Abstract
Aging induces substantial remodeling of glia, including density, morphology, cytokine expression, and phagocytic capacity. Alterations of glial cells, such as hypertrophy of lysosomes, endosomes and peroxisomes, and the progressive accumulation of lipofuscin, lipid droplets, and other debris have also been reported. These abnormalities have been associated with significant declines of microglial processes and reduced ability to survey the surrounding tissue, maintain synapses, and recover from injury. Similarly, aged astrocytes show reduced capacity to support metabolite transportation to neurons. In the setting of reduced glial activity, stressors and/or injury signals can trigger a coordinated action of microglia and astrocytes that may amplify neuroinflammation and contribute to the release of neurotoxic factors. Oxidative stress and proteotoxic aggregates may burst astrocyte-mediated secretion of pro-inflammatory cytokines, thus activating microglia, favoring microgliosis, and ultimately making the brain more susceptible to injury and/or neurodegeneration. Here, we discuss the contribution of microglia and astrocyte oxidative stress to neuroinflammation and neurodegeneration, highlight the pathways that may help gain insights into their molecular mechanisms, and describe the benefits of antioxidant supplementation-based strategies.
1. Introduction
The central nervous system (CNS) is composed of a heterogeneous population of cells that hold unique features and cooperate with neurons for proper CNS function. Neurons are highly specialized cells in charge of transmitting, processing, and storing information and are indicated as “functional cells of the brain”. Non-neuronal cells, including microglia and macroglia (i.e., astrocytes, ependymal cells, and oligodendrocytes), perform other vital functions within the CNS [1].
Owing to its hematopoietic origin, microglia is the primary immune source in the CNS that helps nourish and support neurons by clearing neuronal debris and responding to environmental stimuli [2,3]. Upon infection, trauma, or neurodegeneration, microglial cells become activated, undergo rapid reshaping, including changes in gene expression and function, and are recruited at the site of injury [4,5]. Here, they proliferate and phagocyte damaged cells and cellular debris [4]. As a result of their activation, microglial cells produce high levels of pro-inflammatory mediators, including cytokines (e.g., tumor necrosis factor α (TNFα), interferon-γ (IFN-γ), interleukin-6 (IL-6)), chemokines (e.g., monocyte chemoattractant protein-1 (MCP-1)) [6], reactive oxygen species (ROS), and nitric oxide species (NOS) [7], with cytotoxic effects in case of prolonged production [3]. Anti-inflammatory cytokines (e.g., IL-10, IL-1Rα, and transforming growth factor beta-β (TGF-β)) down-regulate microglia activation [8,9]. As such, microglia has been attributed major roles in neuronal survival and the modulation of neuroinflammation, ultimately contributing to promoting neuronal homeostasis and limiting the onset and progression of neurodegeneration [10].
Astrocytes, similar to microglial cells, are involved in a wide spectrum of functions, including the provision of metabolic substrates to neurons for adequate synaptic activity, synthesis and recycling of neurotransmitters, diffusion of glutamate-induced excitatory signals [11,12,13], and interaction with endothelial cells of the blood–brain barrier [14]. Indeed, since there is no direct contact between neurons and microvessels, some essential substrates (e.g., glucose and oxygen supplied by the cerebral circulation) are delivered to neurons by astrocytes [15].
Aging induces substantial remodeling of glial cells. In particular, changes in microglia density, morphology, cytokine expression, and phagocytic capacity have been observed [5,16,17]. Furthermore, age-related modifications of glial intracellular composition, including hypertrophy of lysosomes, endosomes and peroxisomes, and the progressive accumulation of lipofuscin, lipid droplets, and other debris have been reported [18,19,20,21]. These phenotypic alterations have been associated with a significant decline of microglial function with a reduced ability to survey the surrounding tissue, impaired synapses activities, and poor recovery from injury [3,22]. Aged astrocytes also show reduced capacity to support metabolite delivery to neurons, thus affecting neuronal viability [23]. Moreover, the concentration of glutamate and glutamate-aspartate transporters, and glutamine synthase are reduced in senescent astrocytes, leading to dysfunctional glutamate regulation [24,25].
In the setting of reduced glial activity, several stressors and/or injury signals can trigger a coordinated action of microglia and astrocytes that may amplify neuroinflammation and contribute to the release of neurotoxic factors [26]. In this regard, an overall decline in processes involved in preserving cell’s quality may play a major role. Indeed, oxidative stress and proteotoxic aggregates, which are cleared in physiological conditions, may burst astrocytes-mediated secretion of pro-inflammatory cytokines (e.g., IL-6) [27], thus activating microglia and favoring microgliosis during aging and associated conditions [28]. In the context of persistent stimuli, sustained microglia activation and inflammation can make the brain more susceptible to injury and/or neurodegeneration. A deeper understanding of the pathophysiological mechanisms underlying this age-associated decline in cell quality during neurodegeneration is highly sought after, as it may support the development of novel therapeutic strategies.
Here, we discuss the contribution of microglia and astrocyte oxidative stress to neuroinflammation and neurodegeneration, highlight the pathways that may help gain insights into their molecular mechanisms, and describe the benefits of antioxidant supplementation strategies.
2. Oxidative Stress in Microglia and Astrocytes: The Contribution of Mitochondrial Dysfunction
Age-related mitochondrial dysfunction and declines in processes preserving cell’s quality induce microglia and astrocytes alterations that ultimately compromise their ability to support neuronal health and respond to environmental stimuli. Greater oxidative stress and accumulation of proteotoxic aggregates may burst astrocyte-mediated inflammation [27], thus activating microglia and favoring microgliosis [28].
The integration and regulation of a plethora of signals, including ROS and immune regulation, are pivotal for achieving a cooperation between microglia and astrocytes aimed at preserving brain homeostasis [29]. ROS/RNS, which have long been considered detrimental molecules, are now regarded as relevant signaling factors that modulate CNS activities [30,31]. However, if overproduced, in conditions of cell dysmetabolism, mitochondrial dysfunction, or calcium overload, ROS and RNS can inflict damage to cell’s structures and macromolecules, including lipids, proteins, and DNA [32].
Mitochondria are among the major cellular sources of ROS through the activity of the electron transport chain (ETC) [33]. Plastic and highly interconnected mitochondria occupy the main body of glial cells and the long processes of astrocytes. However, different from neurons, these cells synthesize most of their ATP from glycolysis. The low reliance of astrocytes on oxidative metabolism is partly explained by the organization of their ETCs with a small percentage of complex I-organized supercomplexes [34]. This ETC rearrangement is associated with higher rates of ROS production in astrocytes compared with neuronal ETC [34]. Notwithstanding, well-functioning mitochondria are pivotal in cell’s activities other than metabolic purposes. For instance, mitochondrial biogenesis has been implicated in the regulation of astrocyte maturation and synaptic pruning [35]. Conversely, the deletion of the mitochondrial m-AAA protease in astrocytes of mice has been reported to induce neurodegeneration [36]. On a similar note, dysfunction and fragmentation of mitochondria in microglial cells have also been implicated in neurodegeneration [37]. In particular, the extent of mitochondrial damage and the release of dysfunctional and fragmented organelles have been associated with the ability of triggering neuronal damage and propagating neuronal death via the activation of naïve astrocytes into the pro-inflammatory A1 state [37].
Mitochondrial dysfunction and associated oxidative stress have also been related to the accrual of misfolded proteins in neurodegenerative conditions, among which Parkinson’s disease (PD) is actively investigated [38]. In particular, α-synuclein (α-syn) aggregation has been indicated as a relevant mechanism in both familial and idiopathic forms of PD in which mitochondrial dysfunction seems to play a major role [38]. Indeed, α-syn can be relocated at the mitochondria where it is able to disrupt mitochondrial bioenergetics and interfere with mitochondrial biogenesis [38]. However, mitochondrial impairment may also be an early event in α-syn nucleation and deposition and potentially favors its pathological aggregation [38]. α-syn is highly expressed in neurons; however, astrocytes process this aberrant protein in a more efficient way [39]. Indeed, the exposure of murine astrocyte and neuron co-cultures to oligomeric α-syn induces co-localization of α-syn oligomers in glial cells and promotes the internalization of larger amounts of this protein in astrocytes [39]. Furthermore, following exposure to oligomeric α-syn, aberrant mitochondrial morphology and enhanced cell death were observed [39]. Of note, astrocytes were still able to survive days after α-syn exposure, likely because these cells rely on glycolysis-dependent metabolism, and neuronal cell demise occurred following astrocytic mitochondrial dysfunction and cytokine release [39]. Upon α-syn uptake, astrocytes triggered the clearance of α-syn oligomers via lysosomal degradation. However, in the setting of incomplete α-syn digestion, accrual of intracellular misfolded protein and mitochondrial impairment ensued [39]. Therefore, when the ability of astrocytes to dispose toxic α-syn species is overwhelmed, the persistence of α-syn deposits induces cellular dysfunction [39]. In keeping with a decline in astrocyte quality as a feature of PD pathophysiology is also the severe mitochondrial impairment observed in astrocytes from PTEN-induced kinase 1 (PINK1) knockout mice [40]. Similarly, astrocytes from Parkin-deficient mice showed aberrant mitochondrial activity and were unable to contribute to neuronal differentiation, thus supporting the hypothesis of a role of glia in PD pathogenesis [41]. Finally, the mitochondrial protein deglycase DJ-1, a sensor of oxidative stress, is crucial for preserving astrocyte mitochondrial homeostasis [42].
