
Phospholipid Profiles Are Selectively Altered in the Putamen and White Frontal Cortex of Huntington’s Disease

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Brain Tissue
2.2. Lipid Extraction
2.3. Mass Spectrometry
2.4. Data Processing
2.5. Statistical Analysis
3. Results
3.1. Principal Component Analyses of Control and HD Brain Regions
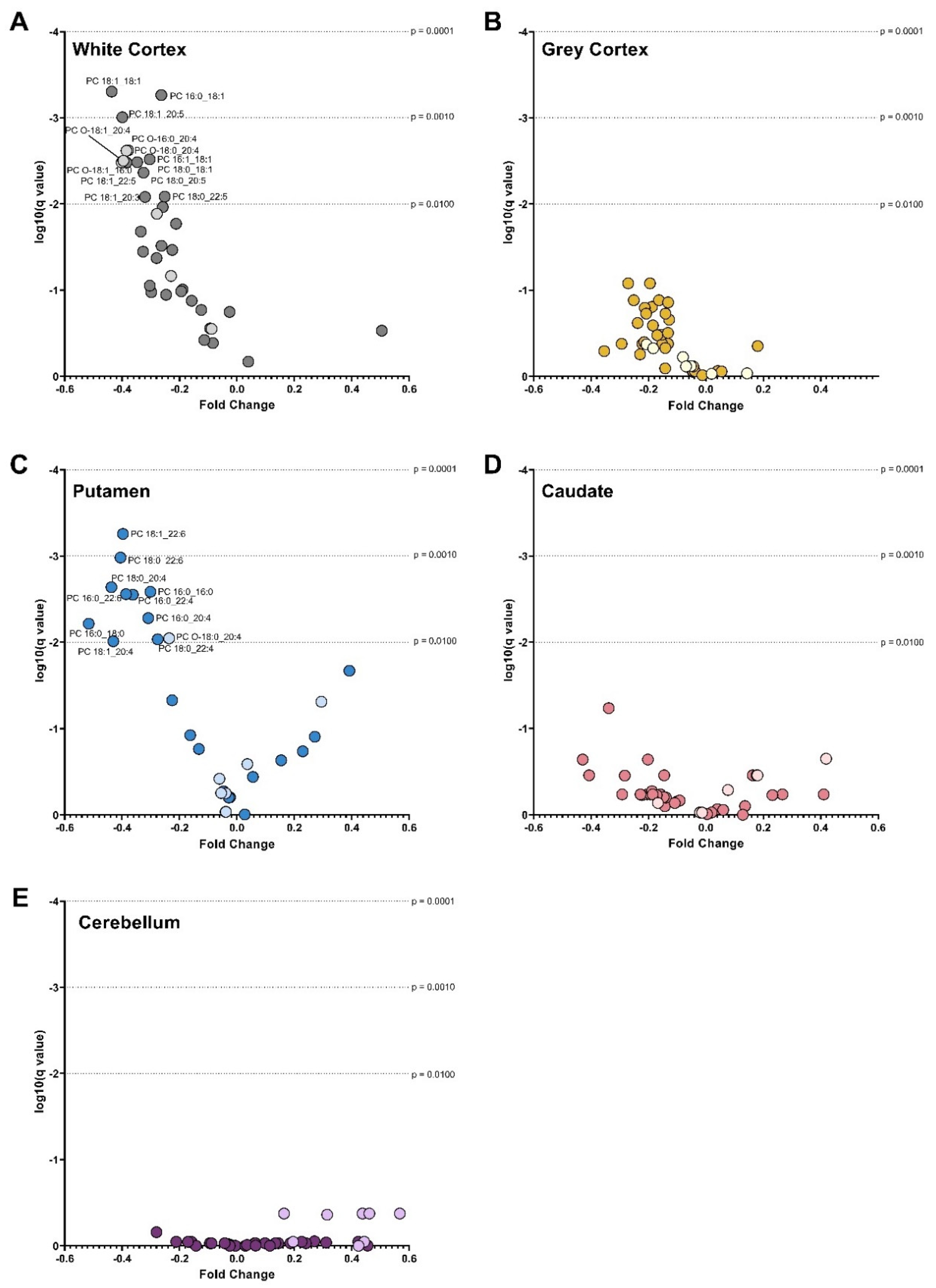
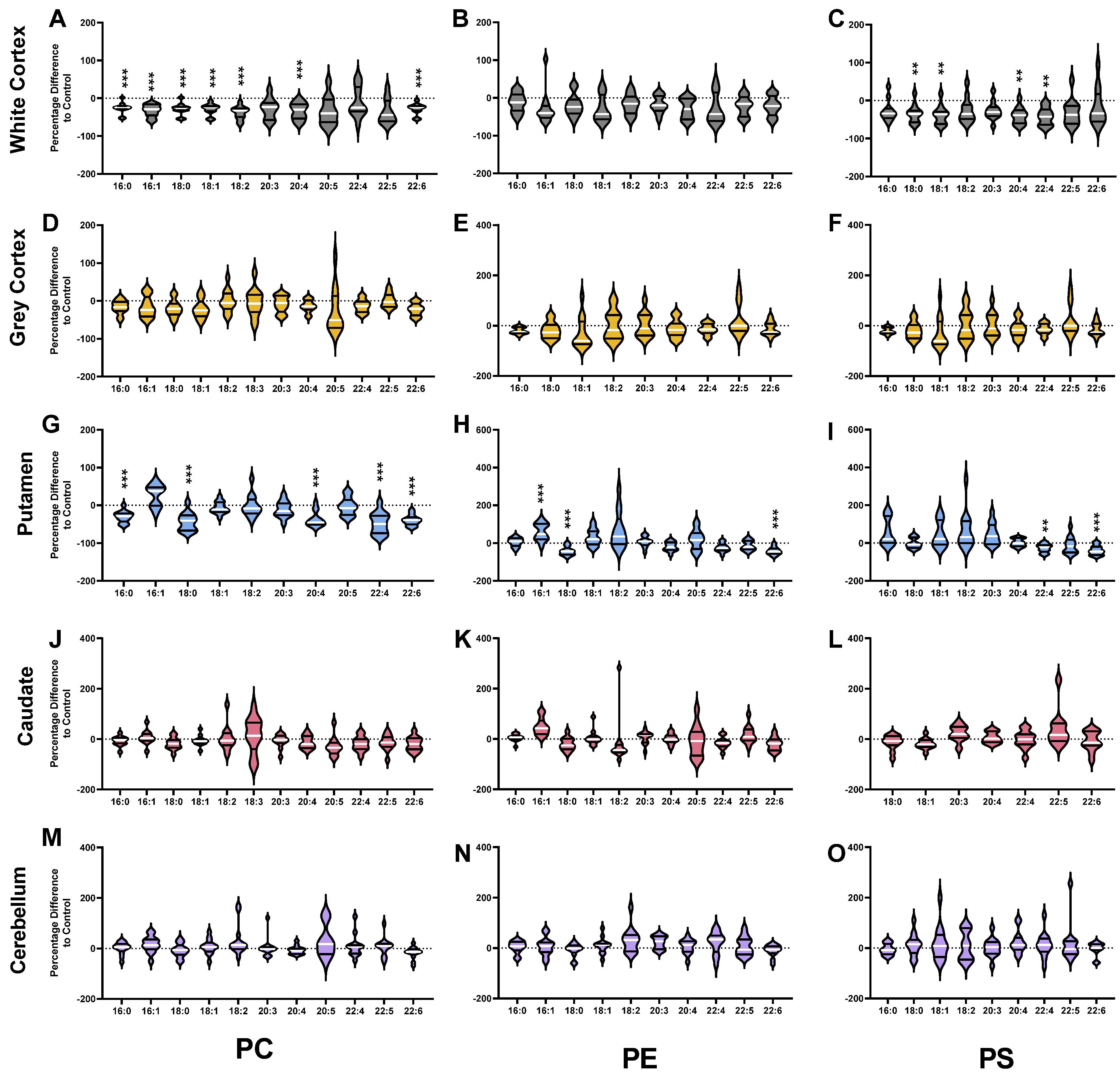
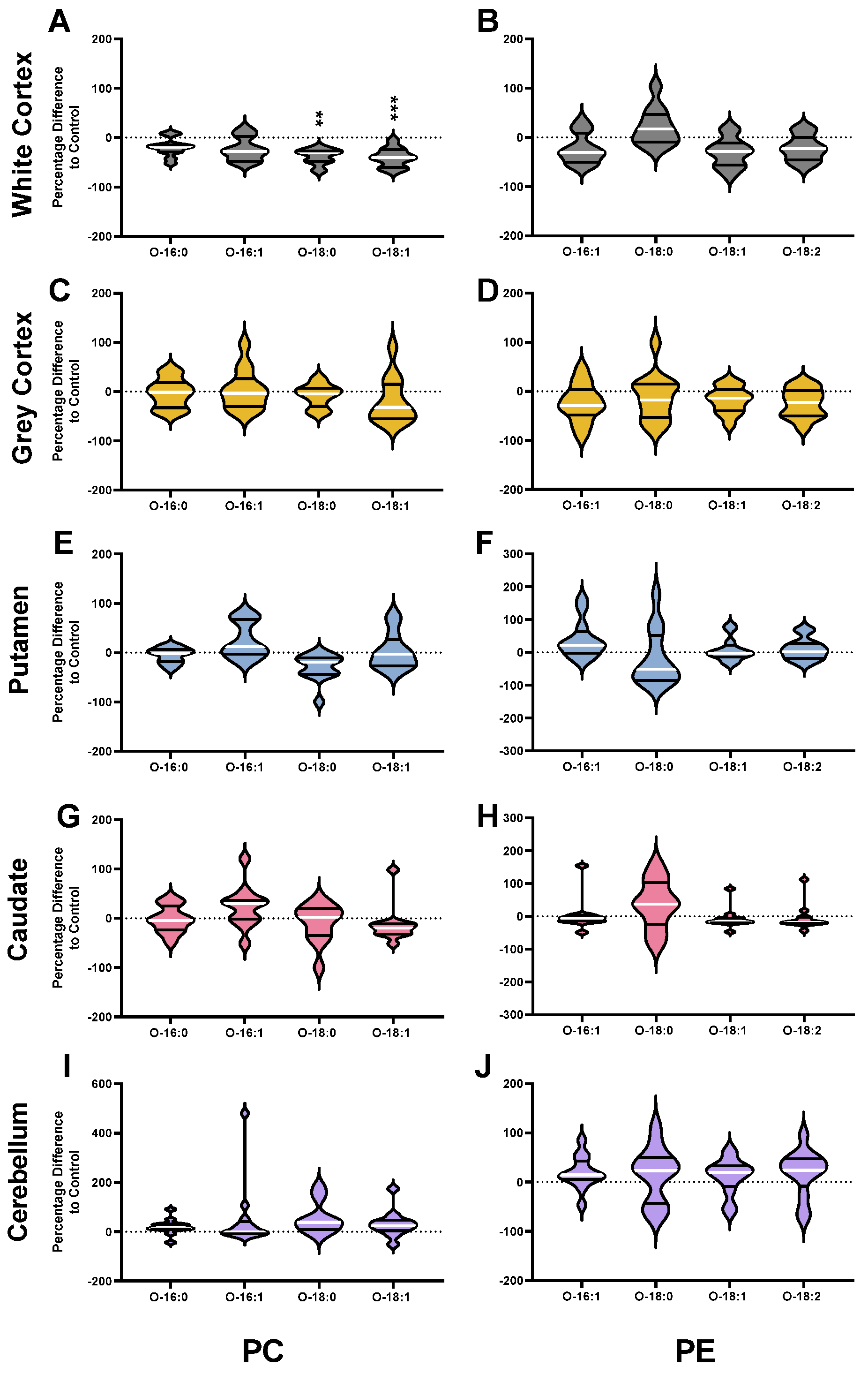
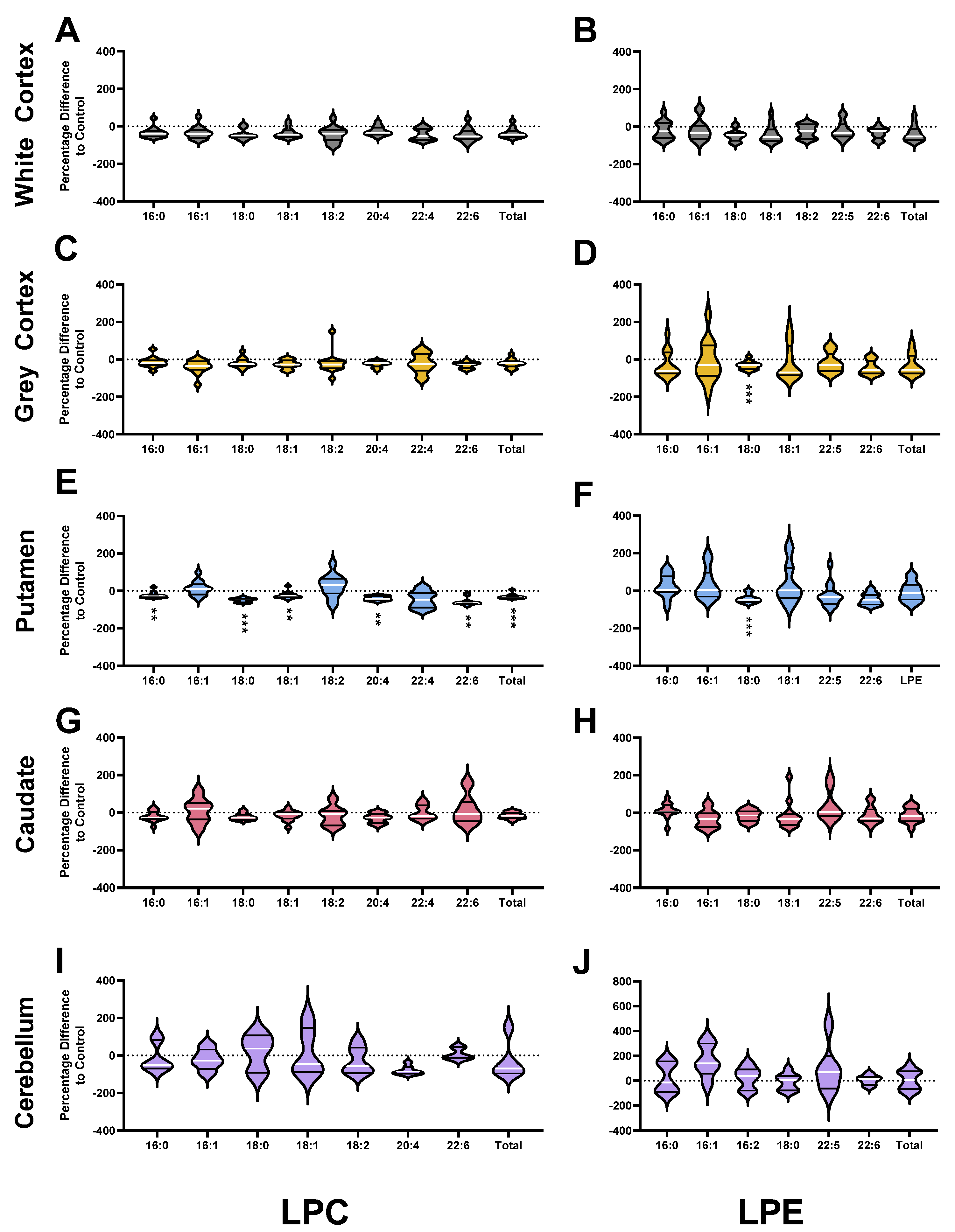
3.2. Phosphatidylcholines
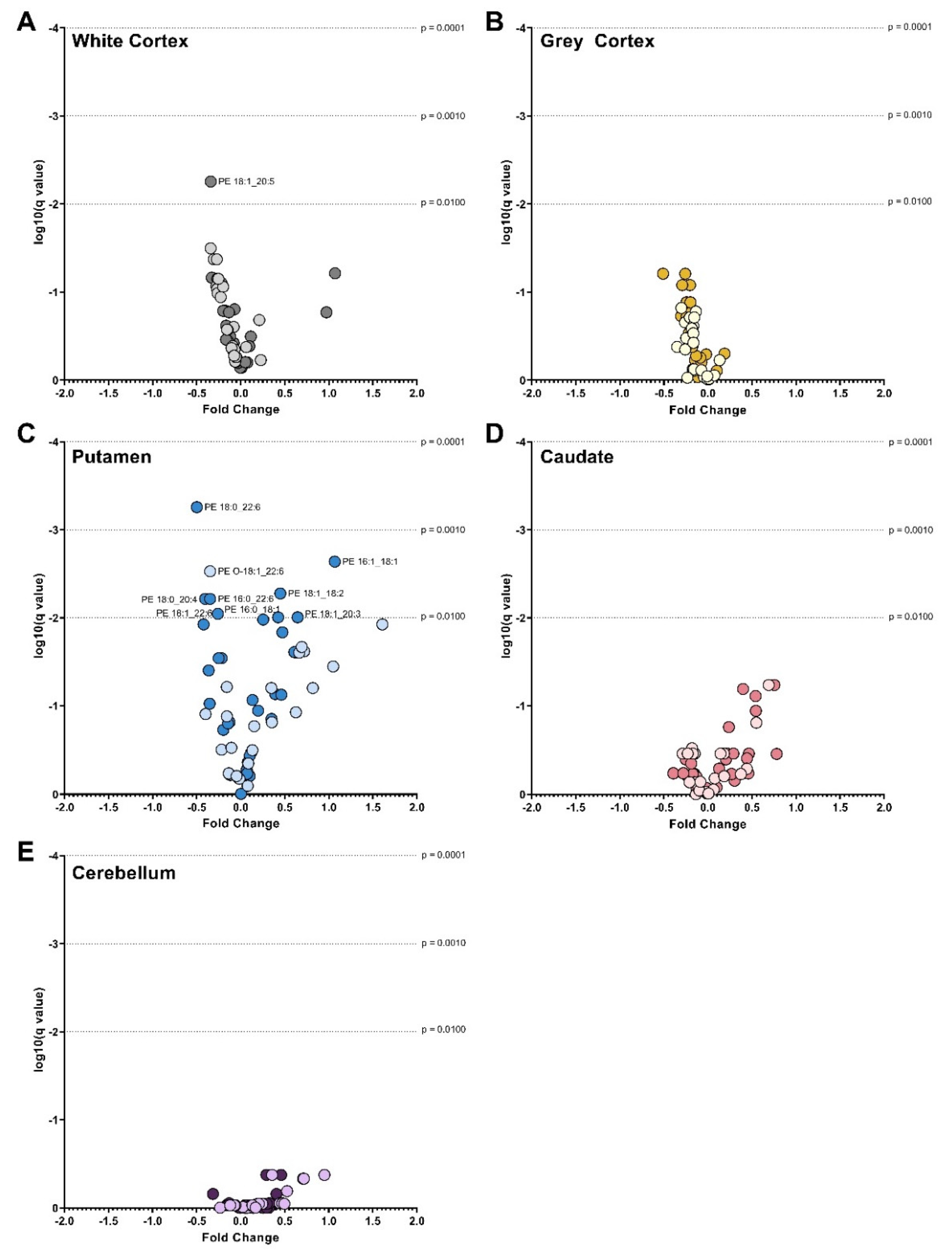
3.3. Phosphatidylethanolamines
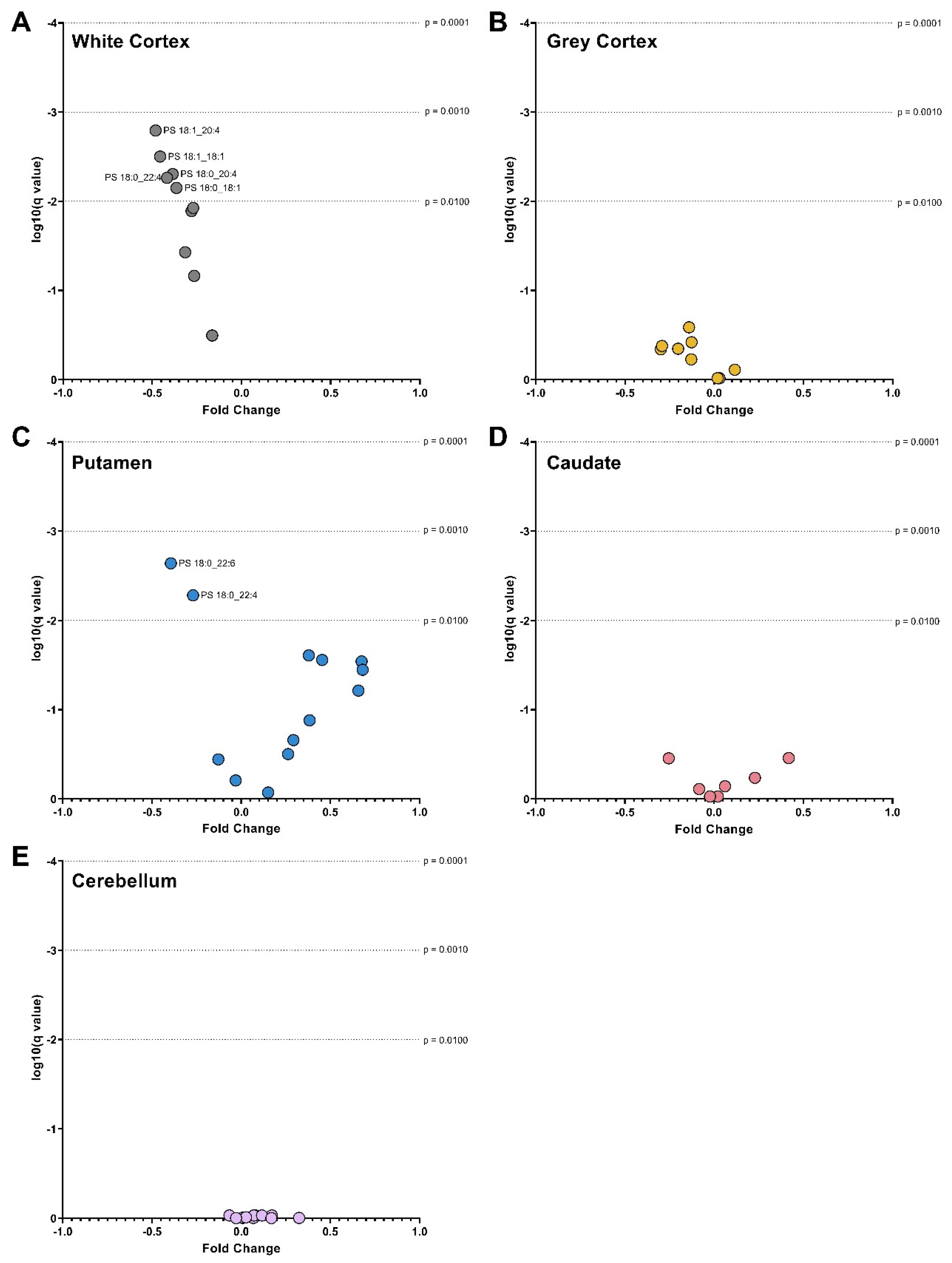
3.4. Phosphatidylserines
3.5. CAG Repeat Length Is Not Related to Neural Phospholipid Abundances in HD Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Macdonald, M. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Kirkwood, S.C.; Su, J.L.; Conneally, P.M.; Foroud, T. Progression of Symptoms in the Early and Middle Stages of Huntington Disease. Arch. Neurol. 2001, 58, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Barnat, M.; Capizzi, M.; Aparicio, E.; Boluda, S.; Wennagel, D.; Kacher, R.; Kassem, R.; Lenoir, S.; Agasse, F.; Braz, B.Y.; et al. Huntington’s disease alters human neurodevelopment. Science 2020, 6505, 787–793. [Google Scholar] [CrossRef]
- Burrus, C.J.; McKinstry, S.U.; Kim, N.; Ozlu, M.I.; Santoki, A.V.; Fang, F.Y.; Ma, A.; Karadeniz, Y.B.; Worthington, A.K.; Dragatsis, I.; et al. Striatal Projection Neurons Require Huntingtin for Synaptic Connectivity and Survival. Cell Rep. 2020, 3, 642–657. [Google Scholar] [CrossRef] [Green