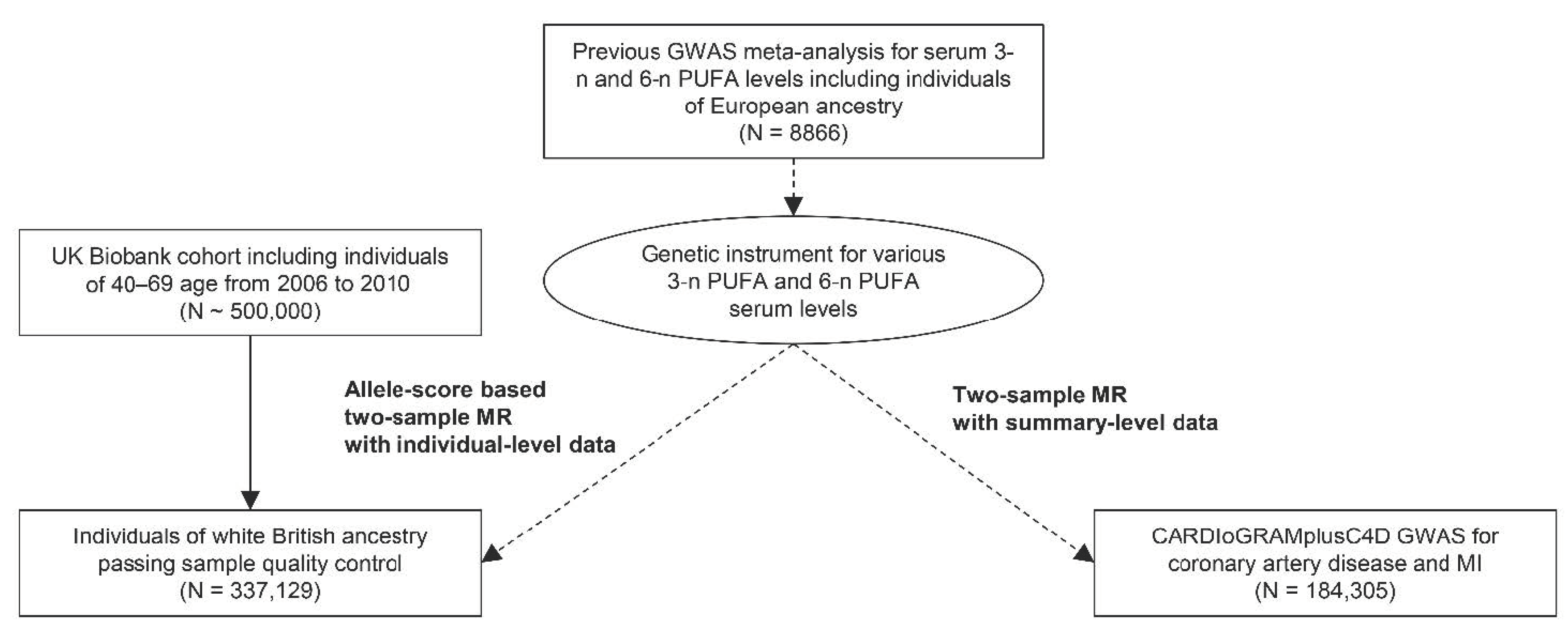
2. Materials and Methods
2.1. Study Setting
2.2. Genetic Instruments
2.3. Allele-Score Based MR with Individual-Level Data in the UK Biobank
2.4. Summary-Level MR with the CARDIoGRAMplusC4D Data
3. Results
3.1. Clinical Characteristics of the UK Biobank Data
3.2. Allele-Score Based MR Results with the UK Biobank Data
3.3. Summary-Level MR Results with the CARDIoGRAMplusC4D Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.-W.; et al. Forecasting life expectancy, years of life lost, and all-Cause and cause-Specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.; Kim, Y.; Lee, Y.; Kang, M.W.; Han, K.; Han, S.S.; Lee, H.; Lee, J.P.; Joo, K.W.; et al. Altered Risk for Cardiovascular Events with Changes in the Metabolic Syndrome Status: A Nationwide Population-Based Study of Approximately 10 Million Persons. Ann. Intern. Med. 2019, 171, 875–884. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Farvid, M.S.; Ding, M.; Pan, A.; Sun, Q.; Chiuve, S.E.; Steffen, L.M.; Willett, W.C.; Hu, F.B. Dietary Linoleic Acid and Risk of Coronary Heart Disease: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. Circulation 2014, 130, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Micha, R.; Wallace, S. Effects on Coronary Heart Disease of Increasing Polyunsaturated Fat in Place of Saturated Fat: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. PLoS Med. 2010, 7, e1000252. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services and US Department of Agriculture. 2015–2020 Dietary Guidelines for Americans, 8th ed.; US Department of Health and Human Services and US Department of Agriculture: Washington, DC, USA, 2015.