Taken as a whole, these findings support a central role of microglia and astrocytes in PD pathophysiology, which is being increasingly appreciated despite a traditional neuro-centric view of the disease. Moreover, the release of mitochondrial-derived damage-associated molecular patterns (DAMPs) as mediators of neurodegeneration is also emerging [43] and will be discussed in more depth in the next section.
3. Mitochondrial-Derived Vesicles: Alleviating Cell’s Oxidative Burden
Dysfunctional mitochondria produce greater amounts of ROS, which have detrimental effects on the cell’s physiology by promoting aberrant protein folding and intracellular accrual of toxic protein aggregates (i.e., Aβ1–42, α-syn, huntingtin, and Tau) [44]. If not disposed promptly, this waste material impacts cellular activities and can spread to neighboring cells and the extracellular environment [44]. The endo-lysosomal system, which is known for delivering portions of plasma membranes to the endosomal compartment for recycling, has recently been recognized as having a role in the orchestration and execution of autophagy, as a complementary mechanism to guarantee cellular quality control. Mitochondrial homeostasis can also be regulated via this pathway, and its dysfunction has been proposed as a mechanism contributing to age-related conditions, including neurodegeneration [44].
Vesicular transport is a highly conserved type of cell/organismal communication spanning different life kingdoms [45]. Among these, bacteria, which are the best studied, use vesicles to regulate quorum sensing and the exchange of signaling molecules that modulate gene expression and coordinate the behavior of bacterial communities [45]. Bacterial vesicles are used to transport proteins over long distance, promote host invasion, form protective biofilms that allow survival and growth of microorganisms in adverse environmental conditions [46,47].
Owing to their endosymbiotic origin, mitochondria hold multiple bacterial features, including a circular genome, the mitochondrial DNA (mtDNA), embedded into the organelle matrix. The double membrane of mitochondria makes them semi-autonomous from the nucleus also via an independent translation machinery. The ability of generating mitochondrial-derived vesicles (MDVs) of about 70–150 nm in diameter carrying mitochondrial proteins indicates that vesicular transport may be an additional bacterial ancestry trait conserved through evolution [48,49,50,51,52].
Upon formation, MDVs can pursue two different fates: (1) they can deliver their cargo to peroxisomes [48,49,52]; or (2) they shuttle mitochondrial proteins along the endocytic pathway via late endosomes/multivesicular bodies (MVBs). Here, mitochondrial components are directed to lysosomes for degradation [48], thereby suggesting the involvement of MDVs in mitochondrial quality control (MQC) [53,54,55,56,57]. An additional route that can be pursued by MVBs is that of cell surface toward which MVBs are directed to fuse their membranes with the plasma membrane and release MDVs as EVs [54].
MDVs directed to the endocytic pathway have been identified among the vesicle subtypes constitutively produced at a high rate in cardiac myoblasts grown in galactose-containing media [48,50], an experimental setting forcing cells to rely on oxidative metabolism and overusing the mitochondrial protein machinery [48,50]. Under these circumstances, MDV generation was activated only minutes after mitochondrial-induced oxidative stress via antimycin-A and xanthine/xanthine oxidase treatment compared to activation of mitophagy hours/days after stressor exposure [48,50,57]. Moreover, a remarkable budding of MDVs was observed following doxorubicin mitochondrial and cardiac toxicity [50]. These results support a role for MDVs as a first line of defense against oxidative stress and indicate MDV generation as a possible source of biomarkers in conditions of mitochondrial distress [53,54].
Recently, Vasam et al. [58], using budding/reconstitution in vitro assays followed by proteomics analyses, identified protein signatures in MDVs generated by cardiac cells under oxidative stress. These vesicles were enriched in ETC constituents, metabolic enzymes of the Krebs cycle and fatty acid metabolism, autophagy-related mediators, proteins with Fe–S clusters, hyper-reactive cysteine residues, and antioxidant systems [58]. Some of these MDV-associated proteins were also identified within EVs, thus indicating that such molecules may reflect mitochondrial stress and possible biomarkers of mitochondrial damage [58]. The proteomic characterization of MDVs under oxidative stress conditions indicates that selective molecule incorporation most likely relies on the proximity of cargo to ROS-emitting sites [58]. Mitochondria produce ROS at multiple sites, including the flavin NADH:ubiquinone oxidoreductase core subunit V1–3 (NDUFV1–3), the Fe–S NADH:ubiquinone oxidoreductase subunit 1–8 (NDUFS1–8) of complex I [59], the subunits of the ubiquinol-cytochrome c reductase of complex III (UQCRFS1, UQCRC1, UQCRC2) [59,60], and the succinate dehydrogenase complex flavoprotein subunit A of complex II (SDHA) [61]. Furthermore, the mitochondrial glycerol-3-phosphate dehydrogenase (GPDH), the electron transport system of fatty acid oxidation [62], and the dihydrolipoamide dehydrogenase of 2-oxoacid dehydrogenase complexes are also important sites of ROS production [63]. The identification of many of these proteins within MDV cargoes and the observation of their increase following antimycin A treatment indicate that these components may be the most susceptible to damage when high levels of ROS are produced.
More encouraging is the fact that these results are in line with pioneer studies aimed at evaluating the profile of MDVs as markers of mitochondrial dysfunction in people with age-associated conditions characterized by decline in MQC pathways [64,65,66]. In particular, low levels of adenosine triphosphate 5A (ATP5A), NDUFS3, and SDHB have been detected in small EVs isolated from serum of older adults with PD or physical frailty and sarcopenia compared with age-matched controls [64,65,66].
Similar to PD, AD is a neurodegenerative condition featured by the accrual of misfolded proteins and their spreading across neurons and glial cells within the CNS. Among the pathogenic mechanisms of AD, an altered amyloid protein precursor (APP) trafficking involving endosomal vesicular transport is actively investigated. Indeed, the endosomal sorting complexes required for transport (ESCRT) pathway is responsible for directing APP sorting into ILVs [67,68]. The characterization of EVs from neuronal cultures revealed uptake of isolated EVs containing Tau and Aβ oligomers by these cells [69,70] with cytotoxic effects and neuronal spreading [70]. Upon EV reception, target cells acquire the ability to regulate further EV uptake and secretion, thereby boosting the spreading of toxic proteins [71]. Finally, the analysis of neuronal EVs isolated from children with Down syndrome (DS) revealed alterations in insulin-signaling/mTOR pathways [72]. Such changes may represent early events in brain dysfunction associated with DS and likely contribute to cognitive decline in this progeroid condition [72]. Although mitochondrial dysfunction has also been recognized as a major contributor to AD [73], additional work is warranted on the possible role of MDVs in this condition.
The analysis of EV trafficking and the characterization of MDVs from different cell sources in conditions characterized by MQC decline may offer novel pathways for biomarker discovery and therapeutic development.
4. Mitochondria, Inflammation, and Astrogliosis
Age-related declines in glial and astrocytic functions have been linked to mitochondrial dysfunction and inflammation. Inflammatory mediators released by activated glial cells can modulate mitochondrial function, thereby establishing a crosstalk between mitochondrial dysfunction and neuroinflammation [43]. In this setting, mitochondrial DAMPs may have the dual role of mediating neurodegeneration and amplifying neuroinflammation [43].
