
Bile Acid-Related Regulation of Mucosal Inflammation and Intestinal Motility: From Pathogenesis to Therapeutic Application in IBD and Microscopic Colitis

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
Abstract
:1. Introduction: Basis of Bile Acids Physiology
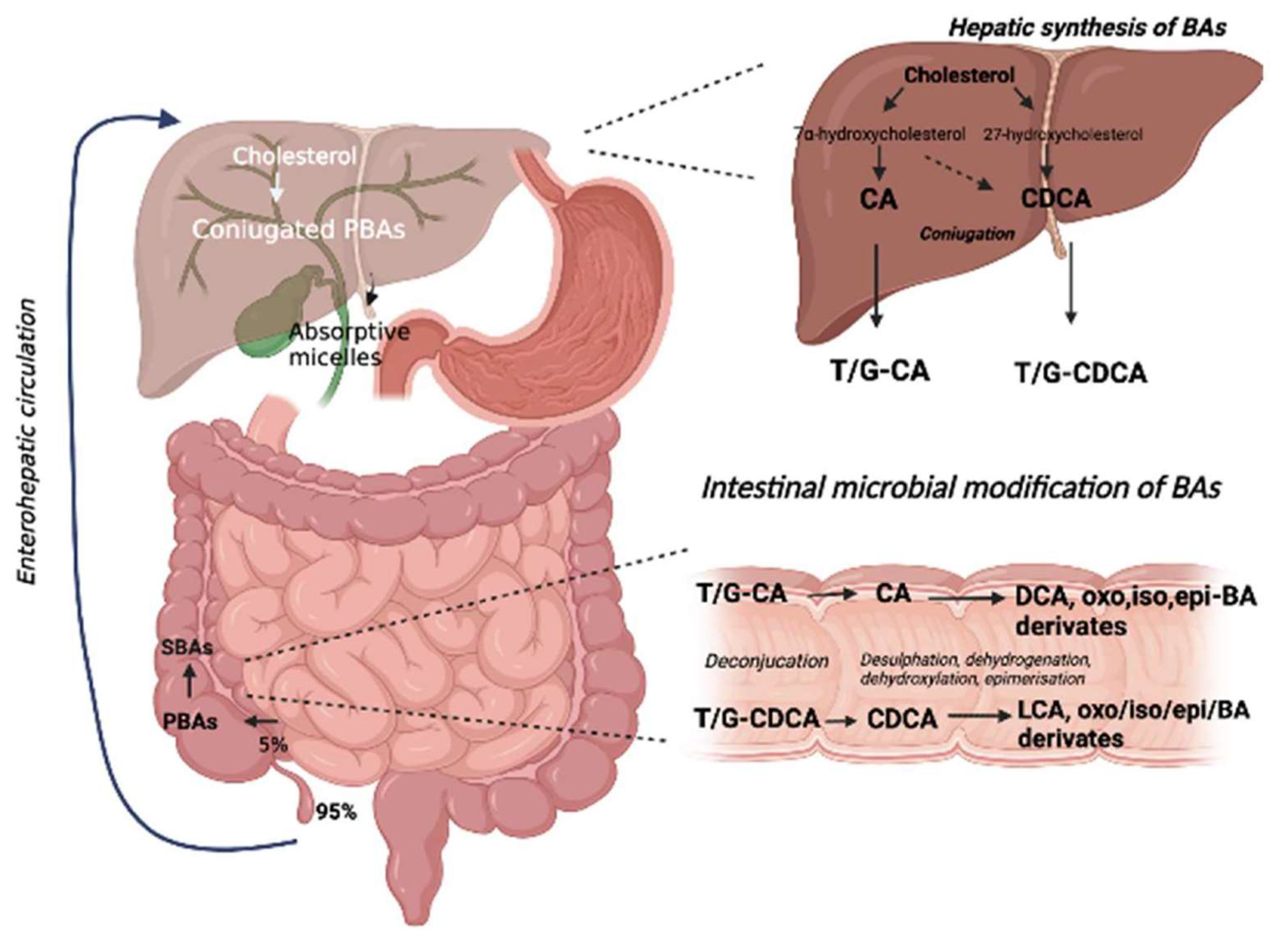
1.1. Bile Production and Composition
- (1).
- The “classic pathway”, mediated by the Cyp7A1 and Cyp8B enzymes, leads to the production of cholic acid; in this pathway, Cyp7A1 is the limiting step.