Version]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 5, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 3, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Ismailoglu, I.; Chen, Q.; Popowski, M.; Yang, L.; Gross, S.S.; Brivanlou, A.H. Huntingtin protein is essential for mitochondrial metabolism, bioenergetics and structure in murine embryonic stem cells. Dev. Biol. 2014, 2, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Ciammola, A.; Sassone, J.; Sciacco, M.; Mencacci, N.E.; Ripolone, M.; Bizzi, C.; Colciago, C.; Moggio, M.; Parati, G.; Silani, V. Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 1, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Toczek, M.; Zielonka, D.; Zukowska, P.; Marcinkowski, J.T.; Slominska, E.; Isalan, M.; Smolenski, R.T.; Mielcarek, M. An impaired metabolism of nucleotides underpins a novel mechanism of cardiac remodeling leading to Huntington’s disease related cardiomyopathy. Biochim. Biophys. Acta 2016, 11, 2147–2157. [Google Scholar] [CrossRef]
- Mielcarek, M.; Inuabasi, L.; Bondulich, M.K.; Muller, T.; Osborne, G.F.; Franklin, S.A.; Smith, D.L.; Neueder, A.; Rosinski, J.; Rattray, I. Dysfunction of the CNS-heart axis in mouse models of Huntington’s disease. PLoS Genet. 2014, 8, e1004550. [Google Scholar] [CrossRef] [PubMed]
- Keum, J.W.; Shin, A.; Gillis, T.; Mysore, J.S.; Abu Elneel, K.; Lucente, D.; Hadzi, T.; Holmans, P.; Jones, L.; Orth, M.; et al. The HTT CAG-Expansion Mutation Determines Age at Death but Not Disease Duration in Huntington Disease. Am. J. Hum. Genet. 2016, 98, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-M.; Correia, K.; Loupe, J.; Kim, K.-H.; Barker, D.; Hong, E.P.; Chao, M.J.; Long, J.; Lucente, D.; Vonsattel, J.P.G.; et al. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e14. [Google Scholar] [CrossRef] [Green Version]