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef]
- Wang, D.D.; Li, Y.; Chiuve, S.E.; Stampfer, M.J.; Manson, J.E.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Association of Specific Dietary Fats with Total and Cause-Specific Mortality. JAMA Intern. Med. 2016, 176, 1134–1145. [Google Scholar] [CrossRef]
- Wu, J.H.Y.; Lemaitre, R.N.; King, I.B.; Song, X.; Psaty, B.M.; Siscovick, D.S.; Mozaffarian, D. Response to Letters Regarding Article, “Circulating Omega-6 Polyunsaturated Fatty Acids and Total and Cause-Specific Mortality: The Cardiovascular Health Study”. Circulation 2015, 132, e25–e26. [Google Scholar] [CrossRef]
- GISSI-Prevenzione investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico). Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GIS-SI-Prevenzione trial. Lancet 1999, 354, 447–455. [Google Scholar] [CrossRef]
- Tavazzi, L.; Maggioni, A.P.; Marchioli, R.; Barlera, S.; Franzosi, M.G.; Latini, R.; Lucci, D.; Nicolosi, G.L.; Porcu, M.; Tognoni, G. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1223–1230. [Google Scholar]
- Abdelhamid, A.S.; Martin, N.; Bridges, C.; Brainard, J.S.; Wang, X.; Brown, T.J.; Hanson, S.; Jimoh, O.F.; Ajabnoor, S.M.; Deane, K.H.; et al. Polyunsaturated fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 11, Cd012345. [Google Scholar]
- Rizos, E.C.; Ntzani, E.E.; Bika, E.; Kostapanos, M.S.; Elisaf, M.S. Association between omega-3 fatty acid supplementation and risk of major cardiovascular disease events: A systematic review and meta-analysis. JAMA 2012, 308, 1024–1033. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Smith, G.D. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.; Kim, Y.; Lee, Y.; Kang, M.W.; Kim, K.; Kim, Y.C.; Han, S.S.; Lee, H.; Lee, J.P.; et al. Short or Long Sleep Duration and CKD: A Mendelian Randomization Study. J. Am. Soc. Nephrol. 2020, 31, 2937–2947. [Google Scholar] [CrossRef] [PubMed]
- Daghlas, I.; Dashti, H.S.; Lane, J.; Aragam, K.G.; Rutter, M.K.; Saxena, R.; Vetter, C. Sleep Duration and Myocardial Infarction. J. Am. Coll. Cardiol. 2019, 74, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.R.; Gill, D.; Davies, N.M.; Taylor, A.E.; Tillmann, T.; Vaucher, J.; Wootton, R.E.; Munafò, M.R.; Hemani, G.; Malik, R.; et al. Understanding the consequences of education inequality on cardiovascular disease: Mendelian randomisation study. BMJ 2019, 365, l1855. [Google Scholar] [CrossRef]
- Tyrrell, J.; Jones, S.E.; Beaumont, R.; Astley, C.M.; Lovell, R.; Yaghootkar, H.; Tuke, M.; Ruth, K.S.; Freathy, R.M.; Hirschhorn, J.N.; et al. Height, body mass index, and socioeconomic status: Mendelian randomisation study in UK Biobank. BMJ 2016, 352, i582. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nat. Cell Biol. 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Lemaitre, R.; Tanaka, T.; Tang, W.; Manichaikul, A.; Foy, M.; Kabagambe, E.; Nettleton, J.; King, I.; Weng, L.-C.; Bhattacharya, S.; et al. Genetic Loci Associated with Plasma Phospholipid n-3 Fatty Acids: A Meta-Analysis of Genome-Wide Association Studies from the CHARGE Consortium. PLoS Genet. 2011, 7, e1002193. [Google Scholar] [CrossRef]
- Guan, W.; Steffen, B.T.; Lemaitre, R.N.; Wu, J.H.; Tanaka, T.; Manichaikul, A.; Foy, M.; Rich, S.S.; Wang, L.; Nettleton, J.A.; et al. Genome-Wide Association Study of Plasma N6 Polyunsaturated Fatty Acids within the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium. Circ. Cardiovasc. Genet. 2014, 7, 321–331. [Google Scholar] [CrossRef]