- (2).
- The “alternative pathway”, mediated by Cyp27 and Cyp7B1, leads to the production of chenodeoxycholic acid [4].
1.2. The Enterohepatic Circulation
2. The Role of BA and Bile Acid Receptors (BARs) in the Pathogenesis of IBD and Dysbiosis
2.1. Bile Acids Metabolism
2.2. Bile Acids Receptors
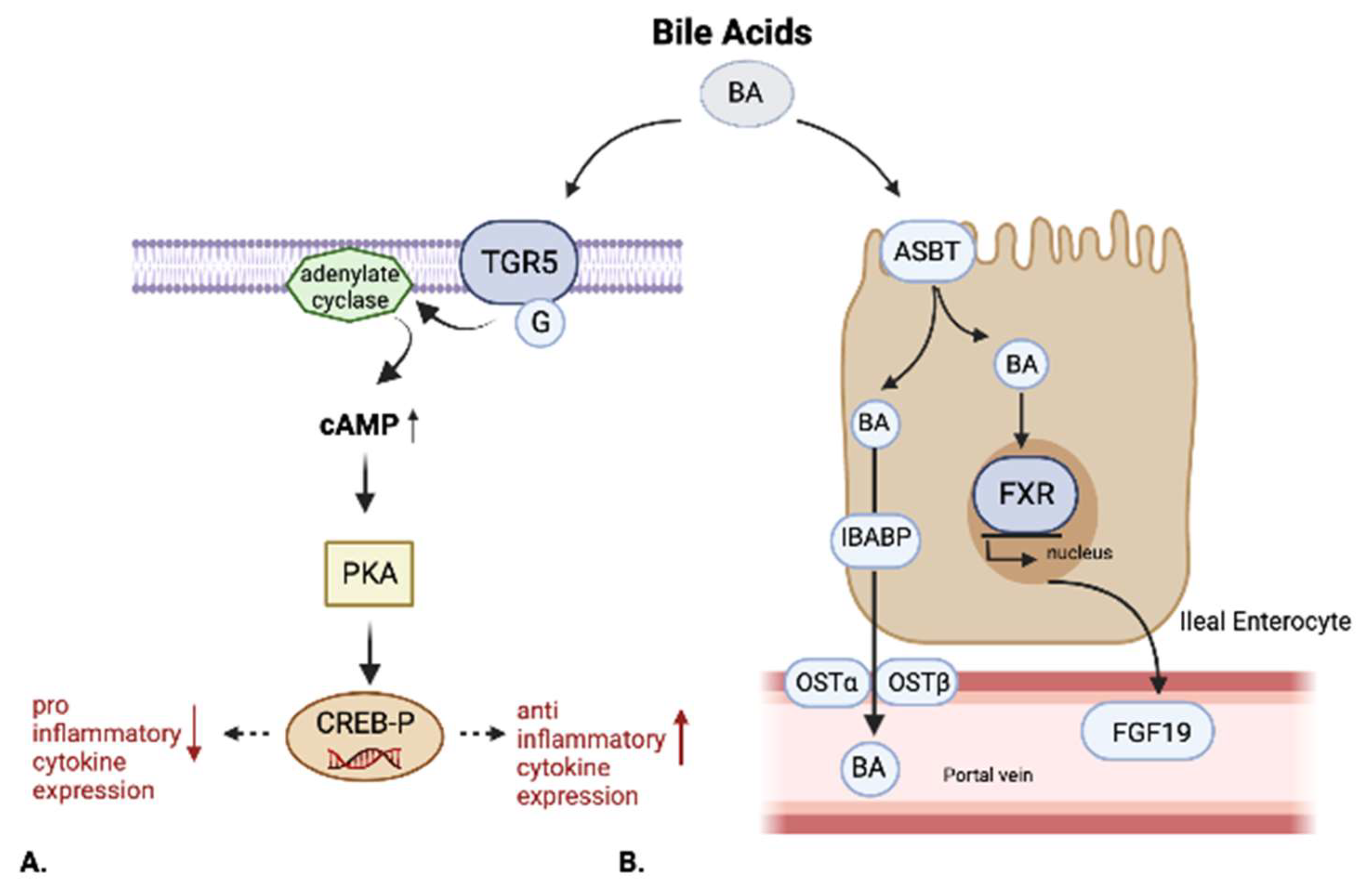
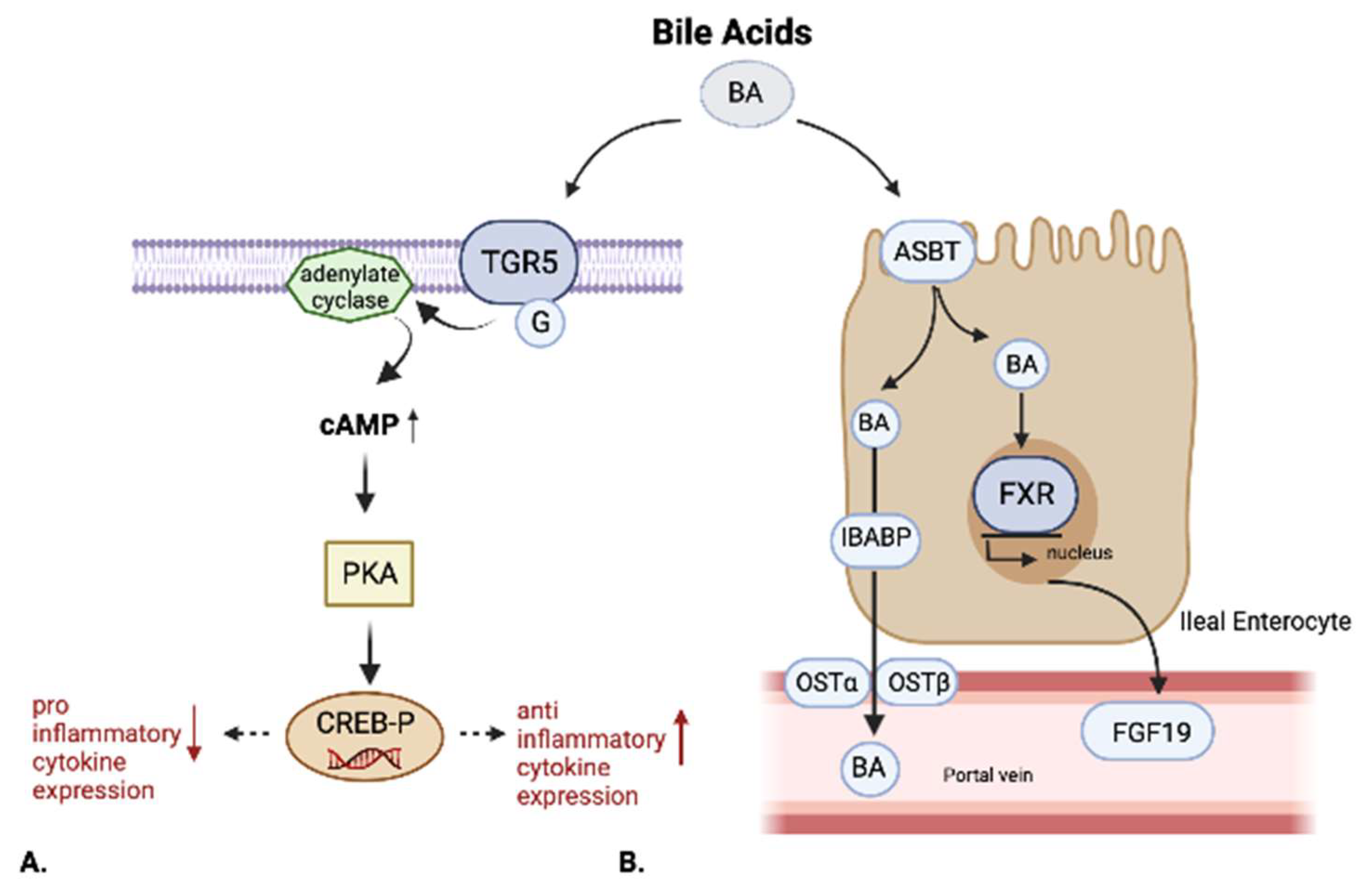
2.3. Farnesoid-X-Receptor (FXR)
2.4. Takeda G-Protein-Coupled Receptor 5 (TGR5)
2.5. Other BARs
3. Bile Acid-Induced Diarrhea (BAD)
3.1. Classification
3.2. Diagnosis
3.3. The seHCAT Test
3.4. Treatment and Therapeutic Perspectives
3.5. The Role of Faecal Microbiota Transplantation (FMT) in Bile Acid Metabolism
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marin, J.J.; Macias, R.; Briz, O.; Banales, J.; Monte, M.J. Bile Acids in Physiology, Pathology and Pharmacology. Curr. Drug Metab. 2015, 17, 4–29. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Albers, C.J.E.M.; Huizenga, J.R.; Krom, R.A.F.; Vonk, R.J.; Gips, C.H. Composition of Human Hepatic Bile. Ann. Clin. Biochem. Int. J. Lab. Med. 1985, 22, 129–132. [Google Scholar] [CrossRef]
- Vaz, F.M.; Ferdinandusse, S. Bile acid analysis in human disorders of bile acid biosynthesis. Mol. Asp. Med. 2017, 56, 10–24. [Google Scholar] [CrossRef]
- Crosignani, A.; Zuin, M.; Allocca, M.; Del Puppo, M. Oxysterols in bile acid metabolism. Clin. Chim. Acta 2011, 412, 2037–2045. [Google Scholar] [CrossRef]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1865, 895–911. [Google Scholar] [CrossRef]
- Devlin, A.S.; A Fischbach, M. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.-J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Sun, L.; Gonzalez, F.J. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 2022, 30, 289–300. [Google Scholar] [CrossRef]
- Stellwag, E.; Hylemon, P. Purification and characterization of bile salt hydrolase from Bacteroides fragilis subsp. fragilis. Biochim. Biophys. Acta (BBA)—Enzym. 1976, 452, 165–176. [Google Scholar] [CrossRef]
- Borriello, S.P.; Owen, R.W. The metabolism of lithocholic acid and lithocholic acid-3-α-sulfate by human fecal bacteria. Lipids 1982, 17, 477–482. [Google Scholar] [CrossRef]
- Stellaard, F.; Lütjohann, D. Dynamics of the enterohepatic circulation of bile acids in healthy humans. Am. J. Physiol. Liver Physiol. 2021, 321, G55–G66. [Google Scholar] [CrossRef]
- Xiao, L.; Pan, G. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: The apical sodium-dependent bile acid transporter (SLC10A2/ASBT). Clin. Res. Hepatol. Gastroenterol. 2017, 41, 509–515. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Aleksunes, L.M. Xenobiotic, Bile Acid, and Cholesterol Transporters: Function and Regulation. Pharmacol. Rev. 2010, 62, 1–96. [Google Scholar] [CrossRef] [Green Version]
