
Storage and Utilization of Glycogen by Mouse Liver during Adaptation to Nutritional Changes Are GLP-1 and PASK Dependent

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Treatments
2.2. Real-Time Polymerase Chain Reaction
2.3. Liver Protein Detection by Western Blot
2.4. Assay of Glucokinase Phosphorylating Activities
2.5. Blood GLP-1 and Glucose Concentrations
2.6. Histological Liver Glycogen Detection by PAS (Periodic Acid-Schiff) Staining
2.7. Statistical Analyses
3. Results
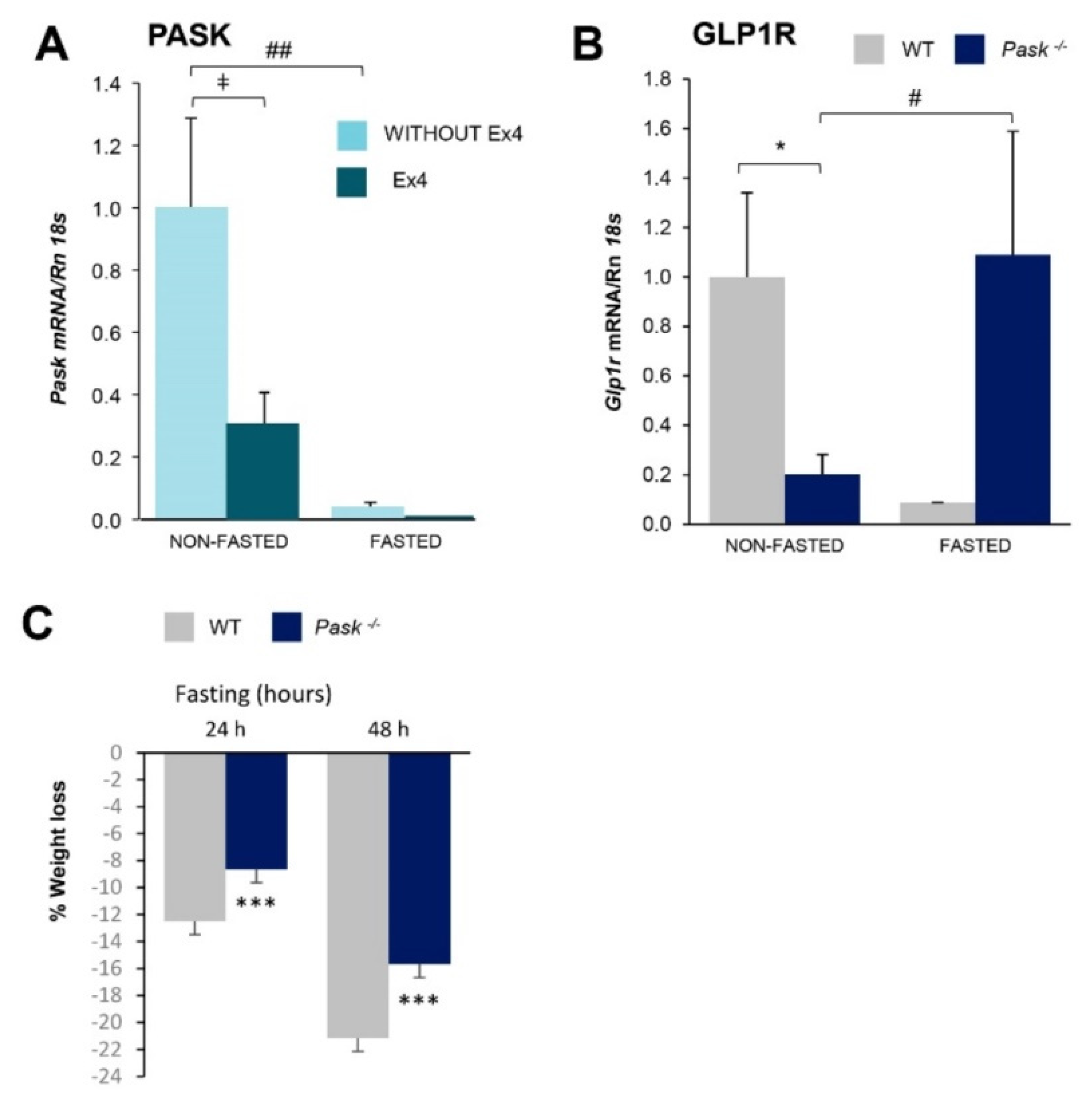
3.1. Exendin-4 and Long Fasting Downregulate Hepatic Pask Gene Expression
3.2. Liver GLP-1 Receptor Expressions and Blood GLP-1 Levels Are PASK-Dependent
3.3. Long Fasting Produces a Lower Weight Loss in PASK-Deficient Mice
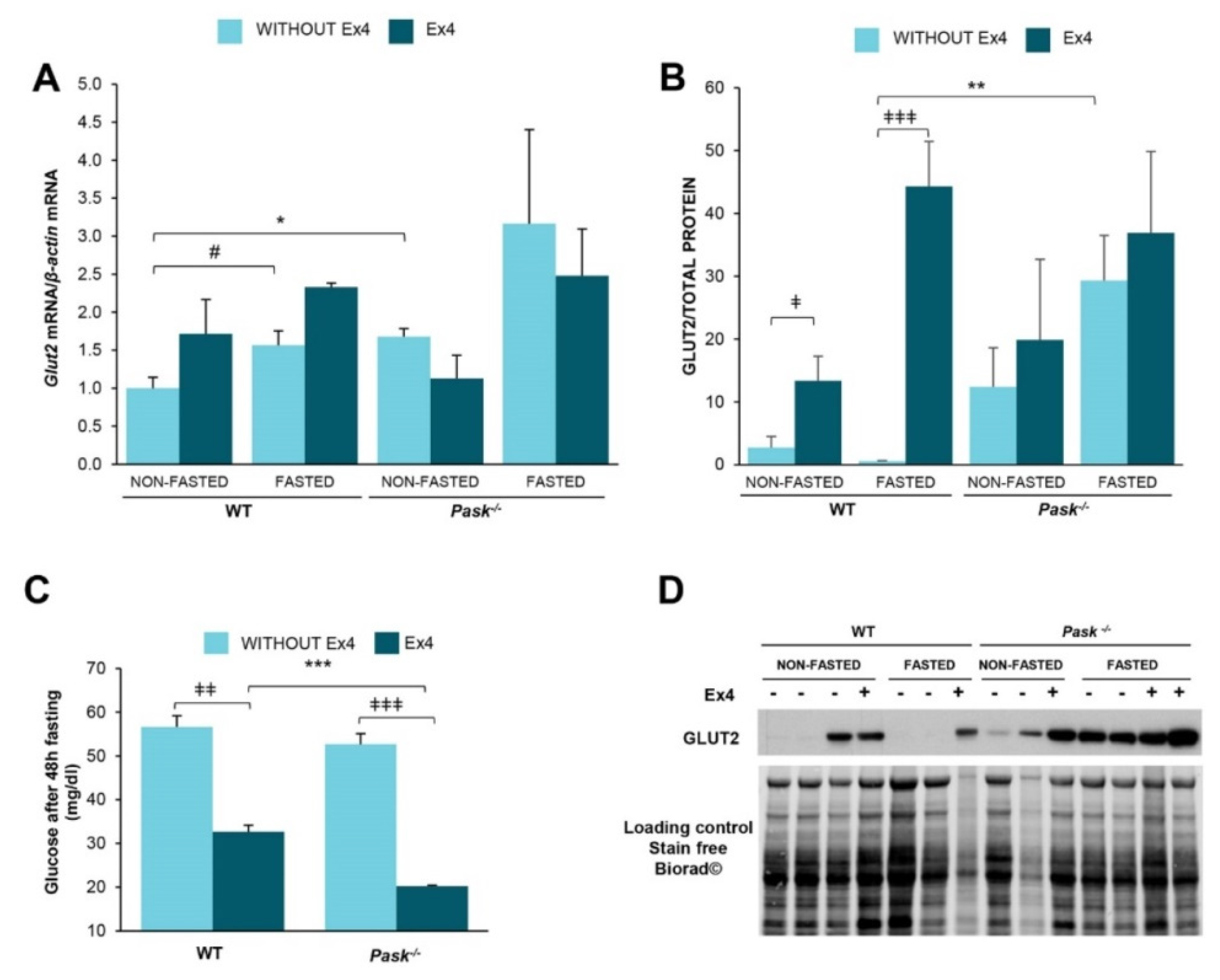
3.4. Exendin-4 Upregulated the GLUT-2 Protein Levels in a PASK-Dependent Way
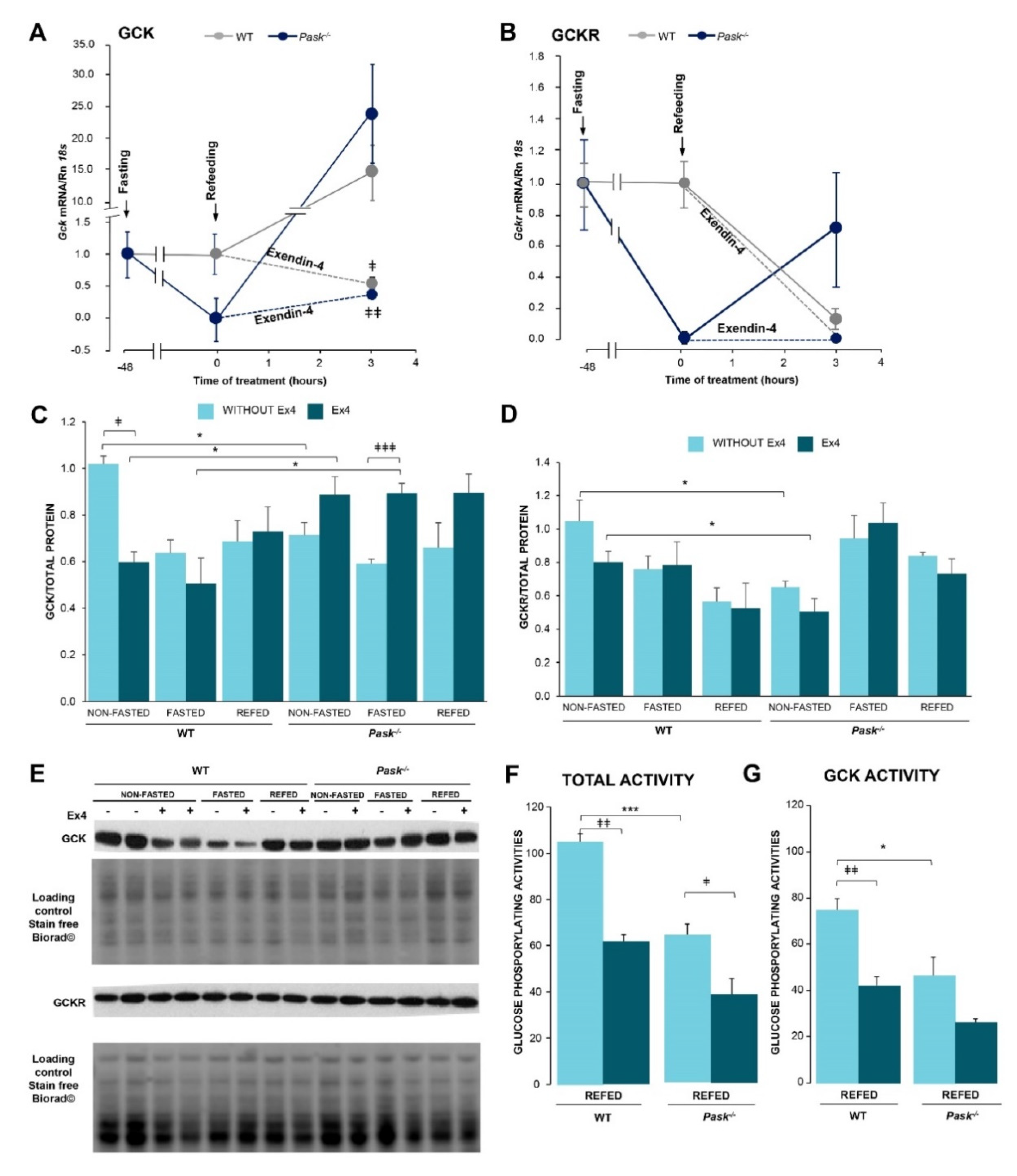
3.5. Exendin-4 Regulated Liver GCK and GCKR Responses to Feeding/Fasting, and This Effect Is PASK-Dependent
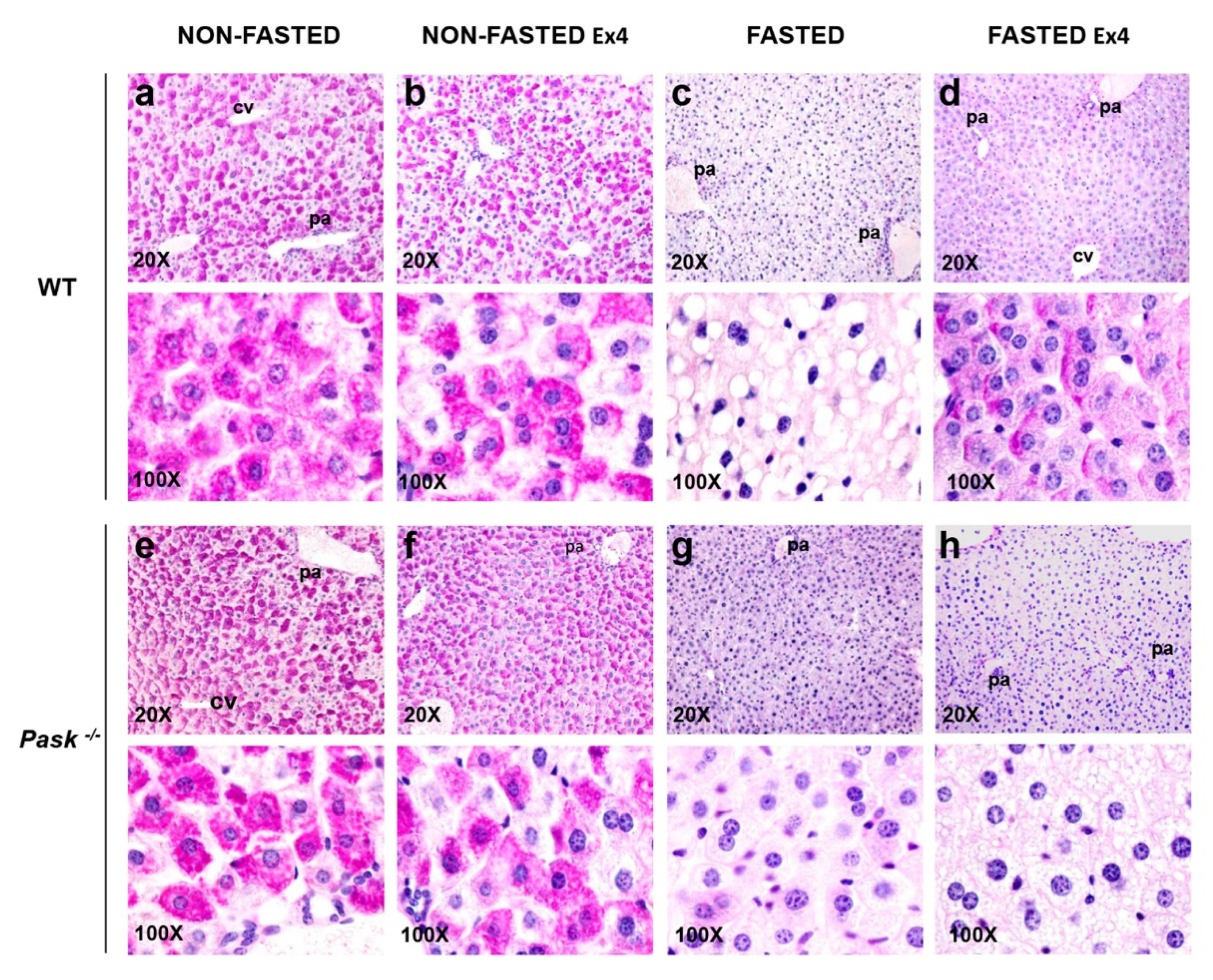
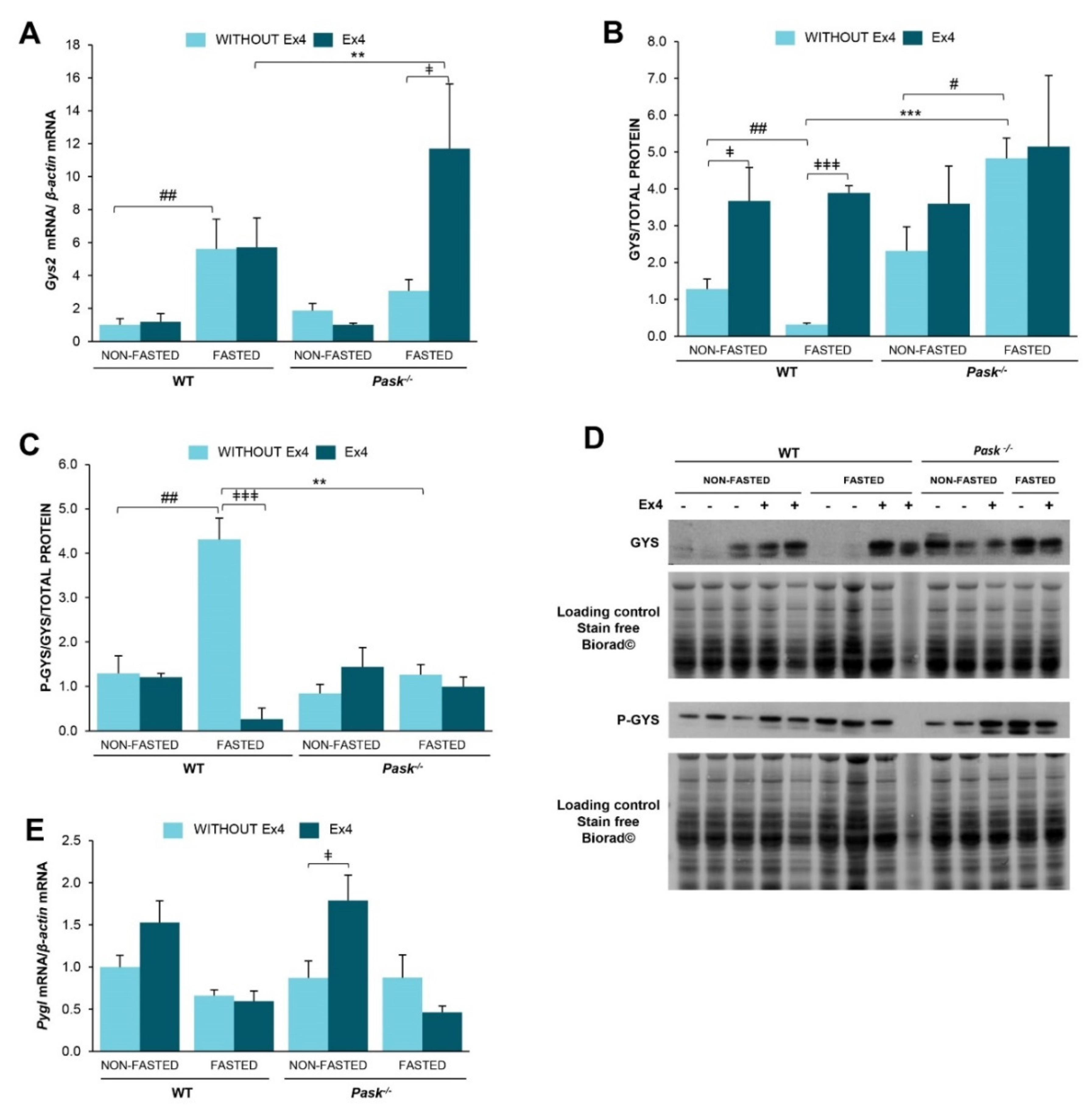
3.6. Exendin-4 and PASK Regulate Glycogen Stores in the Liver in Response to Long Fasting
3.7. The Response of Glycogen-Metabolism Associated Proteins to Long Fasting Was Modified by Exendin-4 and Altered by PASK Deficiency
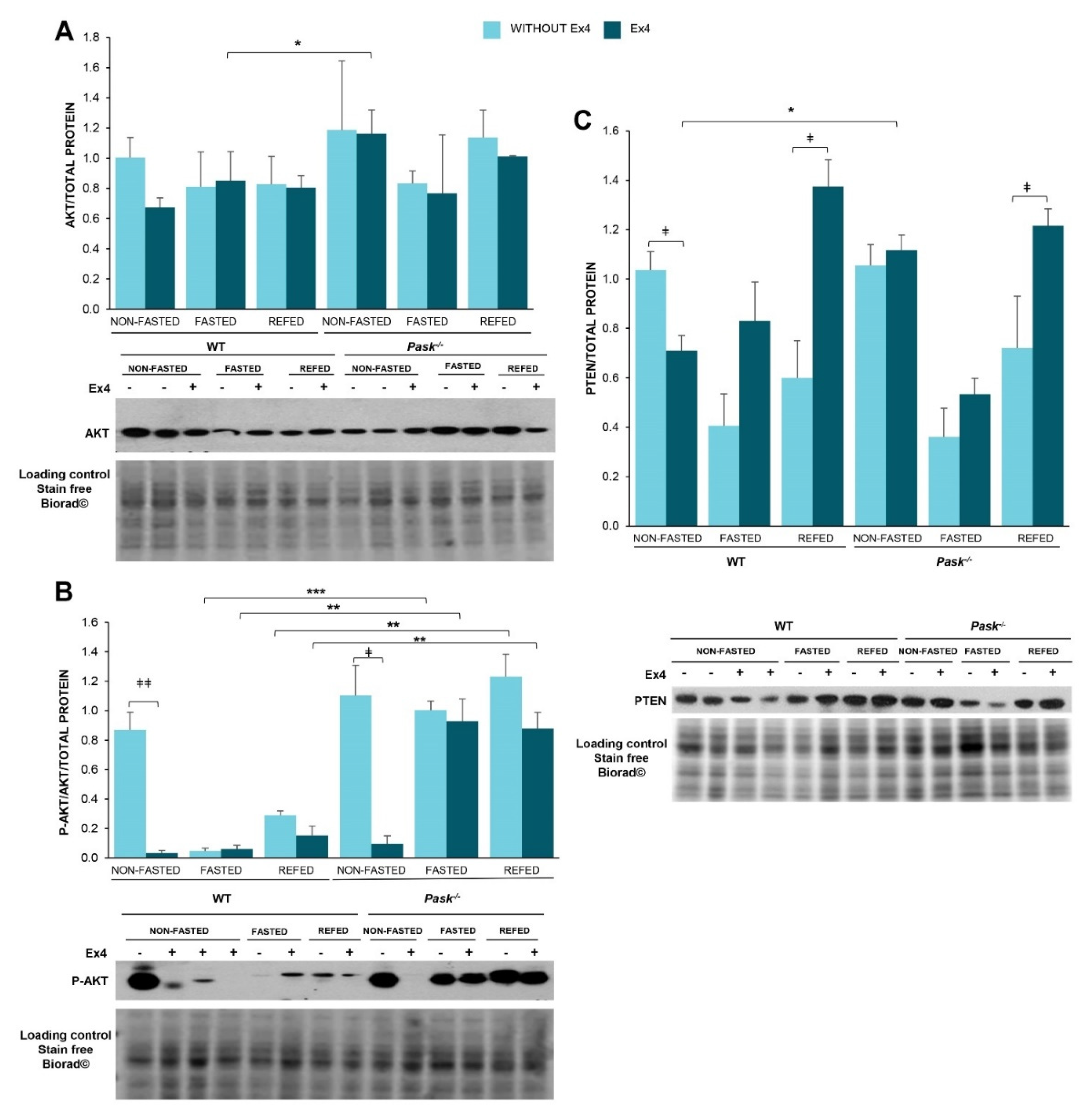
3.8. Exendin-4 Regulated AKT Activity Differently in the Basal State or after Fasting or Refeeding States, and It Is a PASK-Dependent Effect
3.9. Exendin-4-Modulated Expression of Hepatic Metabolic Genes in Response to Fasting and Refeeding, and Some of These Effects Are PASK-Dependent
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roach, P.J. Glycogen and its metabolism. Curr. Mol. Med. 2002, 2, 101–120. [Google Scholar] [CrossRef]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agius, L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem. J. 2008, 414, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.A.; Skurat, A.V.; Probst, B.; de