- Beasley, M.; Groover, S.; Valentine, S.J.; Legleiter, J. Lipid headgroups alter huntingtin aggregation on membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2021, 1863, 183497. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Hensal, K.M.; Umbaugh, C.S.; Chaibva, M.; Legleiter, J. Huntingtin disrupts lipid bilayers in a polyQ-length dependent manner. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Kegel, K.B.; Sapp, E.; Yoder, J.; Cuiffo, B.; Sobin, L.; Kim, Y.J.; Qin, Z.-H.; Hayden, M.; Aronin, N.; Scott, D.L.; et al. Huntingtin Associates with Acidic Phospholipids at the Plasma Membrane. J. Biol. Chem. 2005, 280, 36464–36473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kegel, K.B.; Sapp, E.; Alexander, J.; Valencia, A.; Reeves, P.; Li, X.; Masso, N.; Sobin, L.; Aronin, N.; DiFiglia, M. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 2009, 110, 1585–1597. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000, 106, 1–29. [Google Scholar] [CrossRef]
- Paltauf, F. Ether lipids in biomembranes. Chem. Phys. Lipids 1994, 74, 101–139. [Google Scholar] [CrossRef]
- Rog, T.; Koivuniemi, A. The biophysical properties of ethanolamine plasmalogens revealed by atomistic molecular dynamics simulations. Biochim. Biophys. Acta 2016, 1858, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, R.J.S.; Chouinard-Watkins, R.; Bazinet, R.P. Brain docosahexaenoic acid uptake and metabolism. Mol. Asp. Med. 