- Baars, A.; Oosting, A.; Knol, J.; Garssen, J.; van Bergenhenegouwen, J. The Gut Microbiota as a Therapeutic Target in IBD and Metabolic Disease: A Role for the Bile Acid Receptors FXR and TGR5. Microorganisms 2015, 3, 641–666. [Google Scholar] [CrossRef] [Green Version]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Vantrappen, G.; Ghoos, Y.; Rutgeerts, P.; Janssens, J. Bile acid studies in uncomplicated Crohn’s disease. Gut 1977, 18, 730–735. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Ghoos, Y.; Vantrappen, G. Kinetics of primary bile acids in patients with non-operated Crohn’s disease. Eur. J. Clin. Investig. 1982, 12, 135–143. [Google Scholar] [CrossRef]
- Cummings, J.; James, W.; Wiggins, H. Role of the colon in ileal-resection diarrhœa. Lancet 1973, 301, 344–347. [Google Scholar] [CrossRef]
- Mekhjian, H.S.; Phillips, S.F.; Hofmann, A.F. Colonic absorption of unconjugated bile acids. Am. J. Dig. Dis. 1979, 24, 545–550. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.-A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2012, 62, 531–539. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Chen, W.-D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.-J.; Wang, L. Bile Acid-Activated Receptors: A Review on FXR and Other Nuclear Receptors. Handb. Exp. Pharmacol. 2019, 256, 51–72. [Google Scholar] [CrossRef]
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Tiratterra, E.; Franco, P.; Porru, E.; Katsanos, K.H.; Christodoulou, D.K.; Roda, G. Role of bile acids in inflammatory bowel disease. Ann. Gastroenterol. 2018, 31, 266–272. [Google Scholar] [CrossRef]
- Plass, J.R.M.; Mol, O.; Heegsma, J.; Geuken, M.; Faber, K.N.; Jansen, P.L.M.; Muller, M. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 2002, 35, 589–596. [Google Scholar] [CrossRef]
- Vallim, T.Q.D.A.; Tarling, E.J.; Ahn, H.; Hagey, L.R.; Romanoski, C.E.; Lee, R.G.; Graham, M.J.; Motohashi, H.; Yamamoto, M.; Edwards, P.A. MAFG Is a Transcriptional Repressor of Bile Acid Synthesis and Metabolism. Cell Metab. 2015, 21, 298–311. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, S.-H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef] [Green Version]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef] [Green Version]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The Bile Acid Receptor FXR Is a Modulator of Intestinal Innate Immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, R.M.; Van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Massafra, V.; Ijssennagger, N.; Plantinga, M.; Milona, A.; Pittol, J.M.R.; Boes, M.; van Mil, S.W. Splenic dendritic cell involvement in FXR-mediated amelioration of DSS colitis. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 166–173. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The Bile Acid Sensor FXR Is Required for Immune-Regulatory Activities of TLR-9 in Intestinal Inflammation. PLoS ONE 2013, 8, e54472. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867.e5. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Takabe, K.; Liu, R.; Peng, K.; Wang, X.; Wang, Y.; Hait, N.C.; Wang, X.; Allegood, J.C.; Yamada, A.; et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology 2014, 61, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Ye, J.; Zhang, F.; Su, H.; Yang, X.; He, H.; Liu, F.; Zhu, X.; Wang, L.; Gao, P.; et al. Chenodeoxycholic Acid (CDCA) Protects against the Lipopolysaccharide-Induced Impairment of the Intestinal Epithelial Barrier Function via the FXR-MLCK Pathway. J. Agric. Food Chem. 2019, 67, 8868–8874. [Google Scholar] [CrossRef]
- Klag, T.; Thomas, M.; Ehmann, D.; Courth, L.; Mailänder-Sanchez, D.; Weiss, T.; Dayoub, R.; Abshagen, K.; Vollmar, B.; Thasler, W.E.; et al. β-Defensin 1 Is Prominent in the Liver and Induced During Cholestasis by Bilirubin and Bile Acids via Farnesoid X Receptor and Constitutive Androstane Receptor. Front. Immunol. 2018, 9, 1735. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe 2020, 27, 659–670.e5. [Google Scholar] [CrossRef]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR 5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The Bile Acid Receptor GPBAR-1 (TGR5) Modulates Integrity of Intestinal Barrier and Immune Response to Experimental Colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Alemi, F.; Poole, D.P.; Chiu, J.; Schoonjans, K.; Cattaruzza, F.; Grider, J.R.; Bunnett, N.W.; Corvera, C.U. The Receptor TGR5 Mediates the Prokinetic Actions of Intestinal Bile Acids and Is Required for Normal Defecation in Mice. Gastroenterology 2013, 144, 145–154. [Google Scholar] [CrossRef]
- Hov, J.R.; Keitel, V.; Laerdahl, J.K.; Spomer, L.; Ellinghaus, E.; ElSharawy, A.; Melum, E.; Boberg, K.M.; Manke, T.; Balschun, T.; et al. Mutational Characterization of the Bile Acid Receptor TGR5 in Primary Sclerosing Cholangitis. PLoS ONE 2010, 5, e12403. [Google Scholar] [CrossRef] [Green Version]
- Sawa, S.; Lochner, M.; Satoh-Takayama, N.; Dulauroy, S.; Bérard, M.; A Kleinschek, M.; Cua, D.J.; Di Santo, J.; Eberl, G. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011, 12, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Paik, D.; Yao, L.; Zhang, Y.; Bae, S.; D’Agostino, G.D.; Zhang, M.; Kim, E.; Franzosa, E.A.; Avila-Pacheco, J.; Bisanz, J.E.; et al. Human gut bacteria produce ΤH17-modulating bile acid metabolites. Nature 2022, 603, 907–912. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [Green Version]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-κB target gene expression. Am. J. Physiol. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef]