Among the many mitochondrial-derived DAMPs, ROS have been implicated in triggering sterile inflammation in astrocytes via innate immunity. The activation of the NLR Family Pyrin Domain Containing 3 (NLRP3) protein of the inflammasome and the recruitment of caspase-1 complex at the mitochondria are major players in this response [74]. Via caspase-1 activity, the cleavage of IL-1β and IL-18 precursors occurs, and the release of IL-1β and IL-18 is promoted, which can mediate pyroptosis and cell death [75]. However, a role of astrocytes in mediating antigen presentation and T-cell activation has also been proposed [76].
Regardless of the inflammatory mechanism involved, a persistent activation of inflammation through mitochondrial dysfunction has been reported to induce astrocyte hyperactivation, which further bursts inflammation and may aggravate neuronal damage [77]. This condition is called reactive astrogliosis and is a pathologic feature of several CNS disorders.
Oxidative/nitrosative stress is crucial in mediating astrogliosis via astrocyte-mediated inflammatory response also implicated in neurodegeneration [78]. However, other mitochondrial processes can contribute to inflammation in the setting of astrogliosis. For instance, the uncoupling protein 2 (UCP2) has been reported to regulate astrocyte inflammation via the NLRP3 pathway by modulating levels of mitochondrial ROS [79]. In further support of a mitochondrial-mediated inflammatory astrogliosis is the observation that the release of mtDNA at the cytosolic or extracellular level triggers inflammation via the activation of cyclic GMP—AMP synthase (cGAS)—stimulator of interferon genes (STING) pathway and NLRP3 inflammasome [80]. In this regard, newly synthesized oxidized mtDNA has been reported to bind and activate the NLRP3 complex in the cytosol [81]. In addition, altered protein levels of complex I and depletion of transcription factor A, a histone-like protein for mtDNA, have been associated with α-syn pathology in nigral dopaminergic neurons obtained post mortem from patients with sporadic PD [82]. Moreover, single-nuclei RNA sequencing of midbrain neurons from people with idiopathic PD identified clusters of disease-specific cells and glial activation as pivotal mechanisms involved in PD pathophysiology [83]. However, the causal relationship between mitochondrial dysfunction, α-synucleinopathy, astrogliosis, and neurodegeneration warrants investigation. Preliminary results obtained in astrocytes derived from induced pluripotent stem cells (iPSCs) from healthy people showed that the uptake of high molecular-weight α-syn fibrils conferred a reactive antigen-presenting phenotype to these cells [84]. α-syn exposure of iPSCs also impaired mitochondrial respiration, an effect that was even more pronounced in iPSC-derived astrocytes from PD patients harboring mutations in the mitophagy-related Parkin gene [84]. In a recent study, Joshi et al. [37] showed that the release of dysfunctional and fragmented mitochondria by the microglia was able to trigger neuronal damage and propagate neuronal death via the activation of naïve astrocytes into the pro-inflammatory A1 state [37]. Hence, a crosstalk between microglia and astrocytes rather that a decline in the activity of one of the two cell types alone may be crucial in mediating neuroinflammation and neuronal cell death. Furthermore, the identification of mitochondrial DAMPs as part of innate immunity-driven neuroinflammation may indicate pathways for developing novel therapeutics against neurodegeneration. Indeed, strategies aimed at blunting mitochondrial fragmentation in microglia and therefore inhibiting the release of dysfunctional mitochondria into the brain milieu, without affecting the release of healthy neuroprotective mitochondria, may represent new therapeutic venues (Figure 1).
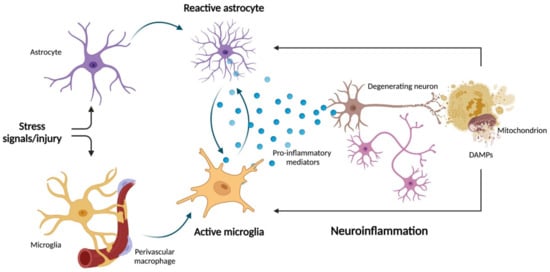
Figure 1.
Schematic representation of microglia–astrocyte crosstalk during neuroinflammation and neurodegeneration. Following stress signals and injuries, aged glial cells and astrocytes may trigger astrogliosis. This process has been linked to crosstalk between mitochondrial dysfunction and neuroinflammation. The release of inflammatory mediators by activated glial cells can impinge on mitochondrial function, which in turn can promote further release of neuronal pro-inflammatory damage-associated molecular patterns (DAMPs). In this setting, mitochondrial DAMPs may have the dual role of mediating neurodegeneration and amplifying neuroinflammation. Created with BioRender.com, accessed on 12 May 2022.
5. Antioxidant Supplementation: A Strategy against Neurodegeneration?
Oxidative stress is a major contributor to brain aging and neurodegeneration. Therefore, supplementation with endogenous or exogenous antioxidants might confer cognitive benefits. Coenzyme Q10 (CoQ10), glutathione (GSH), melatonin, vitamins, polyunsaturated fatty acids (PUFAs), polyphenols, and mitoquinone Q (MitoQ) are among the molecules that have been investigated for the neuroprotective potential and are discussed in the following subsections.
5.1. Coenzyme Q10
CoQ10 is a benzoquinone ring holding a side chain of 10 isoprene units synthetized endogenously. CoQ10 is produced at higher rates in tissues characterized by sustained metabolic activity and high energy demands [85]. This antioxidant compound has been reported to exert its neuroprotective effects by increasing mitochondrial function and promoting lipid reduction, thus protecting the body against the build-up of fats due to unhealthy diet habits [86,87,88]. CoQ10 has also been attributed anti-inflammatory properties through the inhibition of nuclear factor κB (NF-κB) and the release of pro-inflammatory cytokines by endothelial cells of the blood–brain barrier [89]. Moreover, CoQ10 reduces the expression of genes related to endoplasmic reticulum (ER) stress (e.g., calreticulin) and altered ER–mitochondria communication [90].
5.2. Glutathione
GSH is a well-characterized endogenous antioxidant. This compound is a glycine–glutamine–cysteine peptide with a thiol group that allows preserving the cellular redox state through detoxification reactions. GSH is abundant in microglia and has a key role in the expression and regulation of many antioxidant enzymes [91]. A regulated synthesis of GSH in astrocytes is also crucial to replenish the neuronal GSH pool via astrocyte–neuron crosstalk. Studies have shown that a reduction in GSH levels, GSH-S-transferase (GST), superoxide dismutase (SOD) activity, and GSH/oxidized-GSH ratio characterizes brains of patients with AD [92]. GSH limits Aβ-induced mitochondrial membrane depolarization in human cortical neuronal HCN-1A cells [93] and acts as a mitochondrial aconitase activator [94]. Lower levels of GSH and higher oxidative stress have been found in patients with PD [95]. GSH supplementation was found to have a positive effect on the disruption of α-syn aggregates in the brain of a transgenic mouse model overexpressing human α-syn bearing the A53T mutation (prnp.aSyn.A53T) [96]. Finally, co-treatment with γ-glutamylcysteine (GGC), a precursor of GSH, and Aβ40 oligomers of astrocyte cultures increased SOD and GSH peroxidase activity, as well as total cellular antioxidant capacity [97]. GGC supplementation was also shown to increase the levels of anti-inflammatory cytokines and reduce metalloproteinase activity in astrocytes treated with oligomeric Aβ40 [97].
5.3. Melatonin
Another endogenous compound holding antioxidant activity is melatonin, a key hormone in the regulation of circadian rhythm. Melatonin seems to have a protective action in different animal models by maintaining mitochondrial membrane potential, increasing antioxidant enzymatic (e.g., SOD, catalase) and non-enzymatic defenses (i.e., GSH), inhibiting ROS overproduction, increasing ATP production, decreasing calcium concentrations, and enhancing mitochondrial complex I activity [98,99,100,101,102].
5.4. Vitamins
Microglia-mediated response to oxidative stress may become insufficient, and supplementation with natural compounds that improve cellular antioxidant defense may be beneficial. Several compounds extracted from fruits, vegetables, and fish have been attributed a protective role against oxidative damage.
Vitamin E is abundant in sunflower, safflower, and soybean oil, sunflower seeds, almonds, peanuts, beet greens, collard greens, spinach, pumpkin, asparagus, mango, and avocado. Vitamin E preserves biological membranes from oxidation and modulates enzymes that reduce ROS/RNS build-ups [90]. Vitamin E is a family of eight natural forms that include four tocopherols and four tocotrienols divided into α, β, γ, and δ forms. The hydroxyl group of the aromatic ring of vitamin E neutralizes radicals or reactive species by ceasing a hydrogen atom [90]. The administration of α-tocopherol was shown to reverse the altered synaptic plasticity observed in PD mice [103]. Moreover, a recent study indicated that α- and β-tocopherol, δ-tocotrienol, total tocopherols, total tocotrienols, and total vitamin E may be involved in the pathogenesis of AD [104].