2018, 64, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Beal, M.F.; Martin, J.B. Phosphoethanolamine and ethanolamine are decreased in Alzheimer’s disease and Huntington’s disease. Brain Res. 1987, 417, 389–392. [Google Scholar] [CrossRef]
- Hunter, M.; Demarais, N.J.; Faull, R.L.M.; Grey, A.C.; Curtis, M.A. Subventricular zone lipidomic architecture loss in Huntington’s disease. J. Neurochem. 2018, 146, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.R.; Saville, J.T.; Hancock, S.E.; Brown, S.H.; Jenner, A.M.; McLean, C.; Fuller, M.; Newell, K.A.; Mitchell, T.W. The long and the short of Huntington’s disease: How the sphingolipid profile is shifted in the caudate of advanced clinical cases. Brain Commun. 2022, 4, fcab303. [Google Scholar] [CrossRef]
- Kreilaus, F.; Spiro, A.S.; McLean, C.A.; Garner, B.; Jenner, A.M. Evidence for altered cholesterol metabolism in Huntington’s disease post mortem brain tissue: Altered cholesterol metabolism in Huntington’s disease. Neuropathol. Appl. Neurobiol. 2016, 42, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl- tert -butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Abbott, S.K.; Jenner, A.M.; Mitchell, T.W.; Brown, S.H.J.; Halliday, G.M.; Garner, B. An Improved High-Throughput Lipid Extraction Method for the Analysis of Human Brain Lipids. Lipids 2013, 48, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Hancock, S.E.; Friedrich, M.G.; Mitchell, T.W.; Truscott, R.J.W.; Else, P.L. The phospholipid composition of the human entorhinal cortex remains relatively stable over 80 years of adult aging. GeroScience 2017, 39, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Denny, C.A.; Desplats, P.A.; Thomas, E.A.; Seyfried, T.N. Cerebellar lipid differences between R6/1 transgenic mice and humans with Huntington’s disease: Cerebellar lipids in Huntington’s disease. J. Neurochem. 2010, 115, 748–758. [Google Scholar] [CrossRef]
- Phillips, G.R.; Hancock, S.E.; Brown, S.H.J.; Jenner, A.M.; Kreilaus, F.; Newell, K.A.; Mitchell, T.W. Cholesteryl ester levels are elevated in the caudate and putamen of Huntington’s disease patients. Sci. Rep. 2020, 10, 20314. [Google Scholar] [CrossRef] [PubMed]
- Aylward, E.H. Change in MRI striatal volumes as a biomarker in preclinical Huntington’s disease. Brain Res. Bull. 2007, 72, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; McRitchie, D.A.; Macdonald, V.; Double, K.L.; Trent, R.J.; McCusker, E. Regional Specificity of Brain Atrophy in Huntington’s Disease. Exp. Neurol. 1998, 154, 663–672. [Google Scholar] [CrossRef]
- Domenichiello, A.F.; Kitson, A.P.; Bazinet, R.P. Is docosahexaenoic acid synthesis from α-linolenic acid sufficient to supply the adult brain? Prog. Lipid Res. 2015, 59, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.A.; Yoder, E.; Murphy, S.; Dutton, G.R.; Spector, A.A. Astrocytes, not neurons, produce docosahexaenoic acid (22:6 omega-3) and arachidonic acid (20:4 omega-6). J. Neurochem. 1991, 56, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y. Biochemical and Biological Functions of Docosahexaenoic Acid in the Nervous System: Modulation by Ethanol. Chem. Phys. Lipids 2008, 153, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [Green Version]
