
Saccharin and Sucralose Protect the Glomerular Microvasculature In Vitro against VEGF-Induced Permeability


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Line and Reagents
2.2. Cell Viability Assay
2.3. Endothelial Monolayer Permeability
2.4. ELISA Studies for VE-Cadherin and cAMP
2.5. Oxidative Stress Studies
2.6. Gas Chromatography–Mass Spectrometry
2.7. Preparation of GMVEC for GC–MS Analysis
2.8. Statistical Analysis
3. Results
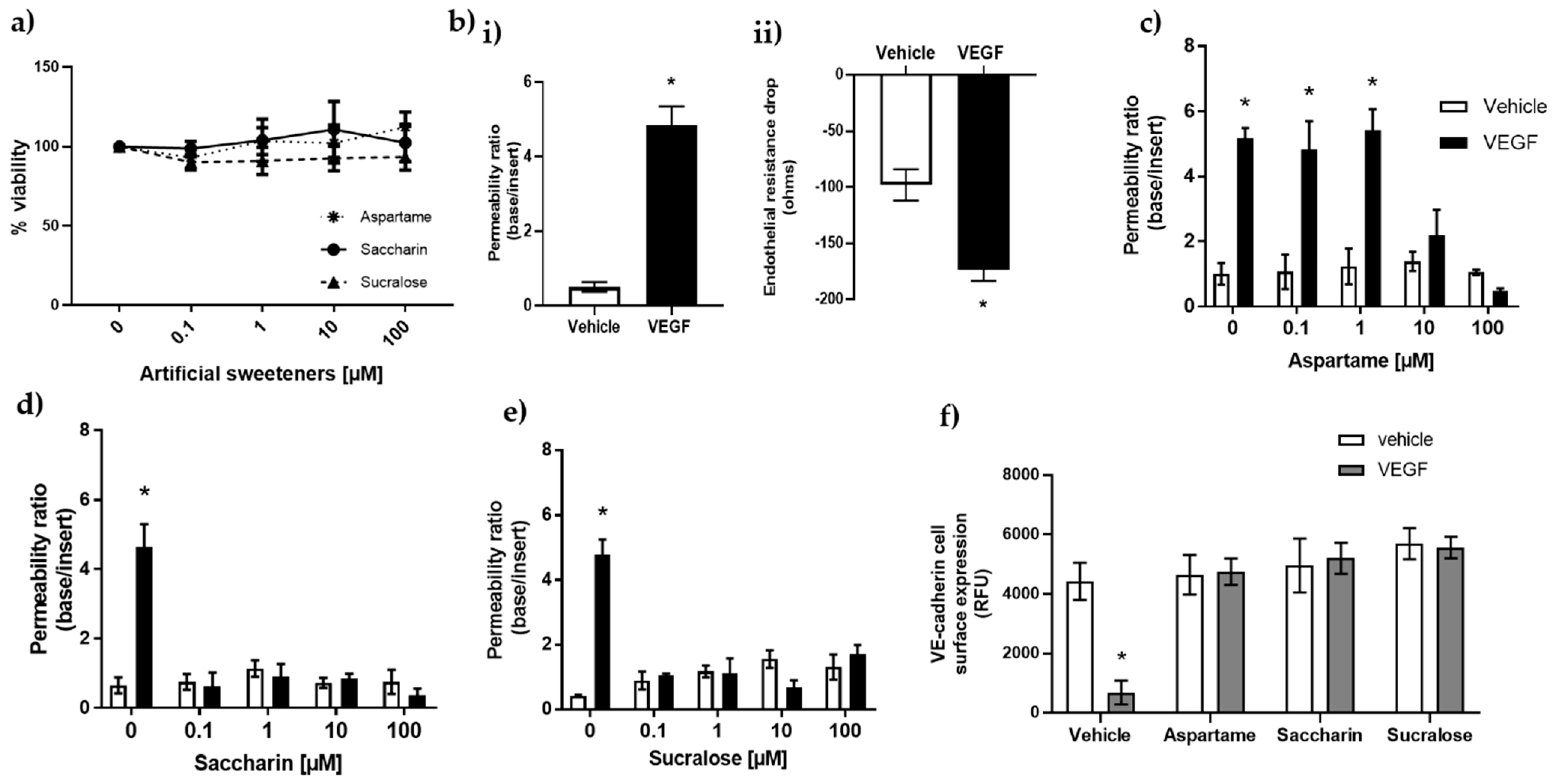
3.1. Artificial Sweeteners Saccharin and Sucralose Attenuate VEGF-Induced Permeability in Glomerular Microvascular Endothelium
3.2. Aspartame, Saccharin, and Sucralose Attenuate VEGF-Induced Permeability via the T1R3 Sweet Taste Receptor
3.3. Artificial Sweeteners Do Not Impact cAMP or Oxidative Stress Pathways in the Glomerular Microvasculature
3.4. Analysis and Detection of Sucralose by GC–MS
3.5. The Artificial Sweetener Sucralose Does Not Enter the Glomerular Endothelium
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martyn, D.; Darch, M.; Roberts, A.; Lee, H.Y.; Yaqiong Tian, T.; Kaburagi, N.; Belmar, P. Low-/No-Calorie Sweeteners: A Review of Global Intakes. Nutrients 2018, 10, 357. [Google Scholar] [CrossRef] [Green Version]
- Gardner, C.; Wylie-Rosett, J.; Gidding, S.S.; Sen, L.M.; Johnson, R.K.; Reader, D.; Lichtenstein, A.H. American Heart Association Nutrition Committee of the Council on Nutrition, Physical Activity and Metabolism, American Diabetes Association. Nonnutritive sweeteners: Current use and health perspectives: A scientific statement from the American Heart Association and the American Diabetes Association. Diabetes Care 2012, 35, 1798–1808. [Google Scholar]
- Fujiwara, S.; Imada, T.; Nakagita, T.; Okada, S.; Nammoku, T.; Abe, K.; Misaka, T. Sweeteners interacting with the transmembrane domain of the human sweet-taste receptor induce sweet-taste synergisms in binary mixtures. Food Chem. 2012, 130, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Mahalapbutr, P.; Darai, N.; Panman, W.; Opasmahakul, A.; Kungwan, N.; Hannongbua, S.; Rungrotmongkol, T. Atomistic mechanisms underlying the activation of the G protein-coupled sweet receptor heterodimer by sugar alcohol recognition. Sci. Rep. 2019, 9, 10205. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Cui, M.; Zhao, B.; Liu, Z.; Snyder, L.A.; Benard, L.M.; Osman, R.; Margolskee, R.F.; Max, M. Lactisole interacts with the transmembrane domains of human T1R3 to inhibit sweet taste. J. Biol. Chem. 2005, 280, 15238–15246. [Google Scholar] [CrossRef] [Green Version]
- Eaton, M.S.; Weinstein, N.; Newby, J.B.; Plattes, M.M.; Foster, H.E.; Arthur, J.W.; Ward, T.D.; Shively, S.R.; Shor, R.; Nathan, J.; et al. Loss of the nutrient sensor TAS1R3 leads to reduced bone resorption. J. Physiol. Biochem. 2018, 74, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Harrington, E.O.; Vang, A.; Braza, J.; Shil, A.; Chichger, H. Activation of the sweet taste receptor, T1R3, by the artificial sweetener sucralose regulates the pulmonary endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L165–L176. [Google Scholar] [CrossRef]