Paoli-Roach, A.; Roach, P.J.; Rutter, J. Control of mammalian glycogen synthase by PAS kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 16596–16601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, E.; Roncero, I.; Chowen, J.A.; Thorens, B.; Blazquez, E. Expression of the glucagon-like peptide-1 receptor gene in rat brain. J. Neurochem. 1996, 66, 920–927. [Google Scholar] [CrossRef]
- Alvarez, E.; Martinez, M.D.; Roncero, I.; Chowen, J.A.; Garcia-Cuartero, B.; Gispert, J.D.; Sanz, C.; Vazquez, P.; Maldonado, A.; de Caceres, J.; et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J. Neurochem. 2005, 92, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.E.; Egan, J.M. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Ther. 2007, 113, 546–593. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Nielsen, L.L. Incretin mimetics and DPP-IV inhibitors for the treatment of type 2 diabetes. Drug Discov. Today 2005, 10, 703–710. [Google Scholar] [CrossRef]
- Villanueva-Penacarrillo, M.L.; Alcantara, A.I.; Clemente, F.; Delgado, E.; Valverde, I. Potent glycogenic effect of GLP-1(7-36)amide in rat skeletal muscle. Diabetologia 1994, 37, 1163–1166. [Google Scholar] [CrossRef] [PubMed]
- Luque, M.A.; Gonzalez, N.; Marquez, L.; Acitores, A.; Redondo, A.; Morales, M.; Valverde, I.; Villanueva-Penacarrillo, M.L. Glucagon-like peptide-1 (GLP-1) and glucose metabolism in human myocytes. J. Endocrinol. 2002, 173, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redondo, A.; Trigo, M.V.; Acitores, A.; Valverde, I.; Villanueva-Penacarrillo, M.L. Cell signalling of the GLP-1 action in rat liver. Mol. Cell. Endocrinol. 2003, 204, 43–50. [Google Scholar] [CrossRef]
- Valverde, I.; Morales, M.; Clemente, F.; Lopez-Delgado, M.I.; Delgado, E.; Perea, A.; Villanueva-Penacarrillo, M.L. Glucagon-like peptide 1: A potent glycogenic hormone. FEBS Lett. 1994, 349, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.X.; Rutter, J. The role of PAS kinase in regulating energy metabolism. IUBMB Life 2008, 60, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Schlafli, P.; Borter, E.; Spielmann, P.; Wenger, R.H. The PAS-domain kinase PASKIN: A new sensor in energy homeostasis. Cell. Mol. Life. Sci. 2009, 66, 876–883. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Rorsman, P. Per-arnt-sim (PAS) domain kinase (PASK) as a regulator of glucagon secretion. Diabetologia 2011, 54, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Carneiro, V.; Pérez-García, A.; Alvarez, E.; Sanz, C. PAS Kinase: A Nutrient and Energy Sensor “Master Key” in the Response to Fasting/Feeding Conditions. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Cardon, C.M.; Swiatek, W.; Cooksey, R.C.; Smith, T.L.; Wilde, J.; Boudina, S.; Abel, E.D.; McClain, D.A.; Rutter, J. PAS kinase is required for normal cellular energy balance. Proc. Natl. Acad. Sci. USA 2007, 104, 15466–15471. [Google Scholar] [CrossRef] [Green Version]
- Perez-Garcia, A.; Dongil, P.; Hurtado-Carneiro, V.; Blazquez, E.; Sanz, C.; Alvarez, E. High-fat diet alters PAS kinase regulation by fasting and feeding in liver. J. Nutr. Biochem. 2018, 57, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Carneiro, V.; Roncero, I.; Egger, S.S.; Wenger, R.H.; Blazquez, E.; Sanz, C.; Alvarez, E. PAS kinase is a nutrient and energy sensor in hypothalamic areas required for the normal function of AMPK and mTOR/S6K1. Mol. Neurobiol. 2014, 50, 314–326. [Google Scholar] [CrossRef]
- Hurtado-Carneiro, V.; Roncero, I.; Blazquez, E.; Alvarez, E.; Sanz, C. PAS Kinase as a Nutrient Sensor in Neuroblastoma and Hypothalamic Cells Required for the Normal Expression and Activity of Other Cellular Nutrient and Energy Sensors. Mol. Neurobiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Xavier, G.; Farhan, H.; Kim, H.; Caxaria, S.; Johnson, P.; Hughes, S.; Bugliani, M.; Marselli, L.; Marchetti, P.; Birzele, F.; et al. Per-arnt-sim (PAS) domain-containing protein kinase is downregulated in human islets in type 2 diabetes and regulates glucagon secretion. Diabetologia 2011, 54, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Perez-Garcia, A.; Dongil, P.; Hurtado-Carneiro, V.; Blazquez, E.; Sanz, C.; Alvarez, E. PAS Kinase deficiency alters the glucokinase function and hepatic metabolism. Sci Rep. 2018, 8, 11091. [Google Scholar] [CrossRef] [PubMed]