- Dyall, S.C. Long-chain omega-3 fatty acids and the brain: A review of the independent and shared effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Lois, C.; González, I.; Izquierdo-García, D.; Zürcher, N.R.; Wilkens, P.; Loggia, M.L.; Hooker, J.M.; Rosas, H.D. Neuroinflammation in Huntington’s Disease: New Insights with 11 C-PBR28 PET/MRI. ACS Chem. Neurosci. 2018, 9, 2563–2571. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Lagarde, M.; Bernoud, N.; Brossard, N.; Lemaitre-Delaunay, D.; Thies, F.; Croset, M.; Lecerf, J. Lysophosphatidylcholine as a preferred carrier form of docosahexaenoic acid to the brain. J. Mol. Neurosci. 2001, 16, 201–204. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Wei-Yi, O.; Horrocks, L.A. Biochemical Aspects of Neurodegeneration in Human Brain: Involvement of Neural Membrane Phospholipids and Phospholipases A2. Neurochem. Res. 2004, 29, 1961–1977. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y.; Akbar, M.; Lau, A.; Edsall, L. Inhibition of Neuronal Apoptosis by Docosahexaenoic Acid (22:6n-3): ROLE OF PHOSPHATIDYLSERINE IN ANTIAPOPTOTIC EFFECT. J. Biol. Chem. 2000, 275, 35215–35223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.S.; Sampson, L.E. Fatty acid and fatty aldehyde composition of the major brain lipids in normal human gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 545–551. [Google Scholar] [CrossRef]
- Morell, P.; Quarles, R.H. Characteristic Composition of Myelin. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Elsevier: New York, NY, USA, 1999. Available online: http://www.ncbi.nlm.nih.gov/books/NBK28221/ (accessed on 4 July 2019).