The precursor of vitamin A, the β-carotene, is found in yellow, orange, and green leafy fruits and vegetables (e.g., carrots, spinach, lettuce, tomatoes, sweet potatoes, broccoli, cantaloupe, pumpkin) and has positive effects against oxidative stress and neurodegeneration [105,106]. Indeed, low β-carotene plasma concentrations have been found in people with AD [107] and PD [108] compared with healthy controls. These findings have recently been confirmed by an in vitro study demonstrating that β-carotene reduces oxidative stress and pro-inflammatory cytokines in mononuclear cells of people with AD [109]. However, proper timing of vitamin E supplementation in relation to the time course of AD pathology should also be considered. Indeed, an association between higher α- and γ- tocopherol levels and lower total and activated microglia density has been identified in the human cortex, suggesting a microglia-mediated beneficial effect on the slowly accumulating AD neuropathology [110]. However, improvements in microglial activation should better be obtained in the early stages of AD, as microglia is crucial for clearing soluble Aβ and creating protective barriers around Aβ plaques [111,112]. In later AD stages, instead, persistent microglial activation can reinforce tau pathology and have negative effects on neurons and synapses [110,113].
Finally, many fruits and vegetables are rich in vitamin C (ascorbic acid). Vitamin-C-rich fruits include citrus fruits, such as oranges, grapefruit, and lemon, kiwi, blackcurrants, strawberries, and guava. As for vegetables, broccoli, cauliflower, cabbage, cooked kale, Brussels sprouts, and Chinese cabbage are good sources of this vitamin. Vitamin C acts as a scavenger of free radicals produced as by-products of cell metabolism and down-regulates the activity of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, thereby reducing ROS production [114]. In addition, vitamin C contributes to the maintenance of both mitochondrial integrity and function by preventing abnormal mitochondria morphology [93,115]. Interestingly, supplementation with vitamin C has been reported to mitigate the degeneration of dopaminergic neurons and locomotor deficits in an animal model of PD [116].
5.5. Polyunsaturated Fatty Acids
Marine-based fish and fish oil are the most important sources of n-3 PUFAs (e.g., docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA)), with a well-known role in neuronal development and growth, as well as in mitochondrial biogenesis and the regulation of genes involved in brain oxidative metabolism [90]. A greater intake of DHA has been associated with partial recovery of the dopaminergic system, suggesting both neuroprotective and neurorestorative capacity in PD patients [90]. In fact, DHA can reduce neuroinflammation, mitochondrial dysfunction, and oxidative stress caused by α-syn alterations [117,118,119]. Preclinical studies in AD models have shown an improvement in mitochondrial function following PUFA treatment, with positive effects on ROS production, cytochrome c release, and caspase-3 activation [120]. A recent study also indicated a favorable effect of a multi-nutrient intervention containing both DHA and EPA in slowing cognitive decline, brain dysfunction and atrophy, and disease progression in AD [121].
5.6. Polyphenols
Fruits, vegetables, cereals, spices, olive oil, and wine are rich in polyphenols, a class of compounds characterized by an aromatic ring with at least one hydroxyl group able to chelate divalent metals (e.g., copper, zinc, and iron) [122]. These compounds can attenuate mitochondrial dysfunction through the regulation of calcium homeostasis, preservation of the membrane potential, and promotion of cytochrome c release into the cytosol during apoptosis [123].
Curcumin is a polyphenol compound derived from the turmeric plant (Curcuma longa L.) and has a protective role in astrocytes, neurons, microglia, and different regions of the CNS, such as hippocampus, mesencephalon, cerebral cortex, and spinal cord [124,125,126], by preventing the production of hydrogen peroxide and nitric oxides [127]. Interestingly, curcumin acts as mitochondrial antiapoptotic agent through the inhibition of caspase-3 and caspase-9 activities and cytochrome c release, and protects mitochondrial integrity and function via reduction in ROS production through amelioration of complex I activity [128,129]. Among synthetic derivative compounds from curcumin, CNB-001 seems to prevent mitochondrial damage induced by rotenone in human neuroblastoma SK-N-SH cells by inhibiting the mitochondrial apoptotic pathway and maintaining mitochondrial structure [130,131]. Moreover, glutamoyl diester of curcumin has been shown to preserve mitochondrial membrane potential and to inhibit ROS production in brain mitochondria of mice with peroxynitrite-induced PD [132].
Resveratrol is a natural polyphenol found in grapes, berries, peanuts and, above all, red wine. Resveratrol modulates mitochondrial bioenergetics in primary fibroblasts cultures from PD patients with parkin mutations (PARK2) by increasing complex I and citrate synthase activity, basal oxygen consumption and ATP production, and reducing lactate content [133]. With regard to AD, resveratrol treatment seems to normalize mitochondrial amount and decrease the abnormal expression of peroxiredoxins and mitochondrial structural genes in Aβ25–35-induced N2a mouse cells [134].
Curcumin and resveratrol bear the weakness of a limited bioavailability. After oral intake, these compounds are rapidly metabolized in the liver and intestine and are promptly disposed by the body. Due to their low bioavailability, curcumin and resveratrol barely cross the blood–brain barrier, which impacts their neuroprotective potential [90]. New strategies have been developed to improve the bioavailability of these polyphenols, such as the use of nanoparticles that can easily reach the blood–brain barrier endothelial cells [135,136,137].
Fruits from Ericaceae are also rich in polyphenols with strong antioxidant properties. Experimental evidence showed that the intake of blueberries, cranberries, and bearberries had a protective effect on the CNS [138]. In particular, an in vitro study reported that blueberries prevented mitochondrial damage associated with Aβ and reduced the accumulation and aggregation of Aβ through NF-κB regulation [139]. In vivo study confirmed the positive effect of cranberries consumption in improving motor coordination and memory in old rats [140].
Several polyphenolic compounds, including flavanols, flavandiols, flavonoids, and phenolic acid, are also found in green tea leaves. In human neuroblastoma SH-SY5Y cell model of 6-hydroxydopamine-induced PD, treatment with green tea polyphenols inhibited the intrinsic apoptotic pathway, reduced ROS production, and ameliorated intracellular calcium concentrations [141]. Finally, treatment with EGb761, an extract from Gingko biloba leaves, improved cognitive performance in people with AD [142].
5.7. Mitoquinone Q
Mitochondrial-targeted antioxidants are a relatively new field of research with promising clinical applications. MitoQ, which is obtained by conjugating the lipophilic triphenylphosphonium cation to coenzyme Q [143], offers the advantage of diffusing through the mitochondrial membranes, thereby accumulating within the organelle [144]. MitoQ was effective in preventing loss of spatial memory and delaying early neuropathology in a triple transgenic mouse model of AD, by preserving mitochondrial membrane potential and reducing apoptosis in cortical neurons [145]. In another animal model of AD, MitoQ attenuated cardiolipin depletion and increased ETC function [146]. Moreover, treatment with MitoQ inhibited the mitochondrial apoptotic pathway in human neuroblastoma SH-SY5Y cell model of 6-hydroxydopamine-induced PD [147,148]. Interestingly, treatment with MitoQ increased the activity of several antioxidant enzymes and attenuated neurological deficits in a mouse model of traumatic brain injury [149].
Although preclinical studies reported promising effects of antioxidant supplementation against neurodegeneration, results from clinical trials using vitamins (vitamin C and E) and CoQ10 in people with PD or AD yielded conflicting results [150,151,152]. Results from these trials indicate that the clinical benefits of antioxidant supplementation may be marginal and likely more evident in people with mild to moderate disease. Further studies are warranted to explore whether specific combinations of supplements given early during the disease course may produce meaningful clinical benefits by targeting multiple processes (i.e., the crosstalk between microglia and astrocytes) rather than a single pathway.
6. Conclusions
The knowledge of the pathophysiological mechanisms associated with neurodegeneration may help develop pharmacological and nutraceutical interventions to counteract cognitive decline. The decline in processes involved in preserving the cell’s quality is a critical event upstream of the accumulation of oxidative damage and proteotoxic aggregates that may burst astrocytes-mediated secretion of pro-inflammatory cytokines and lead to neuroinflammation. In this regard, the analysis of mitochondrial dysfunction and, in particular, MDV trafficking, warrant further investigation. Targeting the crosstalk between microglia and astrocytes has emerged as a novel promising tool to modulate oxidative damage, a relevant pathophysiological mechanism involved in neurodegeneration. In fact, neuroglia can act as a major source of ROS, causing oxidative damage and mediating secondary damage, such as neuroinflammation, excitotoxicity, and blood–brain barrier disruption. On the other hand, ROS affect the phenotype of glial cells by activating astrocytes and promoting polarization of microglia, making interventions to modulate ROS/RNS production a promising, albeit challenging, strategy against neurodegeneration.