- Dongil, P.; Perez-Garcia, A.; Hurtado-Carneiro, V.; Herrero-de-Dios, C.; Blazquez, E.; Alvarez, E.; Sanz, C. Pas Kinase Deficiency Triggers Antioxidant Mechanisms in the Liver. Sci. Rep. 2018, 8, 13810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongil, P.; Perez-Garcia, A.; Hurtado-Carneiro, V.; Herrero-de-Dios, C.; Alvarez, E.; Sanz, C. PAS kinase deficiency reduces aging effects in mice. Aging 2020, 12, 2275–2301. [Google Scholar] [CrossRef] [PubMed]
- Kikani, C.K.; Wu, X.; Fogarty, S.; Kang, S.A.W.; Dephoure, N.; Gygi, S.P.; Sabatini, D.M.; Rutter, J. Activation of PASK by mTORC1 is required for the onset of the terminal differentiation program. Proc. Natl. Acad. Sci. USA 2019, 116, 10382–10391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakkat, J.V.; Kaimala, S.; Sreedharan, S.P.; Jayaprakash, P.; Adeghate, E.A.; Ansari, S.A.; Guccione, E.; Mensah-Brown, E.P.K.; Starling Emerald, B. The metabolic sensor PASK is a histone 3 kinase that also regulates H3K4 methylation by associating with H3K4 MLL2 methyltransferase complex. Nucleic Acids Res. 2019, 47, 10086–10103. [Google Scholar] [CrossRef]
- Katschinski, D.M.; Marti, H.H.; Wagner, K.F.; Shibata, J.; Eckhardt, K.; Martin, F.; Depping, R.; Paasch, U.; Gassmann, M.; Ledermann, B.; et al. Targeted disruption of the mouse PAS domain serine/threonine kinase PASKIN. Mol. Cell Biol. 2003, 23, 6780–6789. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Carneiro, V.; Sanz, C.; Roncero, I.; Vazquez, P.; Blazquez, E.; Alvarez, E. Glucagon-like peptide 1 (GLP-1) can reverse AMP-activated protein kinase (AMPK) and S6 kinase (P70S6K) activities induced by fluctuations in glucose levels in hypothalamic areas involved in feeding behaviour. Mol. Neurobiol. 2012, 45, 348–361. [Google Scholar] [CrossRef]