- Khankari, N.K.; Murff, H.J.; Zeng, C.; Wen, W.; Eeles, R.A.; Easton, D.F.; Kote-Jarai, Z.; Al Olama, A.A.; Benlloch, S.; Muir, K.; et al. Poly-unsaturated fatty acids and prostate cancer risk: A Mendelian randomisation analysis from the PRACTICAL consortium. Br. J. Cancer 2016, 115, 624–631. [Google Scholar] [CrossRef] [PubMed]
- May-Wilson, S.; Sud, A.; Law, P.J.; Palin, K.; Tuupanen, S.; Gylfe, A.; Hänninen, U.A.; Cajuso, T.; Tanskanen, T.; Kondelin, J.; et al. Pro-inflammatory fatty acid profile and colorectal cancer risk: A Mendelian randomisation analysis. Eur. J. Cancer 2017, 84, 228–238. [Google Scholar] [CrossRef]
- Zhao, J.V.; Schooling, C.M. Effect of linoleic acid on ischemic heart disease and its risk factors: A Mendelian randomization study. BMC Med. 2019, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.; Littlejohns, T.J.; Sudlow, C.; Doherty, N.; Adamska, L.; Sprosen, T.; Collins, R.; Allen, N.E. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants with Those of the General Population. Am. J. Epidemiol. 2017, 186, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [PubMed]
- Hanscombe, K.B.; Coleman, J.R.I.; Traylor, M.; Lewis, C.M. ukbtools: An R package to manage and query UK Biobank data. PLoS ONE 2019, 14, e0214311. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar]
- Bowden, J.; Smith, G.D.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Ander, B.P.; Dupasquier, C.M.; Prociuk, M.A.; Pierce, G.N. Polyunsaturated fatty acids and their effects on cardiovascular disease. Exp. Clin. Cardiol. 2003, 8, 164–172. [Google Scholar]
- Sokoła-Wysoczańska, E.; Wysoczański, T.; Wagner, J.; Czyż, K.; Bodkowski, R.; Lochyński, S.; Patkowska-Sokoła, B. Polyunsaturated Fatty Acids and Their Potential Therapeutic Role in Cardiovascular System Disorders—A Review. Nutrients 2018, 10, 1561. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, S.; Miyazaki, T.; Shimada, K.; Sugita, Y.; Shimizu, M.; Murata, A.; Kato, T.; Aikawa, T.; Suda, S.; Shiozawa, T.; et al. Low Docosahexaenoic Acid, Dihomo-Gamma-Linolenic Acid, and Arachidonic Acid Levels Associated with Long-Term Mortality in Patients with Acute Decompensated Heart Failure in Different Nutritional Statuses. Nutrients 2017, 9, 956. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, F.B.; Manson, J.E. Marine Omega-3 Supplementation and Cardiovascular Disease: An Updated Meta-Analysis of 13 Randomized Controlled Trials Involving 127,477 Participants. J. Am. Heart Assoc. 2019, 8, e013543. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.-M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Albert, C.M.; Gordon, D.; Copeland, T.; et al. Marine n-3 Fatty Acids and Prevention of Cardiovascular Disease and Cancer. N. Engl. J. Med. 2019, 380, 23–32. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Pomposiello, S.I.; Alva, M.; Wilde, D.W.; Carretero, O.A. Linoleic acid induces relaxation and hyperpolarization of the pig coronary artery. Hypertension 1998, 31, 615–620. [Google Scholar] [CrossRef]