Author Contributions
Conceptualization, B.A. and A.P.; writing—original draft preparation, B.A., A.P. and E.F.; writing—review and editing, H.J.C.-J. and R.C.; supervision, E.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Università Cattolica del Sacro Cuore (D1.2020); the nonprofit research foundation “Centro Studi Achille e Linda Lorenzon” (N/A). The APC was funded by the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico of Milan, Italian Ministry of Health (Ricerca Corrente 2022).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No data were generated for the present study.
Acknowledgments
The figure was created with BioRender.com, accessed on 12 May 2022.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Von Bernhardi, R.; Heredia, F.; Salgado, N.; Muñoz, P. Microglia Function in the Normal Brain. Adv. Exp. Med. Biol. 2016, 949, 67–92. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J.R. Microglia in Aging and Alzheimer’s Disease: A Comparative Species Review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef] [PubMed]
- Helmut, K.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and Neuroinflammation: A Pathological Perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef][Green Version]
- McGeer, E.G.; Klegeris, A.; McGeer, P.L. Inflammation, the Complement System and the Diseases of Aging. Neurobiol. Aging 2005, 26, 94–97. [Google Scholar] [CrossRef]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of Microglia in CNS Inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammation and the Degenerative Diseases of Aging. Ann. N. Y. Acad. Sci. 2004, 1035, 104–116. [Google Scholar] [CrossRef]
- Kim, J.B.; Yu, Y.M.; Kim, S.W.; Lee, J.K. Anti-inflammatory Mechanism is Involved in Ethyl Pyruvate-Mediated Efficacious Neuroprotection in the Postischemic Brain. Brain Res. 2005, 1060, 188–192. [Google Scholar] [CrossRef]
- Cartier, N.; Lewis, C.A.; Zhang, R.; Rossi, F.M.V. The role of Microglia in Human Disease: Therapeutic Tool or Target? Acta Neuropathol. 2014, 128, 363–380. [Google Scholar] [CrossRef]
- Fellin, T. Communication between Neurons and Astrocytes: Relevance to the Modulation of Synaptic and Network Activity. J. Neurochem. 2009, 108, 533–544. [Google Scholar] [CrossRef]
- Fiacco, T.A.; Agulhon, C.; McCarthy, K.D. Sorting out Astrocyte Physiology from Pharmacology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 151–174. [Google Scholar] [CrossRef]
- Verkhratsky, A. Physiology of Neuronal-Glial Networking. Neurochem. Int. 2010, 57, 332–343. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, Y.; Wang, S.; Huang, Z.; Jin, F.; Zou, Q.; Li, J.; Pu, Y.; Cai, Z. Bioenergetic Impairment in the Neuro-Glia-Vascular Unit: An Emerging Physiopathology during Aging. Aging Dis. 2021, 12, 2080–2095. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The Perivascular Astroglial Sheath Provides a Complete Covering of the Brain Microvessels: An Electron Microscopic 3D Reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Hart, A.D.; Wyttenbach, A.; Hugh Perry, V.; Teeling, J.L. Age Related Changes in Microglial Phenotype Vary Between CNS Regions: Grey Versus White Matter Differences. Brain Behav. Immun. 2012, 26, 754–765. [Google Scholar] [CrossRef]
- Perry, V.H.; Matyszak, M.K.; Fearn, S. Altered Antigen Expression of Microglia in the Aged Rodent CNS. Glia 1993, 7, 60–67. [Google Scholar] [CrossRef]
- O’Neil, S.M.; Witcher, K.G.; McKim, D.B.; Godbout, J.P. Forced Turnover of Aged Microglia Induces an Intermediate Phenotype but does not Rebalance CNS Environmental Cues Driving Priming to Immune Challenge. Acta Neuropathol. Commun. 2018, 6, 129. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 1308. [Google Scholar] [CrossRef]
- Tremblay, M.È.; Zettel, M.L.; Ison, J.R.; Allen, P.D.; Majewska, A.K. Effects of Aging and Sensory Loss on Glial Cells in Mouse Visual and Auditory Cortices. Glia 2012, 60, 541–558. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Zhang, I.; Bisht, K.; Savage, J.C.; Lecours, C.; Parent, M.; Titorenko, V.; Maysinger, D. Remodeling of Lipid Bodies by Docosahexaenoic Acid in Activated Microglial Cells. J. Neuroinflamm. 2016, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Delage, C.I.; Šimončičová, E.; Tremblay, M.È. Microglial Heterogeneity in Aging and Alzheimer’s Disease: Is Sex Relevant? J. Pharmacol. Sci. 2021, 146, 169–181. [Google Scholar] [CrossRef]
- Simpson, J.E.; Ince, P.G.; Lace, G.; Forster, G.; Shaw, P.J.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S.B. Astrocyte Phenotype in Relation to Alzheimer-Type Pathology in the Ageing Brain. Neurobiol. Aging 2010, 31, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Bellaver, B.; dos Santos, J.P.; Leffa, D.T.; Bobermin, L.D.; Roppa, P.H.A.; da Silva Torres, I.L.; Gonçalves, C.A.; Souza, D.O.; Quincozes-Santos, A. Systemic Inflammation as a Driver of Brain Injury: The Astrocyte as an Emerging Player. Mol. Neurobiol. 2018, 55, 2685–2695. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, B.; Liu, Y.; Zhang, J.; Huang, Y.; Cao, P.; Shen, Y.; Lyu, J. Carnosine Modulates Glutamine Synthetase Expression in Senescent Astrocytes Exposed to Oxygen-Glucose Deprivation/Recovery. Brain Res. Bull. 2017, 130, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia in Neurodegenerative Events-An Initiator or a Significant Other? Int. J. Mol. Sci. 2021, 22, 5818. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the Aging Brain Express Characteristics of Senescence-Associated Secretory Phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial Cell Origin and Phenotypes in Health and Disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The Role of Astrocytes in Oxidative Stress of Central Nervous System: A Mixed Blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Mazat, J.P.; Devin, A.; Ransac, S. Modelling Mitochondrial ROS Production by the Respiratory Chain. Cell. Mol. Life Sci. 2020, 77, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef]
- Zehnder, T.; Petrelli, F.; Romanos, J.; De Oliveira Figueiredo, E.C.; Lewis, T.L.; Déglon, N.; Polleux, F.; Santello, M.; Bezzi, P. Mitochondrial Biogenesis in Developing Astrocytes Regulates Astrocyte Maturation and Synapse Formation. Cell Rep. 2021, 35, 108952. [Google Scholar] [CrossRef]
- Murru, S.; Hess, S.; Barth, E.; Almajan, E.R.; Schatton, D.; Hermans, S.; Brodesser, S.; Langer, T.; Kloppenburg, P.; Rugarli, E.I. Astrocyte-Specific Deletion of the Mitochondrial m-AAA Protease Reveals Glial Contribution to Neurodegeneration. Glia 2019, 67, 1526–1541. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New Insights into the Complex Role of Mitochondria in Parkinson’s Disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive Uptake of α-synuclein Oligomers in Astrocytes Results in Sustained Intracellular Deposits and Mitochondrial Damage. Mol. Cell. Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Kim, J.; Jeong, H.K.; Kim, B.; Jou, I.; Park, M.; Chen, L.; Kang, U.J.; Zhuang, X.; Joe, E.H. PINK1 Deficiency Attenuates Astrocyte Proliferation Through Mitochondrial Dysfunction, Reduced AKT and Increased p38 MAPK Activation, and Downregulation of EGFR. Glia 2013, 61, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Linnartz, B.; Mendritzki, S.; Sczepan, T.; Lübbert, M.; Stichel, C.C.; Lübbert, H. Genetic Mouse Models for Parkinson’s Disease Display Severe Pathology in Glial Cell Mitochondria. Hum. Mol. Genet. 2011, 20, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.J.; Ambrosi, G.; Mullett, S.J.; Berman, S.B.; Hinkle, D.A. DJ-1 Knock-Down Impairs Astrocyte Mitochondrial Function. Neuroscience 2011, 196, 251–264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, M.; Liu, N.; Qin, Z.; Wang, Y. Mitochondrial-Derived Damage-Associated Molecular Patterns Amplify Neuroinflammation in Neurodegenerative Diseases. Acta Pharmacol. Sin. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Deatheragea, B.L.; Cooksona, B.T. Membrane Vesicle Release in Bacteria, Eukaryotes, and Archaea: A Conserved yet Underappreciated Aspect of Microbial Life. Infect. Immun. 2012, 80, 1948–1957. [Google Scholar] [CrossRef]