- Gilda, J.E.; Gomes, A.V. Stain-Free total protein staining is a superior loading control to beta-actin for Western blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [Green Version]
- Roncero, I.; Alvarez, E.; Vazquez, P.; Blazquez, E. Functional glucokinase isoforms are expressed in rat brain. J. Neurochem. 2000, 74, 1848–1857. [Google Scholar] [CrossRef]
- Bain, S.C.; Mosenzon, O.; Arechavaleta, R.; Bogdanski, P.; Comlekci, A.; Consoli, A.; Deerochanawong, C.; Dungan, K.; Faingold, M.C.; Farkouh, M.E.; et al. Cardiovascular safety of oral semaglutide in patients with type 2 diabetes: Rationale, design and patient baseline characteristics for the PIONEER 6 trial. Diabetes Obes. Metab. 2019, 21, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Semplici, F.; Vaxillaire, M.; Fogarty, S.; Semache, M.; Bonnefond, A.; Fontes, G.; Philippe, J.; Meur, G.; Diraison, F.; Sessions, R.B.; et al. Human mutation within Per-Arnt-Sim (PAS) domain-containing protein kinase (PASK) causes basal insulin hypersecretion. J. Biol. Chem. 2011, 286, 44005–44014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatek, W.; Parnell, K.M.; Nickols, G.A.; Scharschmidt, B.F.; Rutter, J. Validation of PAS Kinase, a Regulator of Hepatic Fatty Acid and Triglyceride Synthesis, as a Therapeutic Target for Nonalcoholic Steatohepatitis. Hepatol. Commun. 2020, 4, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Fisher, S.J.; Kahn, C.R. Insulin signaling is required for insulin’s direct and indirect action on hepatic glucose production. J. Clin. Investig. 2003, 111, 463–468. [Google Scholar] [CrossRef]
- Kim, S.P.; Ellmerer, M.; Van Citters, G.W.; Bergman, R.N. Primacy of hepatic insulin resistance in the development of the metabolic syndrome induced by an isocaloric moderate-fat diet in the dog. Diabetes 2003, 52, 2453–2460. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, D.; Eriksson, K.F.; Orho-Melander, M.; Fredriksson, J.; Ahlqvist, G.; Groop, L. Parallel manifestation of insulin resistance and beta cell decompensation is compatible with a common defect in Type 2 diabetes. Diabetologia 2004, 47, 782–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef]