- Lizunkova, P.; Enuwosa, E.; Chichger, H. Activation of the sweet taste receptor T1R3 by sucralose attenuates VEGF-induced vasculogenesis in a cell model of the retinal microvascular endothelium. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, B. Receptors and transduction in taste. Nature 2001, 413, 219–225. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nagasawa, M.; Yamada, S.; Hara, A.; Mogami, H.; Nikolaev, V.O.; Lohse, M.J.; Shigemura, N.; Ninomiya, Y.; Kojima, I. Sweet taste receptor expressed in pancreatic beta-cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS ONE 2009, 4, e5106. [Google Scholar] [CrossRef] [Green Version]
- Gilbertson, T.A.; Damak, S.; Margolskee, R.F. The molecular physiology of taste transduction. Curr. Opin. Neurobiol. 2000, 10, 519–527. [Google Scholar] [CrossRef]
- Foster, S.R.; Porrello, E.R.; Stefani, M.; Smith, N.J.; Molenaar, P.; dos Remedios, C.G.; Thomas, W.G.; Ramialison, M. Cardiac gene expression data and in silico analysis provide novel insights into human and mouse taste receptor gene regulation. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 1009–1027. [Google Scholar] [CrossRef]
- Elliott, R.A.; Kapoor, S.; Tincello, D.G. Expression and distribution of the sweet taste receptor isoforms T1R2 and T1R3 in human and rat bladders. J. Urol. 2011, 186, 2455–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shil, A.; Olusanya, O.; Ghufoor, Z.; Forson, B.; Marks, J.; Chichger, H. Artificial Sweeteners Disrupt Tight Junctions and Barrier Function in the Intestinal Epithelium through Activation of the Sweet Taste Receptor, T1R3. Nutrients 2020, 12, 1862. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Gui, D.; Zheng, L.; Zhai, R.; Wang, F.; Wang, N. Mechanistic Insight and Management of Diabetic Nephropathy: Recent Progress and Future Perspective. J. Diabetes Res. 2017, 2017, 1839809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lee, K.; Ni, Z.; He, J.C. Diabetic Kidney Disease: Challenges, Advances, and Opportunities. Kidney Dis. 2020, 6, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Groop, P.H.; Thomas, M.C.; Moran, J.L.; Waden, J.; Thorn, L.M.; Makinen, V.P.; Rosengard-Barlund, M.; Saraheimo, M.; Hietala, K.; Heikkila, O.; et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes 2009, 58, 1651–1658. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, A.; Forbes, J.M. Diabetic kidney disease: A role for advanced glycation end-product receptor 1 (AGE-R1)? Glycoconj. J. 2016, 33, 645–652. [Google Scholar] [CrossRef]