- Li, Z.; Clarke, A.J.; Beveridge, T.J. Gram-Negative Bacteria Produce Membrane Vesicles which are Capable of Killing Other Bacteria. J. Bacteriol. 1998, 180, 5478–5483. [Google Scholar] [CrossRef]
- Kadurugamuwa, J.L.; Beveridge, T.J. Membrane Vesicles Derived from Pseudomonas Aeruginosa and Shigella Flexneri can be Integrated into the Surfaces of Other Gram-Negative Bacteria. Microbiology 1999, 145, 2051–2060. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A Vesicular Transport Pathway Shuttles Cargo from Mitochondria to Lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of Mitochondria Derived Vesicle Formation Demonstrates Selective Enrichment of Oxidized Cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef]
- Cadete, V.J.J.; Deschênes, S.; Cuillerier, A.; Brisebois, F.; Sugiura, A.; Vincent, A.; Turnbull, D.; Picard, M.; McBride, H.M.; Burelle, Y. Formation of Mitochondrial-Derived Vesicles is an Active and Physiologically Relevant Mitochondrial Quality Control Process in the Cardiac System. J. Physiol. 2016, 594, 5343–5362. [Google Scholar] [CrossRef]
- McLelland, G.L.; Lee, S.A.; McBride, H.M.; Fon, E.A. Syntaxin-17 Delivers PINK1/Parkin-Dependent Mitochondrial Vesicles to the Endolysosomal System. J. Cell Biol. 2016, 214, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-Selected Transport from the Mitochondria to Peroxisomes Is Mediated by Vesicular Carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Cadete, V.J.J.; Vasam, G.; Menzies, K.J.; Burelle, Y. Mitochondrial Quality Control in the Cardiac System: An Integrative View. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A New Pathway for Mitochondrial Quality Control: Mitochondrial-Derived Vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- McLelland, G.-L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 Function in a Vesicular Trafficking Pathway Regulating Mitochondrial Quality Control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Vasam, G.; Nadeau, R.; Cadete, V.J.J.; Lavallée-Adam, M.; Menzies, K.J.; Burelle, Y. Proteomics Characterization of Mitochondrial-Derived Vesicles Under Oxidative Stress. FASEB J. 2021, 35, e21278. [Google Scholar] [CrossRef]
- Treberg, J.R.; Quinlan, C.L.; Brand, M.D. Evidence for Two Sites of Superoxide Production by Mitochondrial NADH-Ubiquinone Oxidoreductase (Complex I). J. Biol. Chem. 2011, 286, 27103–27110. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The Mechanism of Superoxide Production by the antimycin-Inhibited Mitochondrial Q-Cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Perevoshchikova, I.V.; Quinlan, C.L.; Orr, A.L.; Gerencser, A.A.; Brand, M.D. Sites of Superoxide and Hydrogen Peroxide Production During Fatty Acid Oxidation in Rat Skeletal Muscle Mitochondria. Free Radic. Biol. Med. 2013, 61, 298–309. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-Oxoacid Dehydrogenase Complexes in Mitochondria Can Produce Superoxide/Hydrogen Peroxide at Much Higher Rates Than Complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef]
- Picca, A.; Beli, R.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Bucci, C.; Guerra, F.; Marzetti, E. Older Adults with Physical Frailty and Sarcopenia Show Increased Levels of Circulating Small Extracellular Vesicles with a Specific Mitochondrial Signature. Cells 2020, 9, 973. [Google Scholar] [CrossRef]
- Marzetti, E.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Primiano, A.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; et al. Circulating Mitochondrial-Derived Vesicles, Inflammatory Biomarkers and Amino Acids in Older Adults with Physical Frailty and Sarcopenia: A Preliminary BIOSPHERE Multi-Marker Study Using Sequential and Orthogonalized Covariance Selection–Linear Discriminant Analysis. Front. Cell Dev. Biol. 2020, 8, 564417. [Google Scholar] [CrossRef]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-Phosphate Regulates Sorting and Processing of Amyloid Precursor Protein through the Endosomal System. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef]
- Edgar, J.R.; Willén, K.; Gouras, G.K.; Futter, C.E. ESCRTs Regulate Amyloid Precursor Protein Sorting in Multivesicular Bodies and Intracellular Amyloid-Β Accumulation. J. Cell Sci. 2015, 128, 2520–2528. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of Microglia and Inhibition of Exosome Synthesis Halt Tau Propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s Disease Pathology Propagation by Exosomes Containing Toxic Amyloid-Beta Oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Durisic, N.; Sullivan, R.; Götz, J. Exosomes Taken Up by Neurons Hijack the Endosomal Pathway to Spread to Interconnected Neurons. Acta Neuropathol. Commun. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Picca, A.; Montanari, E.; Calvani, R.; Marini, F.; Matassa, R.; Tramutola, A.; Villani, A.; Familiari, G.; Di Domenico, F.; et al. Aberrant Crosstalk Between Insulin Signaling and mTOR in Young Down Syndrome Individuals Revealed by Neuronal-Derived Extracellular Vesicles. Alzheimer’s Dement. 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Ureña-Peralta, J.R.; Morillo-Bargues, M.J.; De La Cruz, J.O.; Guerri, C. Role of Mitochondria ROS Generation in Ethanol-Induced NLRP3 Inflammasome Activation and Cell Death in Astroglial Cells. Front. Cell. Neurosci. 2014, 8, 216. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Dong, Y.; Benveniste, E.N. Immune Function of Astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Rizor, A.; Pajarillo, E.; Johnson, J.; Aschner, M.; Lee, E. Astrocytic Oxidative/Nitrosative Stress Contributes to Parkinson’s Disease Pathogenesis: The Dual Role of Reactive Astrocytes. Antioxidants 2019, 8, 265. [Google Scholar] [CrossRef]
- Du, R.H.; Wu, F.F.; Lu, M.; Shu, X.d.; Ding, J.H.; Wu, G.; Hu, G. Uncoupling Protein 2 Modulation of the NLRP3 Inflammasome in Astrocytes and its Implications in Depression. Redox Biol. 2016, 9, 178–187. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.j.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Grünewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef]
- Smajić, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-Cell Sequencing of Human Midbrain Reveals Glial Activation and a Parkinson-Specific Neuronal State. Brain 2021, 145, 964–978. [Google Scholar] [CrossRef]
- Russ, K.; Teku, G.; Bousset, L.; Redeker, V.; Piel, S.; Savchenko, E.; Pomeshchik, Y.; Savistchenko, J.; Stummann, T.C.; Azevedo, C.; et al. TNF-α and α-synuclein Fibrils Differently Regulate Human Astrocyte Immune Reactivity and Impair Mitochondrial Respiration. Cell Rep. 2021, 34, 108895. [Google Scholar] [CrossRef]
- Sharma, A.; Fonarow, G.C.; Butler, J.; Ezekowitz, J.A.; Felker, G.M. Coenzyme Q10 and Heart Failure: A State-of-the-Art Review. Circ. Heart Fail. 2016, 9, e002639. [Google Scholar] [CrossRef]