- Wu, X.; Romero, D.; Swiatek, W.I.; Dorweiler, I.; Kikani, C.K.; Sabic, H.; Zweifel, B.S.; McKearn, J.; Blitzer, J.T.; Nickols, G.A.; et al. PAS kinase drives lipogenesis through SREBP-1 maturation. Cell Rep. 2014, 8, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scrocchi, L.A.; Marshall, B.A.; Cook, S.M.; Brubaker, P.L.; Drucker, D.J. Identification of glucagon-like peptide 1 (GLP-1) actions essential for glucose homeostasis in mice with disruption of GLP-1 receptor signaling. Diabetes 1998, 47, 632–639. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Tschop, M.H.; Woods, S.C.; Morton, G.J.; Myers, M.G.; D’Alessio, D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 2013, 503, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H. Central insulin-mediated regulation of hepatic glucose production [Review]. Endocr. J. 2016, 63, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Wang, J.; Wu, S.; Yuan, L.; Zhao, X.; Liu, C.; Xie, J.; Jia, Y.; Lai, Y.; Zhao, A.Z.; et al. Duodenal GLP-1 signaling regulates hepatic glucose production through a PKC-delta-dependent neurocircuitry. Cell Death Dis. 2017, 8, e2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86. [Google Scholar] [CrossRef]
- Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 474–485. [Google Scholar] [CrossRef]
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002, 277, 9520–9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Yu, X.; Murao, K.; Imachi, H.; Li, J.; Muraoka, T.; Masugata, H.; Zhang, G.X.; Kobayashi, R.; Ishida, T.; et al. Exendin-4 regulates GLUT2 expression via the CaMKK/CaMKIV pathway in a pancreatic beta-cell line. Metabolism 2011, 60, 579–585. [Google Scholar] [CrossRef]