- Chen, T.; Lin, R.; Jin, S.; Chen, R.; Xue, H.; Ye, H.; Huang, Z. The Sphingosine-1-Phosphate/Sphingosine-1-Phosphate Receptor 2 Axis in Intestinal Epithelial Cells Regulates Intestinal Barrier Function During Intestinal Epithelial Cells–CD4+T-Cell Interactions. Cell. Physiol. Biochem. 2018, 48, 1188–1200. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; DeLuca, H.F. Where is the vitamin D receptor? Arch. Biochem. Biophys. 2012, 523, 123–133. [Google Scholar] [CrossRef]
- Bhalla, A.K.; Amento, E.P.; Krane, S.M. Differential effects of 1,25-dihydroxyvitamin D3 on human lymphocytes and monocyte/macrophages: Inhibition of interleukin-2 and augmentation of interleukin-1 production. Cell. Immunol. 1986, 98, 311–322. [Google Scholar] [CrossRef]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Chen, S.; Lipsky, P.E. Modulatory Effects of 1,25-Dihydroxyvitamin D3 on Human B Cell Differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef] [Green Version]
- Lemire, J.M.; Adams, J.S.; Sakai, R.; Jordan, S.C. 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J. Clin. Investig. 1984, 74, 657–661. [Google Scholar] [CrossRef] [Green Version]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.J.; O’Garra, A. 1α,25-Dihydroxyvitamin D3 Has a Direct Effect on Naive CD4+ T Cells to Enhance the Development of Th2 Cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef] [Green Version]
- Gorman, S.; Kuritzky, L.A.; Judge, M.A.; Dixon, K.M.; McGlade, J.P.; Mason, R.S.; Finlay-Jones, J.J.; Hart, P.H. Topically Applied 1,25-Dihydroxyvitamin D3 Enhances the Suppressive Activity of CD4+CD25+ Cells in the Draining Lymph Nodes. J. Immunol. 2007, 179, 6273–6283. [Google Scholar] [CrossRef] [Green Version]
- Penna, G.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Berti, E.; Colonna, M.; Adorini, L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood 2005, 106, 3490–3497. [Google Scholar] [CrossRef]
- Tang, J.; Zhou, R.; Luger, D.; Zhu, W.; Silver, P.B.; Grajewski, R.S.; Su, S.-B.; Chan, C.-C.; Adorini, L.; Caspi, R.R. Calcitriol Suppresses Antiretinal Autoimmunity through Inhibitory Effects on the Th17 Effector Response. J. Immunol. 2009, 182, 4624–4632. [Google Scholar] [CrossRef]
- Daniel, C.; Sartory, N.A.; Zahn, N.; Radeke, H.H.; Stein, J.M. Immune Modulatory Treatment of Trinitrobenzene Sulfonic Acid Colitis with Calcitriol Is Associated with a Change of a T Helper (Th) 1/Th17 to a Th2 and Regulatory T Cell Profile. J. Pharmacol. Exp. Ther. 2008, 324, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chen, Y.; Golan, M.A.; Annunziata, M.L.; Du, J.; Dougherty, U.; Kong, J.; Musch, M.; Huang, Y.; Pekow, J.; et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J. Clin. Investig. 2013, 123, 3983–3996. [Google Scholar] [CrossRef] [Green Version]