- Bonda, M.; Perrin, V.; Vileno, B.; Runne, H.; Kretlow, A.; Forró, L.; Luthi-Carter, R.; Miller, L.M.; Jeney, S. Synchrotron Infrared Microspectroscopy Detecting the Evolution of Huntington’s Disease Neuropathology and Suggesting Unique Correlates of Dysfunction in White versus Gray Brain Matter. Anal. Chem. 2011, 83, 7712–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourbon-Teles, J.; Bells, S.; Jones, D.K.; Coulthard, E.; Rosser, A.; Metzler-Baddeley, C. Myelin Breakdown in Human Huntington’s Disease: Multi-Modal Evidence from Diffusion MRI and Quantitative Magnetization Transfer. Neuroscience 2019, 403, 79–92. [Google Scholar] [CrossRef]
- Casella, C.; Lipp, I.; Rosser, A.; Jones, D.K.; Metzler-Baddeley, C. A Critical Review of White Matter Changes in Huntington’s Disease. Mov. Disord. 2020, 35, 1302–1311. [Google Scholar] [CrossRef]
- Ginsberg, L.; Rafique, S.; Xuereb, J.H.; Rapoport, S.I.; Gershfeld, N.L. Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res. 1995, 698, 223–226. [Google Scholar] [CrossRef]
- Broniec, A.; Klosinski, R.; Pawlak, A.; Wrona-Krol, M.; Thompson, D.; Sarna, T. Interaction of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free Radic. Biol. Med. 2011, 50, 892–898. [Google Scholar] [CrossRef] [Green Version]
- Sindelar, P.J.; Guan, Z.; Dallner, G.; Ernster, L. The protective role of plasmalogens in iron-induced lipid peroxidation. Free Radic. Biol. Med. 1999, 26, 318–324. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Farooqui, T.; Horrocks, L.A. Catabolism of Plasmalogens in Brain. In Metabolism and Functions of Bioactive Ether Lipids in the Brain; Springer: New York, NY, USA, 2008; pp. 39–65. [Google Scholar] [CrossRef]
- Vial, D.; Piomelli, D. Dopamine D2 receptors potentiate arachidonate release via activation of cytosolic, arachidonate-specific phospholipase A2. J. Neurochem. 1995, 64, 2765–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felder, C.C.; Kanterman, R.Y.; Ma, A.L.; Axelrod, J. Serotonin stimulates phospholipase A2 and the release of arachidonic acid in hippocampal neurons by a type 2 serotonin receptor that is independent of inositolphospholipid hydrolysis. Proc. Natl. Acad. Sci. USA 1990, 87, 2187–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumuis, A.; Sebben, M.; Haynes, L.; Pin, J.-P.; Bockaert, J. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature 1988, 336, 6194. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B.; Handelin, B.; Balfour, R.; Anderson, K.D.; Markel, D.S.; Tourtellotte, W.W.; Reiner, A. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington’s disease. N. Engl. J. Med. 1990, 322, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.J.; Reynolds, G.P.; Hewitt, L.M.; Slater, P. Brain serotonin receptors in Huntington’s disease. Neurochem. Int. 1986, 9, 431–435. [Google Scholar] [CrossRef]
- Sian, J.; Youdim, M.B.H.; Riederer, P.; Gerlach, M. Huntington’s Disease. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Elsevier: New York, NY, USA, 1999. Available online: https://www.ncbi.nlm.nih.gov/books/NBK27968/ (accessed on 30 October 2020).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid | Brain Region | CONTROL | HD | PD (%) | p |
|---|---|---|---|---|---|
| PC | White Cortex | 25,402.41 ± 1254.05 (11) | 18,182.46 ± 1155.50 (11) | −28.42 | <0.0001 *** |
| Grey Cortex | 19,323.14 ± 1077.29 (13) | 15,835.15 ± 1038.53 (13) | −18.05 | <0.0001 *** | |
| Putamen | 36,248.62 ± 1480.35 (12) | 25,940.88 ± 1415.30 (13) | −28.44 | <0.0001 *** | |
| Caudate | 18,214.87 ± 1187.31 (13) | 16,558.68 ± 1019.03 (12) | −9.09 | 0.0656 | |
| Cerebellum | 13,285.99 ± 1251.90 (12) | 13,420.99 ± 902.97 (13) | 1.02 | 0.8806 | |