- Iynedjian, P.B.; Marie, S.; Wang, H.; Gjinovci, A.; Nazaryan, K. Liver-specific enhancer of the glucokinase gene. J. Biol. Chem. 1996, 271, 29113–29120. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, M.A.; Andreone, T.L.; Printz, R.L.; Koch, S.; Granner, D.K. Rat glucokinase gene: Structure and regulation by insulin. Proc. Natl. Acad. Sci. USA 1989, 86, 4838–4842. [Google Scholar] [CrossRef] [Green Version]
- Iynedjian, P.B. Mammalian glucokinase and its gene. Biochem. J. 1993, 293 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Iizuka, K.; Horikawa, Y. ChREBP: A glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr. J. 2008, 55, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef]
- Van Dijk, T.H.; van der Sluijs, F.H.; Wiegman, C.H.; Baller, J.F.; Gustafson, L.A.; Burger, H.J.; Herling, A.W.; Kuipers, F.; Meijer, A.J.; Reijngoud, D.J. Acute inhibition of hepatic glucose-6-phosphatase does not affect gluconeogenesis but directs gluconeogenic flux toward glycogen in fasted rats. A pharmacological study with the chlorogenic acid derivative S4048. J. Biol. Chem. 2001, 276, 25727–25735. [Google Scholar] [CrossRef] [Green Version]
- Gannon, M.C.; Nuttall, F.Q. Effect of feeding, fasting, and diabetes on liver glycogen synthase activity, protein, and mRNA in rats. Diabetologia 1997, 40, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Wititsuwannakul, D.; Kim, K.H. Immunological studies of liver glycogen synthase. Relative significance of covalent modification and changes in the rate of synthesis and degradation. J. Biol. Chem. 1979, 254, 3562–3569. [Google Scholar] [CrossRef]
- Ros, S.; Garcia-Rocha, M.; Dominguez, J.; Ferrer, J.C.; Guinovart, J.J. Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. J. Biol. Chem. 2009, 284, 6370–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, G.I.; Rothman, D.L.; Jue, T.; Stein, P.; DeFronzo, R.A.; Shulman, R.G. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 1990, 322, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Tantiwong, P.; Stuenaes, J.T.; Molina-Carrion, M.; DeFronzo, R.A.; Sakamoto, K.; Musi, N. Effect of acute exercise on glycogen synthase in muscle from obese and diabetic subjects. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E82–E89. [Google Scholar] [CrossRef] [Green Version]