- Gubatan, J.; Mitsuhashi, S.; Zenlea, T.; Rosenberg, L.; Robson, S.; Moss, A.C. Low Serum Vitamin D During Remission Increases Risk of Clinical Relapse in Patients With Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2016, 15, 240–246.e1. [Google Scholar] [CrossRef] [Green Version]
- Zator, Z.A.; Cantu, S.M.; Konijeti, G.G.; Nguyen, D.D.; Sauk, J.; Yajnik, V.; Ananthakrishnan, A.N. Pretreatment 25-Hydroxyvitamin D Levels and Durability of Anti–Tumor Necrosis Factor–α Therapy in Inflammatory Bowel Diseases. J. Parenter. Enter. Nutr. 2013, 38, 385–391. [Google Scholar] [CrossRef]
- Xue, L.-N.; Xu, K.-Q.; Zhang, W.; Wang, Q.; Wu, J.; Wang, X.-Y. Associations Between Vitamin D Receptor Polymorphisms and Susceptibility to Ulcerative Colitis and Crohnʼs Disease. Inflamm. Bowel Dis. 2013, 19, 54–60. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Hagey, L.R. Bile Acids: Chemistry, Pathochemistry, Biology, Pathobiology, and Therapeutics. Cell. Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef]
- Camilleri, M. Bile Acid Diarrhea: Prevalence, Pathogenesis, and Therapy. Gut Liver 2015, 9, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Oduyebo, I.; Camilleri, M. Bile acid disease. Curr. Opin. Gastroenterol. 2017, 33, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, R.-G.; Fan, L.; Liu, J.; Cheng, Y.; Chang, Z.-P.; Wu, B.; Shao, Y. Bile acid malabsorption is associated with diarrhea in acute phase of colitis. Can. J. Physiol. Pharmacol. 2018, 96, 1328–1336. [Google Scholar] [CrossRef]
- Bampton, P.A.; Dinning, P.; Kennedy, M.L.; Lubowski, D.Z.; Cook, I.J. The proximal colonic motor response to rectal mechanical and chemical stimulation. Am. J. Physiol. Liver Physiol. 2002, 282, G443–G449. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, L.; Borsboom, D.; Devriese, S.; Van Welden, S.; Holvoet, T.; Devisscher, L.; Hindryckx, P.; De Vos, M.; Laukens, D. Tauroursodeoxycholic acid protects bile acid homeostasis under inflammatory conditions and dampens Crohn’s disease-like ileitis. Lab. Investig. 2017, 97, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Giaretta, P.R.; Rech, R.R.; Guard, B.C.; Blake, A.B.; Blick, A.K.; Steiner, J.M.; Lidbury, J.A.; Cook, A.K.; Hanifeh, M.; Spillmann, T.; et al. Comparison of intestinal expression of the apical sodium-dependent bile acid transporter between dogs with and without chronic inflammatory enteropathy. J. Veter.-Intern. Med. 2018, 32, 1918–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Martins, R.; Sullivan, M.C.; Friedman, E.S.; Misic, A.M.; El-Fahmawi, A.; De Martinis, E.C.P.; O’Brien, K.; Chen, Y.; Bradley, C.; et al. Diet-induced remission in chronic enteropathy is associated with altered microbial community structure and synthesis of secondary bile acids. Microbiome 2019, 7, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyutakov, I.; Nakov, R.; Valkov, H.; Vatcheva-Dobrevska, R.; Vladimirov, B.; Penchev, P. Serum Levels of Fibroblast Growth Factor 19 Correlate with the Severity of Diarrhea and Independently from Intestinal Inflammation in Patients with Inflammatory Bowel Disease or Microscopic Colitis. Turk. J. Gastroenterol. 2021, 32, 374–381. [Google Scholar] [CrossRef]