- Retnakaran, R.; Cull, C.A.; Thorne, K.I.; Adler, A.I.; Holman, R.R.; UKPDS Study Group. Risk factors for renal dysfunction in type 2 diabetes: U.K. Prospective Diabetes Study 74. Diabetes 2006, 55, 1832–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H. Diabetes microvascular complications-A clinical update. Diabetes Metab. Syndr. 2017, 11 (Suppl. 1), S133–S139. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; Gao, L.; Siu, P.M.; Lai, C.W. Diabetic nephropathy and endothelial dysfunction: Current and future therapies, and emerging of vascular imaging for preclinical renal-kinetic study. Life Sci. 2016, 166, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fioretto, P.; Mauer, M. Histopathology of diabetic nephropathy. Semin. Nephrol. 2007, 27, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, I.; Agrawal, U.; Amitabh, V.; Jain, A.K.; Saxena, S. Thickness of glomerular and tubular basement membranes in preclinical and clinical stages of diabetic nephropathy. Indian J. Nephrol. 2008, 18, 64–69. [Google Scholar] [PubMed]
- Osterby, R.; Tapia, J.; Nyberg, G.; Tencer, J.; Willner, J.; Rippe, B.; Torffvit, O. Renal structures in type 2 diabetic patients with elevated albumin excretion rate. APMIS 2001, 109, 751–761. [Google Scholar] [CrossRef]
- Saito, Y.; Kida, H.; Takeda, S.; Yoshimura, M.; Yokoyama, H.; Koshino, Y.; Hattori, N. Mesangiolysis in diabetic glomeruli: Its role in the formation of nodular lesions. Kidney Int. 1988, 34, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, K.; Mauer, M.; International Diabetic Nephropathy Study Group. The early natural history of nephropathy in type 1 diabetes: II. Early renal structural changes in type 1 diabetes. Diabetes 2002, 51, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.E.; Vranes, D.; Youssef, S.; Stacker, S.A.; Cox, A.J.; Rizkalla, B.; Casley, D.J.; Bach, L.A.; Kelly, D.J.; Gilbert, R.E. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 1999, 48, 2229–2239. [Google Scholar] [CrossRef]
- Braun, L.; Kardon, T.; Reisz-Porszasz, Z.S.; Banhegyi, G.; Mandl, J. The regulation of the induction of vascular endothelial growth factor at the onset of diabetes in spontaneously diabetic rats. Life Sci. 2001, 69, 2533–2542. [Google Scholar] [CrossRef]
- Hohenstein, B.; Hausknecht, B.; Boehmer, K.; Riess, R.; Brekken, R.A.; Hugo, C.P. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 2006, 69, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- Vriese, A.S.; Tilton, R.G.; Elger, M.; Stephan, C.C.; Kriz, W.; Lameire, N.H. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J. Am. Soc. Nephrol. 2001, 12, 993–1000. [Google Scholar] [CrossRef]
- Lafayette, R.A.; McCall, B.; Li, N.; Chu, L.; Werner, P.; Das, A.; Glassock, R. Incidence and relevance of proteinuria in bevacizumab-treated patients: Pooled analysis from randomized controlled trials. Am. J. Nephrol. 2014, 40, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, W.; Wang, Z.; Nie, W.; Guo, Y.; Huang, L. GC–MS Determination of Sucralose in Splenda. Chromatographia 2007, 66, 935–939. [Google Scholar] [CrossRef]