- Skurat, A.V.; Wang, Y.; Roach, P.J. Rabbit skeletal muscle glycogen synthase expressed in COS cells. Identification of regulatory phosphorylation sites. J. Biol. Chem. 1994, 269, 25534–25542. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Accili, D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011, 14, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood GLP-1 Concentration (pM) | ||
|---|---|---|
| Nutritional State | WT | Pask −/− |
| NON-FASTED | 4.13 ± 1.15 | 0.78 ± 0.38 * |
| FASTED (48H) | 1.21± 0.82 | 4.29 ± 2.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-García, A.; Hurtado-Carneiro, V.; Herrero-De-Dios, C.; Dongil, P.; García-Mauriño, J.E.; Sánchez, M.D.; Sanz, C.; Álvarez, E. Storage and Utilization of Glycogen by Mouse Liver during Adaptation to Nutritional Changes Are GLP-1 and PASK Dependent. Nutrients 2021, 13, 2552. https://doi.org/10.3390/nu13082552
Pérez-García A, Hurtado-Carneiro V, Herrero-De-Dios C, Dongil P, García-Mauriño JE, Sánchez MD, Sanz C, Álvarez E. Storage and Utilization of Glycogen by Mouse Liver during Adaptation to Nutritional Changes Are GLP-1 and PASK Dependent. Nutrients. 2021; 13(8):2552. https://doi.org/10.3390/nu13082552
Chicago/Turabian StylePérez-García, Ana, Verónica Hurtado-Carneiro, Carmen Herrero-De-Dios, Pilar Dongil, José Enrique García-Mauriño, María Dolores Sánchez, Carmen Sanz, and Elvira Álvarez. 2021. "Storage and Utilization of Glycogen by Mouse Liver during Adaptation to Nutritional Changes Are GLP-1 and PASK Dependent" Nutrients 13, no. 8: 2552. https://doi.org/10.3390/nu13082552
APA StylePérez-García, A., Hurtado-Carneiro, V., Herrero-De-Dios, C., Dongil, P., García-Mauriño, J. E., Sánchez, M. D., Sanz, C., & Álvarez, E. (2021). Storage and Utilization of Glycogen by Mouse Liver during Adaptation to Nutritional Changes Are GLP-1 and PASK Dependent. Nutrients, 13(8), 2552. https://doi.org/10.3390/nu13082552