- Gallagher, H.; Williams, J.O.; Ferekidis, N.; Ismail, A.; Chan, Y.-H.; Michael, D.R.; Guschina, I.A.; Tyrrell, V.J.; O’Donnell, V.B.; Harwood, J.L.; et al. Dihomo-γ-linolenic acid inhibits several key cellular processes associated with atherosclerosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2538–2550. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Huang, Y.S. Gamma linolenic acid: An antiinflammatory omega-6 fatty acid. Curr. Pharm. Biotechnol. 2006, 7, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. CRP CHD Genetics Collaboration Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 2011, 40, 755–764. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.S.; Thompson, J.R. Beyond Mendelian randomization: How to interpret evidence of shared genetic predictors. J. Clin. Epidemiol. 2016, 69, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Butterworth, A.; Malarstig, A.; Thompson, S.G. Use of Mendelian randomisation to assess potential benefit of clinical intervention. BMJ 2012, 345, e7325. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Phenotype | SNP | Effect Allele | Other Allele | Effect Allele Frequency | Beta | Standard Error |
|---|---|---|---|---|---|---|
| Eicosapentaenoic acid | rs3798713 | C | G | 0.43 | 0.035 | 0.005 |
| rs174538 | A | G | 0.72 | 0.083 | 0.005 | |
| Docosapentaenoic acid | rs780094 | T | C | 0.41 | 0.017 | 0.003 |
| rs3734398 | T | C | 0.43 | 0.04 | 0.003 | |
| rs174547 | T | C | 0.67 | 0.075 | 0.003 | |
| Docosahexaenoic acid | rs2236212 | C | G | 0.57 | 0.113 | 0.014 |
| Linoleic Acid | rs10740118 | G | C | 0.56 | 0.2484 | 0.0431 |
| rs174547 | C | T | 0.32 | 1.4737 | 0.0417 | |
| rs16966952 | A | G | 0.31 | 0.3512 | 0.0439 | |
| Gamma-linolenic acid | rs174547 | T | C | 0.67 | 0.0156 | 0.0009 |
| rs16966952 | G | A | 0.69 | 0.0061 | 0.0009 | |
| Dihomo-gamma-linolenic acid | rs174547 | C | T | 0.33 | 0.355 | 0.0136 |
| rs16966952 | G | A | 0.69 | 0.22 | 0.013 | |
| Arachidonic acid | rs174547 | T | C | 0.68 | 1.6909 | 0.0253 |
| rs16966952 | G | A | 0.69 | 0.1989 | 0.0314 | |
| Adrenic acid | rs174547 | T | C | 0.67 | 0.0483 | 0.0019 |
| Characteristics | Total (N = 337,129) | Males (N = 156,106) | Females (N = 181,023) |
|---|---|---|---|
| Age (years) | 58 [51; 63] | 59 [51; 64] | 58 [51; 63] |
| Body mass index | 26.7 [24.1; 29.8] | 27.3 [25.0; 30.0] | 26.1 [23.4; 29.6] |
| Obesity (body mass index ≥30 kg/m2) | 81,022 (24.1%) | 39,328 (25.3%) | 41,694 (23.1%) |
| Smoking history | |||
| Non-smoker | 183,636 (55%) | 76,356 (49%) | 107,280 (60%) |
| Ex-smoker | 118,399 (35%) | 60,835 (39%) | 57,564 (32%) |
| Current-smoker | 33,921 (10%) | 18,360 (12%) | 15,561 (9%) |
| Hypertension | 70,018 (20.9%) | 38,538 (24.9%) | 31,480 (17.5%) |
| Systolic BP (mmHg) | 136.5 [125.0; 149.5] | 139.5 [129.0; 152.0] | 133.5 [121.5; 147.5] |
| Diastolic BP (mmHg) | 82.0 [75.5; 89.0] | 84.0 [77.5; 90.5] | 80.0 [73.5; 87.0] |
| Diabetes mellitus | 16,178 (5%) | 10,012 (6%) | 6166 (3%) |
| Hemoglobin A1c (mmol/L) | 35.1 [32.7; 37.7] | 35.2 [32.7; 37.9] | 35.1 [32.7; 37.6] |
| Medications for dyslipidemia | 58,531 (18%) | 35,832 (23%) | 22,699 (12.6%) |
| Triglycerides (mmol/L) | 1.5 [1.1; 2.2] | 1.7 [1.2; 2.5] | 1.3 [1.0; 1.9] |
| High-density lipoprotein (mmol/L) | 1.4 [1.2; 1.7] | 1.2 [1.1; 1.5] | 1.6 [1.3; 1.8] |
| Low-density lipoprotein (mmol/L) | 3.5 [3.0; 4.1] | 3.5 [2.9; 4.1] | 3.6 [3.0; 4.2] |
| Aspartate aminotransferase (U/L) | 20.2 [15.4; 27.4] | 23.8 [18.4; 31.8] | 17.5 [13.9; 23.0] |
| Alanine aminotransferase (U/L) | 24.4 [21.0; 28.8] | 26.1 [22.6; 30.9] | 23.0 [20.0; 26.8] |
| Creatinine (mmol/L) | 70.5 [61.6; 81.0] | 80.0 [72.6; 88.3] | 63.2 [57.1; 70.0] |
| Estimated glomerular filtration rate (mL/min/1.73 m2) | 92.5 [82.6; 99.5] | 92.2 [82.6; 99.3] | 92.9 [82.6; 99.8] |