- International Conference on Harmonization (ICH) Topic Q 2 (R1). Validation of Analytical Procedures: Text and Methodology Step 5 Note for Guidance on Validation of Analytical Procedures. 2005. Available online: https://www.gmp-compliance.org/files/guidemgr/Q2(R1).pdf (accessed on 15 April 2021).
- Gunduz, D.; Troidl, C.; Tanislav, C.; Rohrbach, S.; Hamm, C.; Aslam, M. Role of PI3K/Akt and MEK/ERK Signalling in cAMP/Epac-Mediated Endothelial Barrier Stabilisation. Front. Physiol. 2019, 10, 1387. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, B.A.; Carakostas, M.C.; Moore, N.H.; Poulos, S.P.; Renwick, A.G. Biological fate of low-calorie sweeteners. Nutr. Rev. 2016, 74, 670–689. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.; Renwick, A.G.; Sims, J.; Snodin, D.J. Sucralose metabolism and pharmacokinetics in man. Food Chem. Toxicol. 2000, 38 (Suppl. 2), S31–S41. [Google Scholar] [CrossRef]
- Sylvetsky, A.C.; Bauman, V.; Blau, J.E.; Garraffo, H.M.; Walter, P.J.; Rother, K.I. Plasma concentrations of sucralose in children and adults. Toxicol. Environ. Chem. 2017, 99, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, I.M.; Shephard, N.W.; Merritt, R.J.; Hildick-Smith, G. Repeated dose study of sucralose tolerance in human subjects. Food Chem. Toxicol. 2000, 38 (Suppl. 2), S123–S129. [Google Scholar] [CrossRef]
- Nakagita, T.; Ishida, A.; Matsuya, T.; Kobayashi, T.; Narukawa, M.; Hirokawa, T.; Hashimoto, M.; Misaka, T. Structural insights into the differences among lactisole derivatives in inhibitory mechanisms against the human sweet taste receptor. PLoS ONE 2019, 14, e0213552. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.J.; Lin, C.; Liu, X.; Antonetti, D.A. The EPAC-Rap1 pathway prevents and reverses cytokine-induced retinal vascular permeability. J. Biol. Chem. 2018, 293, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Monaghan-Benson, E.; Burridge, K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J. Biol. Chem. 2009, 284, 25602–25611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE 2000, 2000, pe1. [Google Scholar] [CrossRef] [PubMed]
- Bastiaan-Net, S.; van den Berg-Somhorst, D.; Ariens, R.; Paques, M.; Mes, J. A novel functional screening assay to monitor sweet taste receptor activation in vitro. Flavour Fragr. J. 2018, 33, 173–183. [Google Scholar] [CrossRef]
- Ruiz-Matute, A.I.; Hernandez-Hernandez, O.; Rodriguez-Sanchez, S.; Sanz, M.L.; Martinez-Castro, I. Derivatization of carbohydrates for GC and GC-MS analyses. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1226–1240. [Google Scholar] [CrossRef]
- Scientific Working Group for Forensic Toxicology. Scientific Working Group for Forensic Toxicology (SWGTOX) Standard Practices for Method Validation in Forensic Toxicology. J. Anal. Toxicol. 2013, 37, 452–474. [Google Scholar] [CrossRef]