| Ether PC | White Cortex | 1979.21 ± 148.62 (11) | 1228.02 ± 104.97 (11) | −37.95 | 0.4327 |
| Grey Cortex | 561.59 ± 62.31 (13) | 490.65 ± 61.45 (13) | −12.63 | 0.9358 | |
| Putamen | 758.35 ± 122.87 (12) | 760.10 ± 59.79 (13) | 0.23 | 0.9984 | |
| Caudate | 545.78 ± 75.12 (13) | 503.68 ± 50.50 (12) | −7.71 | 0.9626 | |
| Cerebellum | 368.99 ± 31.65 (12) | 492.14 ± 44.46 (13) | 33.38 | 0.8910 | |
| PE | White Cortex | 6946.47 ± 480.46 (12) | 6022.46 ± 596.55 (12) | −13.3 | 0.3135 |
| Grey Cortex | 10,430.49 ± 491.76 (13) | 8081.10 ± 489.24 (13) | −22.52 | 0.0078 ns | |
| Putamen | 12,304.40 ± 670.80 (12) | 8771.73 ± 673.31 (13) | −28.71 | <0.0001 *** | |
| Caudate | 8034.76 ± 837.44 (13) | 7050.75 ± 476.51 (12) | −12.25 | 0.2737 | |
| Cerebellum | 5829.33 ± 612.97 (12) | 5625.40 ± 413.87 (13) | −3.5 | 0.8205 | |
| Ether PE | White Cortex | 14,046.65 ± 1311.70 (12) | 10,273.73 ± 1127.64 (12) | −26.86 | <0.0001 *** |
| Grey Cortex | 5382.45 ± 467.61 (13) | 4303.00 ± 427.34 (13) | −20.06 | 0.2204 | |
| Putamen | 5678.41 ± 324.75 (12) | 6363.84 ± 573.87 (13) | 12.07 | 0.4457 | |
| Caudate | 4080.83 ± 535.07 (13) | 3889.34 ± 416.19 (12) | −4.69 | 0.8313 | |
| Cerebellum | 4207.76 ± 463.00 (12) | 4848.44 ± 426.84 (13) | 15.23 | 0.4760 | |
| PS | White Cortex | 20,523.65 ± 1554.27 (12) | 13,021.57 ± 1375.81 (12) | −36.55 | <0.0001 *** |
| Grey Cortex | 7393.91 ± 747.16 (13) | 5871.27 ± 711.59 (13) | −20.59 | 0.0841 | |
| Putamen | 7645.28 ± 378.45 (12) | 7935.62 ± 626.60 (13) | 3.8 | 0.7466 | |
| Caudate | 5106.90 ± 339.73 (12) | 4502.53 ± 407.56 (12) | −11.83 | 0.5097 | |
| Cerebellum | 3542.65 ± 406.07 (12) | 3916.13 ± 427.30 (13) | 10.54 | 0.6777 | |
| LPC | White Cortex | 124.85 ± 12.68 (12) | 79.38 ± 9.14 (13) | −36.41 | 0.9597 |
| Grey Cortex | 100.86 ± 4.71 (13) | 78.88 ± 6.40 (13) | −21.78 | 0.9801 | |
| Putamen | 144.80 ± 8.99 (13) | 100.77 ± 5.05 (13) | −30.41 | 0.9601 | |
| Caudate | 133.10 ± 6.65 (13) | 111.74 ± 6.44 (13) | −16.05 | 0.9806 | |
| Cerebellum | 22.52 ± 6.75 (11) | 15.95 ± 6.95 (10) | −29.16 | 0.9947 | |
| LPE | White Cortex | 1153.67 ± 158.31 (12) | 729.78 ± 138.85 (13) | −36.74 | 0.6372 |
| Grey Cortex | 510.41 ± 66.15 (13) | 360.13 ± 80.77 (13) | −29.44 | 0.8645 | |
| Putamen | 574.38 ± 67.27 (13) | 541.14 ± 81.68 (13) | −5.79 | 0.9699 | |
| Caudate | 317.12 ± 30.20 (13) | 271.58 ± 34.41 (13) | −14.36 | 0.9588 | |
| Cerebellum | 78.65 ± 22.48 (11) | 82.90 ± 17.78 (12) | 5.4 | 0.9964 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillips, G.R.; Hancock, S.E.; Jenner, A.M.; McLean, C.; Newell, K.A.; Mitchell, T.W. Phospholipid Profiles Are Selectively Altered in the Putamen and White Frontal Cortex of Huntington’s Disease. Nutrients 2022, 14, 2086. https://doi.org/10.3390/nu14102086
Phillips GR, Hancock SE, Jenner AM, McLean C, Newell KA, Mitchell TW. Phospholipid Profiles Are Selectively Altered in the Putamen and White Frontal Cortex of Huntington’s Disease. Nutrients. 2022; 14(10):2086. https://doi.org/10.3390/nu14102086
Chicago/Turabian StylePhillips, Gabrielle R., Sarah E. Hancock, Andrew M. Jenner, Catriona McLean, Kelly A. Newell, and Todd W. Mitchell. 2022. "Phospholipid Profiles Are Selectively Altered in the Putamen and White Frontal Cortex of Huntington’s Disease" Nutrients 14, no. 10: 2086. https://doi.org/10.3390/nu14102086
APA StylePhillips, G. R., Hancock, S. E., Jenner, A. M., McLean, C., Newell, K. A., & Mitchell, T. W. (2022). Phospholipid Profiles Are Selectively Altered in the Putamen and White Frontal Cortex of Huntington’s Disease. Nutrients, 14(10), 2086. https://doi.org/10.3390/nu14102086