- Ibrahim Fouad, G. Combination of Omega 3 and Coenzyme Q10 Exerts Neuroprotective Potential Against Hypercholesterolemia-Induced Alzheimer’s-like Disease in Rats. Neurochem. Res. 2020, 45, 1142–1155. [Google Scholar] [CrossRef]
- Raizner, A.E. Coenzyme Q10. Methodist Debakey Cardiovasc. J. 2019, 15, 185–191. [Google Scholar] [CrossRef]
- Mason, S.A.; Trewin, A.J.; Parker, L.; Wadley, G.D. Antioxidant Supplements and Endurance Exercise: Current Evidence and Mechanistic Insights. Redox Biol. 2020, 35, 101471. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Song, C.Y.; Yuan, Y.; Eber, A.; Rodriguez, Y.; Levitt, R.C.; Takacs, P.; Yang, Z.; Goldberg, R.; Candiotti, K.A. Diabetic Neuropathic Pain Development in Type 2 Diabetic Mouse Model and The Prophylactic and Therapeutic Effects of Coenzyme Q10. Neurobiol. Dis. 2013, 58, 169–178. [Google Scholar] [CrossRef]
- Oppedisano, F.; Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Scicchitano, M.; Scarano, F.; Bosco, F.; Macrì, R.; et al. The Potential for Natural Antioxidant Supplementation in the Early Stages of Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 2618. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef]
- Mandal, P.K.; Tripathi, M.; Sugunan, S. Brain Oxidative Stress: Detection and Mapping of Anti-Oxidant Marker “Glutathione” in Different Brain Regions of Healthy Male/Female, MCI and Alzheimer Patients Using Non-Invasive Magnetic Resonance Spectroscopy. Biochem. Biophys. Res. Commun. 2012, 417, 43–48. [Google Scholar] [CrossRef]
- Medina, S.; Martínez, M.; Hernanz, A. Antioxidants Inhibit the Human Cortical Neuron Apoptosis Induced by Hydrogen Peroxide, Tumor Necrosis Factor Alpha, Dopamine and Beta-Amyloid Peptide 1-42. Free Radic. Res. 2002, 36, 1179–1184. [Google Scholar] [CrossRef]
- Raukas, M.; Rebane, R.; Mahlapuu, R.; Jefremov, V.; Zilmer, K.; Karelson, E.; Bogdanovic, N.; Zilmer, M. Mitochondrial Oxidative Stress Index, Activity of Redox-Sensitive Aconitase and Effects of Endogenous Anti- and Pro-Oxidants on its Activity in Control, Alzheimer’s Disease and Swedish Familial Alzheimer’s Disease Brain. Free Radic. Res. 2012, 46, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Coles, L.D.; Tuite, P.J.; Öz, G.; Mishra, U.R.; Kartha, R.V.; Sullivan, K.M.; Cloyd, J.C.; Terpstra, M. Repeated-Dose Oral N-Acetylcysteine in Parkinson’s Disease: Pharmacokinetics and Effect on Brain Glutathione and Oxidative Stress. J. Clin. Pharmacol. 2018, 58, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Carboni, E.; Tatenhorst, L.; Tönges, L.; Barski, E.; Dambeck, V.; Bähr, M.; Lingor, P. Deferiprone Rescues Behavioral Deficits Induced by Mild Iron Exposure in a Mouse Model of Alpha-Synuclein Aggregation. Neuromol. Med. 2017, 19, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Zarka, M.; Jugder, B.E.; Welch, J.; Jayasena, T.; Chan, D.K.Y.; Sachdev, P.; Bridge, W. The Precursor to Glutathione (GSH), γ-Glutamylcysteine (GGC), Can Ameliorate Oxidative Damage and Neuroinflammation Induced by Aβ 40 Oligomers in Human Astrocytes. Front. Aging Neurosci. 2019, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Patki, G.; Lau, Y.S. Melatonin Protects Against Neurobehavioral and Mitochondrial Deficits in a Chronic Mouse Model of Parkinson’s Disease. Pharmacol. Biochem. Behav. 2011, 99, 704–711. [Google Scholar] [CrossRef]
- Saravanan, K.S.; Sindhu, K.M.; Mohanakumar, K.P. Melatonin Protects Against Rotenone-Induced Oxidative Stress in a Hemiparkinsonian Rat Model. J. Pineal Res. 2007, 42, 247–253. [Google Scholar] [CrossRef]
- Sousa, S.C.; Castilho, R.F. Protective Effect of Melatonin on Rotenone plus Ca2+-Induced Mitochondrial Oxidative Stress and PC12 Cell Death. Antioxid. Redox Signal. 2005, 7, 1110–1116. [Google Scholar] [CrossRef]
- Dabbeni-Sala, F.; Santo, S.; Franceschini, D.; Skaper, S.D.; Giusti, P. Melatonin Protects Against 6-OHDA-Induced Neurotoxicity in Rats: A Role for Mitochondrial Complex I Activity. FASEB J. 2001, 15, 164–170. [Google Scholar] [CrossRef]
- Absi, E.; Ayala, A.; Machado, A.; Parrado, J. Protective Effect of Melatonin Against the 1-Methyl-4-Phenylpyridinium-Induced Inhibition of Complex I of the Mitochondrial Respiratory Chain. J. Pineal Res. 2000, 29, 40–47. [Google Scholar] [CrossRef]
- Schirinzi, T.; Martella, G.; Imbriani, P.; Di Lazzaro, G.; Franco, D.; Colona, V.L.; Alwardat, M.; Salimei, P.S.; Mercuri, N.B.; Pierantozzi, M.; et al. Dietary Vitamin E as a Protective Factor for Parkinson’s Disease: Clinical and Experimental Evidence. Front. Neurol. 2019, 10, 148. [Google Scholar] [CrossRef]
- Casati, M.; Boccardi, V.; Ferri, E.; Bertagnoli, L.; Bastiani, P.; Ciccone, S.; Mansi, M.; Scamosci, M.; Rossi, P.D.; Mecocci, P.; et al. Vitamin E and Alzheimer’s disease: The Mediating Role of Cellular Aging. Aging Clin. Exp. Res. 2020, 32, 459–464. [Google Scholar] [CrossRef]
- Grodstein, F.; Kang, J.H.; Glynn, R.J.; Cook, N.R.; Gaziano, J.M. A Randomized Trial of Beta Carotene Supplementation and Cognitive Function in Men: The Physicians’ Health Study II. Arch. Intern. Med. 2007, 167, 2184–2190. [Google Scholar] [CrossRef]
- Kang, J.H.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Albert, C.M.; Grodstein, F. Vitamin E, Vitamin C, Beta Carotene, and Cognitive Function Among Women with or at Risk of Cardiovascular Disease: The Women’s Antioxidant and Cardiovascular Study. Circulation 2009, 119, 2772–2780. [Google Scholar] [CrossRef]
- Boccardi, V.; Arosio, B.; Cari, L.; Bastiani, P.; Scamosci, M.; Casati, M.; Ferri, E.; Bertagnoli, L.; Ciccone, S.; Rossi, P.D.; et al. Beta-Carotene, Telomerase Activity and Alzheimer’s Disease in Old Age Subjects. Eur. J. Nutr. 2020, 59, 119–126. [Google Scholar] [CrossRef]
- Kim, J.H.; Hwang, J.; Shim, E.; Chung, E.J.; Jang, S.H.; Koh, S.B. Association of Serum Carotenoid, Retinol, and Tocopherol Concentrations with the Progression of Parkinson’s Disease. Nutr. Res. Pract. 2017, 11, 114–120. [Google Scholar] [CrossRef]
- De Oliveira, B.F.; Veloso, C.A.; Nogueira-Machado, J.A.; de Moraes, E.N.; dos Santos, R.R.; Cintra, M.T.G.; Chaves, M.M. Ascorbic Acid, Alpha-Tocopherol, and Beta-Carotene Reduce Oxidative Stress and Proinflammatory Cytokines in Mononuclear Cells of Alzheimer’s Disease Patients. Nutr. Neurosci. 2012, 15, 244–251. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in A P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of Microglia and Astrocytes: A Roadway to Neuroinflammation and Alzheimer’s Disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Ülker, S.; McKeown, P.P.; Bayraktutan, U. Vitamins Reverse Endothelial Dysfunction through Regulation of eNOS and NAD(P)H Oxidase Activities. Hypertension 2003, 41, 534–539. [Google Scholar] [CrossRef]
- Kook, S.Y.; Lee, K.M.; Kim, Y.; Cha, M.Y.; Kang, S.; Baik, S.H.; Lee, H.; Park, R.; Mook-Jung, I. High-Dose of Vitamin C Supplementation Reduces Amyloid Plaque Burden and Ameliorates Pathological Changes in the Brain of 5XFAD Mice. Cell Death Dis. 2014, 5, e1083. [Google Scholar] [CrossRef]
- Tran, H.H.; Dang, S.N.A.; Nguyen, T.T.; Huynh, A.M.; Dao, L.M.; Kamei, K.; Yamaguchi, M.; Dang, T.T.P. Drosophila Ubiquitin C-Terminal Hydrolase Knockdown Model of Parkinson’s Disease. Sci. Rep. 2018, 8, 4468. [Google Scholar] [CrossRef]
- Janssen, C.I.F.; Kiliaan, A.J. Long-Chain Polyunsaturated Fatty Acids (LCPUFA) from Genesis to Senescence: The Influence of LCPUFA on Neural Development, Aging, and Neurodegeneration. Prog. Lipid Res. 2014, 53, 1–17. [Google Scholar] [CrossRef]