- Fisher, M.; Spilias, D.C.; Tong, L.K. Diarrhoea after laparoscopic cholecystectomy: Incidence and main determinants. ANZ J. Surg. 2008, 78, 482–486. [Google Scholar] [CrossRef]
- Sauter, G.H.; Moussavian, A.C.; Meyer, G.; Steitz, H.O.; Parhofer, K.G.; Jüngst, D. Bowel Habits and Bile Acid Malabsorption in The Months After Cholecystectomy. Am. J. Gastroenterol. 2002, 97, 1732–1735. [Google Scholar] [CrossRef]
- Oelkers, P.; Kirby, L.C.; Heubi, J.E.; Dawson, P.A. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J. Clin. Investig. 1997, 99, 1880–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camilleri, M.; Carlson, P.; BouSaba, J.; McKinzie, S.; Vijayvargiya, P.; Magnus, Y.; Sannaa, W.; Wang, X.J.; Chedid, V.; Zheng, T.; et al. Comparison of biochemical, microbial and mucosal mRNA expression in bile acid diarrhoea and irritable bowel syndrome with diarrhoea. Gut 2022. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68 (Suppl. S3), s1–s106. [Google Scholar] [CrossRef] [Green Version]
- Greco, A.; Caviglia, G.P.; Brignolo, P.; Ribaldone, D.G.; Reggiani, S.; Sguazzini, C.; Smedile, A.; Pellicano, R.; Resegotti, A.; Astegiano, M.; et al. Glucose breath test and Crohn’s disease: Diagnosis of small intestinal bacterial overgrowth and evaluation of therapeutic response. Scand. J. Gastroenterol. 2015, 50, 1376–1381. [Google Scholar] [CrossRef]
- Barratt, H.S.; Kalantzis, C.; Polymeros, D.; Forbes, A. Functional symptoms in inflammatory bowel disease and their potential influence in misclassification of clinical status. Aliment. Pharmacol. Ther. 2005, 21, 141–147. [Google Scholar] [CrossRef]
- Boschetti, G.; Laidet, M.; Moussata, D.; Stefanescu, C.; Roblin, X.; Phelip, G.; Cotte, E.; Passot, G.; Francois, Y.; Drai, J.; et al. Levels of Fecal Calprotectin Are Associated With the Severity of Postoperative Endoscopic Recurrence in Asymptomatic Patients With Crohn’s Disease. Am. J. Gastroenterol. 2015, 110, 865–872. [Google Scholar] [CrossRef]
- Wright, E.K.; Kamm, M.A.; De Cruz, P.; Hamilton, A.; Ritchie, K.J.; Krejany, E.O.; Leach, S.; Gorelik, A.; Liew, D.; Prideaux, L.; et al. Measurement of Fecal Calprotectin Improves Monitoring and Detection of Recurrence of Crohn’s Disease After Surgery. Gastroenterology 2015, 148, 938–947.e1. [Google Scholar] [CrossRef] [Green Version]