| Number of prevalent/incident MI cases | 12,812 (4%) | 9878 (6%) | 2934 (2%) |
| Genetically Predicted PUFA Level by Allele–Scores (1 Standard Deviation Increase) | Main Analysis a | Sensitivity Analysis Adjusted for Phenotypical Covariates b | ||
|---|---|---|---|---|
| Adjusted OR (95% CI) | P | Adjusted OR (95% CI) | P | |
| n-3 PUFAs | ||||
| Eicosapentaenoic acid | 0.973 (0.956–0.991) | 0.003 | 0.969 (0.949–0.989) | 0.002 |
| Docosapentaenoic acid | 1.027 (1.009–1.046) | 0.004 | 1.029 (1.008–1.050) | 0.006 |
| Docosahexaenoic acid | 1.000 (0.982–1.018) | 0.986 | 1.003 (0.982–1.023) | 0.804 |
| n-6 PUFAs | ||||
| Linoleic acid | 0.975 (0.957–0.992) | 0.005 | 0.967 (0.947–0.987) | 0.001 |
| Gamma-linolenic acid | 1.022 (1.003–1.040) | 0.020 | 1.028 (1.007–1.049) | 0.009 |
| Dihomo-gamma-linolenic acid | 0.972 (0.955–0.990) | 0.002 | 0.969 (0.950–0.989) | 0.003 |
| Arachidonic acid | 1.027 (1.009–1.046) | 0.004 | 1.034 (1.013–1.056) | 0.001 |
| Adrenic acid | 1.004 (0.986–1.022) | 0.672 | 1.008 (0.987–1.029) | 0.458 |
| Genetically Predicted PUFA Level | For Coronary Artery Disease | For Myocardial Infarction | ||
|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |
| n-3 PUFAs | ||||
| Eicosapentaenoic acid | 0.781 (0.626–0.975) | 0.029 | 0.793 (0.537–1.172) | 0.245 |
| Docosapentaenoic acid a | 1.215 (0.971–1.522) | 0.089 | 1.227 (0.954–1.578) | 0.110 |
| Docosahexaenoic acid | 1.000 (0.851–1.175) | 0.999 | 1.057 (0.883–1.264) | 0.548 |
| n-6 PUFAs | ||||
| Linoleic acid | 0.987 (0.975–1.000) | 0.055 | 0.986 (0.972–1.000) | 0.053 |
| Penalised weighted median b | 0.986 (0.974–0.999) | 0.035 | 0.984 (0.970–0.999) | 0.033 |
| MR-Egger b | 0.979 (0.958–1.000) | 0.024 | 0.975 (0.951–0.999) | 0.022 |
| Gamma-linolenic acid | 2.541 (0.783–8.244) | 0.120 | 2.960 (0.790–11.097) | 0.107 |
| Dihomo-gamma-linolenic acid | 0.940 (0.897–0.985) | 0.010 | 0.932 (0.884–0.983) | 0.009 |
| Arachidonic acid | 1.012 (1.000–1.024) | 0.042 | 1.014 (1.001–1.027) | 0.037 |
| Adrenic acid a | 1.587 (1.054–2.391) | 0.027 | 1.700 (1.073–2.693) | 0.024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Lee, S.; Kim, Y.; Lee, Y.; Kang, M.W.; Kim, K.; Kim, Y.C.; Han, S.S.; Lee, H.; Lee, J.P.; et al. Causal Effects of Serum Levels of n-3 or n-6 Polyunsaturated Fatty Acids on Coronary Artery Disease: Mendelian Randomization Study. Nutrients 2021, 13, 1490. https://doi.org/10.3390/nu13051490
Park S, Lee S, Kim Y, Lee Y, Kang MW, Kim K, Kim YC, Han SS, Lee H, Lee JP, et al. Causal Effects of Serum Levels of n-3 or n-6 Polyunsaturated Fatty Acids on Coronary Artery Disease: Mendelian Randomization Study. Nutrients. 2021; 13(5):1490. https://doi.org/10.3390/nu13051490
Chicago/Turabian StylePark, Sehoon, Soojin Lee, Yaerim Kim, Yeonhee Lee, Min Woo Kang, Kwangsoo Kim, Yong Chul Kim, Seung Seok Han, Hajeong Lee, Jung Pyo Lee, and et al. 2021. "Causal Effects of Serum Levels of n-3 or n-6 Polyunsaturated Fatty Acids on Coronary Artery Disease: Mendelian Randomization Study" Nutrients 13, no. 5: 1490. https://doi.org/10.3390/nu13051490
APA StylePark, S., Lee, S., Kim, Y., Lee, Y., Kang, M. W., Kim, K., Kim, Y. C., Han, S. S., Lee, H., Lee, J. P., Joo, K. W., Lim, C. S., Kim, Y. S., & Kim, D. K. (2021). Causal Effects of Serum Levels of n-3 or n-6 Polyunsaturated Fatty Acids on Coronary Artery Disease: Mendelian Randomization Study. Nutrients, 13(5), 1490. https://doi.org/10.3390/nu13051490