- Coulombe, K.; Kerdiles, O.; Tremblay, C.; Emond, V.; Lebel, M.; Boulianne, A.S.; Plourde, M.; Cicchetti, F.; Calon, F. Impact of DHA Intake in a Mouse Model of Synucleinopathy. Exp. Neurol. 2018, 301, 39–49. [Google Scholar] [CrossRef]
- Canerina-Amaro, A.; Pereda, D.; Diaz, M.; Rodriguez-Barreto, D.; Casañas-Sánchez, V.; Heffer, M.; Garcia-Esparcia, P.; Ferrer, I.; Puertas-Avendaño, R.; Marin, R. Differential Aggregation and Phosphorylation of Alpha Synuclein in Membrane Compartments Associated with Parkinson Disease. Front. Neurosci. 2019, 13, 382. [Google Scholar] [CrossRef]
- Eckert, G.P.; Lipka, U.; Muller, W.E. Omega-3 Fatty Acids in Neurodegenerative Diseases: Focus on Mitochondria. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 105–114. [Google Scholar] [CrossRef]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T. 36-month LipiDiDiet Multinutrient Clinical Trial in Prodromal Alzheimer’s Disease. Alzheimer’s Dement. 2021, 17, 29–40. [Google Scholar] [CrossRef]
- Hider, R.C.; Liu, Z.D.; Khodr, H.H. Metal Chelation of Polyphenols. Methods Enzymol. 2001, 335, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Valero, T. Mitochondrial Biogenesis: Pharmacological Approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef] [PubMed]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and Neurodegenerative Diseases. Biofactors 2013, 39, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.; Al-Suhaimi, E.A.; Wahid, F.; Shehzad, O.; Shehzad, A. Therapeutic Potential of Curcumin for Multiple Sclerosis. Neurol. Sci. 2018, 39, 207–214. [Google Scholar] [CrossRef]
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral Bioavailability of Curcumin: Problems and Advancements. J. Drug Target. 2016, 24, 694–702. [Google Scholar] [CrossRef]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, Y.; Li, X.; Ross, C.A.; Smith, W.W. Curcumin Protects Against A53T Alpha-Synuclein-Induced Toxicity in a PC12 Inducible Cell Model for Parkinsonism. Pharmacol. Res. 2011, 63, 439–444. [Google Scholar] [CrossRef]
- Jagatha, B.; Mythri, R.B.; Vali, S.; Bharath, M.M.S. Curcumin Treatment Alleviates the Effects of Glutathione Depletion in vitro and in vivo: Therapeutic Implications for Parkinson’s Disease Explained via in Silico Studies. Free Radic. Biol. Med. 2008, 44, 907–917. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Elangovan, N.; Manigandan, K.; Singh, S.; Shukla, S. CNB-001 a Novel Curcumin Derivative, Guards Dopamine Neurons in MPTP Model of Parkinson’s disease. Biomed Res. Int. 2014, 2014, 236182. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Tamilselvam, K.; Manivasagam, T.; Elangovan, N. Neuroprotective Effect of CNB-001, a Novel Pyrazole Derivative of Curcumin on Biochemical and Apoptotic Markers Against Rotenone-Induced SK-N-SH Cellular Model of Parkinson’s Disease. J. Mol. Neurosci. 2013, 51, 863–870. [Google Scholar] [CrossRef]
- Mythri, R.B.; Harish, G.; Dubey, S.K.; Misra, K.; Srinivas Bharath, M.M. Glutamoyl Diester of the Dietary Polyphenol Curcumin Offers Improved Protection Against Peroxynitrite-Mediated Nitrosative Stress and Damage of Brain Mitochondria in vitro: Implications for Parkinson’s disease. Mol. Cell. Biochem. 2011, 347, 135–143. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of Resveratrol on Mitochondrial Function: Implications in Parkin-Associated Familiar Parkinson’s Disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Rege, S.D.; Geetha, T.; Griffin, G.D.; Broderick, T.L.; Babu, J.R. Neuroprotective Effects of Resveratrol in Alzheimer Disease Pathology. Front. Aging Neurosci. 2014, 6, 1–27. [Google Scholar] [CrossRef]
- Gao, H. Progress and Perspectives on Targeting Nanoparticles for Brain Drug Delivery. Acta Pharm. Sin. B 2016, 6, 268–286. [Google Scholar] [CrossRef]
- Neves, A.R.; Queiroz, J.F.; Costa Lima, S.A.; Figueiredo, F.; Fernandes, R.; Reis, S. Cellular Uptake and Transcytosis of Lipid-Based Nanoparticles Across the Intestinal Barrier: Relevance for Oral Drug Delivery. J. Colloid Interface Sci. 2016, 463, 258–265. [Google Scholar] [CrossRef]
- Neves, A.R.; Queiroz, J.F.; Reis, S. Brain-Targeted Delivery of Resveratrol using Solid Lipid Nanoparticles Functionalized with Apolipoprotein E. J. Nanobiotechnol. 2016, 14, 27. [Google Scholar] [CrossRef]
- Miller, M.G.; Hamilton, D.A.; Joseph, J.A.; Shukitt-Hale, B. Dietary Blueberry Improves Cognition Among Older Adults in a Randomized, Double-Blind, Placebo-Controlled Trial. Eur. J. Nutr. 2018, 57, 1169–1180. [Google Scholar] [CrossRef]
- Fuentealba, J.; Dibarrart, A.J.; Fuentes-Fuentes, M.C.; Saez-Orellana, F.; Quiñones, K.; Guzmán, L.; Perez, C.; Becerra, J.; Aguayo, L.G. Synaptic Failure and Adenosine Triphosphate Imbalance Induced by Amyloid-Β Aggregates are Prevented by Blueberry-Enriched Polyphenols Extract. J. Neurosci. Res. 2011, 89, 1499–1508. [Google Scholar] [CrossRef]
- Shukitt-Hale, B.; Cheng, V.; Joseph, J.A. Effects of Blackberries on Motor and Cognitive Function in Aged Rats. Nutr. Neurosci. 2009, 12, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Bezard, E.; Zhao, B. Protective Effect of Green Tea Polyphenols on the SH-SY5Y Cells Against 6-OHDA Induced Apoptosis through ROS-NO Pathway. Free Radic. Biol. Med. 2005, 39, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Le Bars, P.L.; Velasco, F.M.; Ferguson, J.M.; Dessain, E.C.; Kieser, M.; Hoerr, R. Influence of the Severity of Cognitive Impairment on the Effect of the Gnkgo Biloba Extract EGb 761 in Alzheimer’s Disease. Neuropsychobiology 2002, 45, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective Targeting of a Redox-Active Ubiquinone to Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Cochemé, H.M.; Smith, R.A.J.; Murphy, M.P. Interactions of Mitochondria-Targeted and Untargeted Ubiquinones with the Mitochondrial Respiratory Chain and Reactive Oxygen Species. Implications for the Use of Exogenous Ubiquinones as Therapies and Experimental Tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef]
- Mcmanus, M.J.; Murphy, M.P.; Franklin, J.L. The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The Mitochondria-Targeted Antioxidant MitoQ Extends Lifespan and Improves Healthspan of a Transgenic Caenorhabditis Elegans Model of Alzheimer Disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; del Mar Arroyo-Jimenez, M.; Jordán, J.; Galindo, M.F. The Mitochondria-Targeted Anti-oxidant MitoQ Reduces Aspects of Mitochondrial Fission in the 6-OHDA Cell Model of Parkinson’s Disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef]
- Chun, H.S.; Low, W.C. Ursodeoxycholic Acid Suppresses Mitochondria-Dependent Programmed Cell Death Induced by Sodium Nitroprusside in SH-SY5Y Cells. Toxicology 2012, 292, 105–112. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-Targeted Antioxidant MitoQ Provides Neuroprotection and Reduces Neuronal Apoptosis in Experimental Traumatic Brain Injury Possibly via the Nrf2-ARE Pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer Disease: A Randomized Clinical Trial with Cerebrospinal Fluid Biomarker Measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef]
- Flint Beal, M.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A Randomized Clinical Trial of High-Dosage Coenzyme Q10 in Early Parkinson Disease: No Evidence of Benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of Vitamin E and Memantine on Functional Decline in Alzheimer Disease: The TEAM-AD VA Cooperative Randomized Trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).