- Lopez-Sanchez, R.D.C.; Lara-Diaz, V.J.; Aranda-Gutierrez, A.; Martinez-Cardona, J.A.; Hernandez, J.A. HPLC Method for Quantification of Caffeine and Its Three Major Metabolites in Human Plasma Using Fetal Bovine Serum Matrix to Evaluate Prenatal Drug Exposure. J Anal. Methods Chem. 2018, 2018, 2085059. [Google Scholar] [CrossRef] [Green Version]
- Hayman, S.R.; Leung, N.; Grande, J.P.; Garovic, V.D. VEGF inhibition, hypertension, and renal toxicity. Curr. Oncol. Rep. 2012, 14, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Oh, J.H.; Seo, J.A.; Lee, K.W.; Kim, S.G.; Choi, K.M.; Baik, S.H.; Choi, D.S.; Kang, Y.S.; Han, S.Y.; et al. Vascular endothelial growth factor (VEGF) and soluble VEGF receptor FLT-1 in diabetic nephropathy. Kidney Int. 2005, 67, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Lv, C.; Yuan, Q.; Wang, Q. Levels of Serum 25(OH)VD3, HIF-1alpha, VEGF, vWf, and IGF-1 and Their Correlation in Type 2 Diabetes Patients with Different Urine Albumin Creatinine Ratio. J. Diabetes Res. 2016, 2016, 1925424. [Google Scholar] [CrossRef] [Green Version]
- Sato, W.; Tanabe, K.; Kosugi, T.; Hudkins, K.; Lanaspa, M.A.; Zhang, L.; Campbell-Thompson, M.; Li, Q.; Long, D.A.; Alpers, C.E.; et al. Selective stimulation of VEGFR2 accelerates progressive renal disease. Am. J. Pathol. 2011, 179, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.B. Rethinking glomerular basement membrane thickening in diabetic nephropathy: Adaptive or pathogenic? Am. J. Physiol. Ren. Physiol. 2016, 311, F831–F843. [Google Scholar] [CrossRef] [Green Version]
- Flyvbjerg, A.; Dagnaes-Hansen, F.; De Vriese, A.S.; Schrijvers, B.F.; Tilton, R.G.; Rasch, R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes 2002, 51, 3090–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, S.H.; Ziyadeh, F.N.; Wang, A.; Pyagay, P.E.; Kanwar, Y.S.; Chen, S. Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J. Am. Soc. Nephrol. 2006, 17, 3093–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Wu, S.; Dahut, W.L.; Parikh, C.R. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: Systematic review and meta-analysis. Am. J. Kidney Dis. 2007, 49, 186–193. [Google Scholar] [CrossRef]
- Patel, T.V.; Morgan, J.A.; Demetri, G.D.; George, S.; Maki, R.G.; Quigley, M.; Humphreys, B.D. A preeclampsia-like syndrome characterized by reversible hypertension and proteinuria induced by the multitargeted kinase inhibitors sunitinib and sorafenib. J. Natl. Cancer Inst. 2008, 100, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, E.S.; Khankin, E.V.; Choueiri, T.K.; Dhawan, M.S.; Rogers, M.J.; Karumanchi, S.A.; Humphreys, B.D. Suppression of the nitric oxide pathway in metastatic renal cell carcinoma patients receiving vascular endothelial growth factor-signaling inhibitors. Hypertension 2010, 56, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Schiano, C.; Grimaldi, V.; Franzese, M.; Fiorito, C.; De Nigris, F.; Donatelli, F.; Soricelli, A.; Salvatore, M.; Napoli, C. Non-nutritional sweeteners effects on endothelial vascular function. Toxicol. In Vitro 2020, 62, 104694. [Google Scholar] [CrossRef]
- Zhou, L.; Huang, W.; Xu, Y.; Gao, C.; Zhang, T.; Guo, M.; Liu, Y.; Ding, J.; Qin, L.; Xu, Z.; et al. Sweet Taste Receptors Mediated ROS-NLRP3 Inflammasome Signaling Activation: Implications for Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 7078214. [Google Scholar] [CrossRef]
- Masuda, K.; Koizumi, A.; Nakajima, K.; Tanaka, T.; Abe, K.; Misaka, T.; Ishiguro, M. Characterization of the modes of binding between human sweet taste receptor and low-molecular-weight sweet compounds. PLoS ONE 2012, 7, e35380. [Google Scholar] [CrossRef]
- Risdon, S.; Meyer, G.; Marziou, A.; Riva, C.; Roustit, M.; Walther, G. Artificial sweeteners impair endothelial vascular reactivity: Preliminary results in rodents. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 843–846. [Google Scholar] [CrossRef]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.R.; Parlee, S.D.; Learman, B.S.; Mori, H.; Scheller, E.L.; Cawthorn, W.P.; Ning, X.; Gallagher, K.; Tyrberg, B.; Assadi-Porter, F.M.; et al. Artificial sweeteners stimulate adipogenesis and suppress lipolysis independently of sweet taste receptors. J. Biol. Chem. 2013, 288, 32475–32489. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | ||||||||
| siRNA | Permeability Ratio (Base/Insert) | |||||||
| Scrambled | T1R3 | |||||||
| Treatment: | Vehicle | Aspartame | Saccharin | Sucralose | Vehicle | Aspartame | Saccharin | Sucralose |
| Vehicle | 1.77 ± 0.25 | 1.99 ± 0.57 | 1.88 ± 0.73 | 1.71 ± 0.58 | 1.95 ± 0.38 | 2.06 ± 0.61 | 1.89 ± 0.67 | 1.59 ± 0.73 |
| VEGF | 5.91 ± 0.62 * | 2.05 ± 0.41 | 2.01 ± 0.60 | 1.89 ± 0.36 | 5.79 ± 0.55 * | 1.83 ± 0.71 | 2.13 ± 0.82 | 1.98 ± 0.40 |
| (b) | ||||||||
| siRNA | VE-Cadherin Cell-Surface Expression (r.f.u.) | |||||||
| Scrambled | T1R3 | |||||||
| Treatment: | Vehicle | Aspartame | Saccharin | Sucralose | Vehicle | Aspartame | Saccharin | Sucralose |
| Vehicle | 4038 ± 693 | 4126 ± 827 | 4150 ± 1017 | 4061 ± 904 | 4183 ± 1027 | 5199 ± 757 | 4289 ± 863 | 4890 ± 1007 |
| VEGF | 971 ± 183 * | 4090 ± 938 | 4281 ± 1105 | 4135 ± 1036 | 891 ± 130 * | 4937 ± 812 | 3980 ± 1170 | 4136 ± 1021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enuwosa, E.; Gautam, L.; King, L.; Chichger, H. Saccharin and Sucralose Protect the Glomerular Microvasculature In Vitro against VEGF-Induced Permeability. Nutrients 2021, 13, 2746. https://doi.org/10.3390/nu13082746
Enuwosa E, Gautam L, King L, Chichger H. Saccharin and Sucralose Protect the Glomerular Microvasculature In Vitro against VEGF-Induced Permeability. Nutrients. 2021; 13(8):2746. https://doi.org/10.3390/nu13082746
Chicago/Turabian StyleEnuwosa, Emmanuella, Lata Gautam, Linda King, and Havovi Chichger. 2021. "Saccharin and Sucralose Protect the Glomerular Microvasculature In Vitro against VEGF-Induced Permeability" Nutrients 13, no. 8: 2746. https://doi.org/10.3390/nu13082746