- Vijayvargiya, P.; Camilleri, M.; Carlson, P.; Lueke, A.; O’Neill, J.; Burton, D.; Busciglio, I.; Donato, L. Performance characteristics of serum C4 and FGF19 measurements to exclude the diagnosis of bile acid diarrhoea in IBS-diarrhoea and functional diarrhoea. Aliment. Pharmacol. Ther. 2017, 46, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Pattni, S.S.; Brydon, W.G.; Dew, T.; Johnston, I.M.; Nolan, J.D.; Srinivas, M.; Basumani, P.; Bardhan, K.D.; Walters, J.R.F. Fibroblast growth factor 19 in patients with bile acid diarrhoea: A prospective comparison of FGF19 serum assay and SeHCAT retention. Aliment. Pharmacol. Ther. 2013, 38, 967–976. [Google Scholar] [CrossRef]
- Battat, R.; Duijvestein, M.; Casteele, N.V.; Singh, S.; Dulai, P.S.; Valasek, M.A.; Mimms, L.; McFarland, J.; Hester, K.D.; Renshaw, M.; et al. Serum Concentrations of 7α-hydroxy-4-cholesten-3-one Are Associated with Bile Acid Diarrhea in Patients With Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2018, 17, 2722–2730.e4. [Google Scholar] [CrossRef]
- Fernández-Bañares, F.; Esteve, M.; Salas, A.; Forné, M.; Espinós, J.C.; Martín-Comín, J.; Viver, J.M. Bile Acid Malabsorption in Microscopic Colitis and in Previously Unexplained Functional Chronic Diarrhea. Am. J. Dig. Dis. 2001, 46, 2231–2238. [Google Scholar] [CrossRef]
- Lyutakov, I.; Lozanov, V.; Sugareva, P.; Valkov, H.; Penchev, P. Serum 7-alfa-hydroxy-4-cholesten-3-one and fibroblast growth factor-19 as biomarkers diagnosing bile acid malabsorption in microscopic colitis and inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2020, 33, 380–387. [Google Scholar] [CrossRef] [PubMed]
- García, A.B.; Palma, F.P.; Martínez, S.G.; Candau, M.D.B.; Vinardell, M.P. 75 Se-Homocholic acid taurine scintigraphy (75 SeHCAT®), a standard benchmark test in bile acid malabsorption? Rev. Esp. Med. Nucl. Imagen Mol. 2019, 38, 305–311. [Google Scholar] [CrossRef]
- Nyhlin, H.; Merrick, M.V.; A Eastwood, M. Bile acid malabsorption in Crohn’s disease and indications for its assessment using SeHCAT. Gut 1994, 35, 90–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ung, K.-A.; Gillberg, R.; Kilander, A.; Abrahamsson, H. Role of bile acids and bile acid binding agents in patients with collagenous colitis. Gut 2000, 46, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Role of Bile Acids in Lymphocytic Colitis | Request PDF. Available online: https://www.researchgate.net/publication/11376040_Role_of_bile_acids_in_lymphocytic_colitis (accessed on 23 April 2022).
- Murray, I.; Murray, L.K.; Woolson, K.L.; Sherfi, H.; Dixon, I.; Palmer, J.; Sulkin, T. Incidence and predictive factors for positive 75SeHCAT test: Improving the diagnosis of bile acid diarrhoea. Scand. J. Gastroenterol. 2017, 52, 698–703. [Google Scholar] [CrossRef]
- Riemsma, R.; Al, M.; Ramos, I.C.; Deshpande, S.N.; Armstrong, N.; Lee, Y.-C.; Ryder, S.; Noake, C.; Krol, M.; Oppe, M.; et al. SeHCAT [tauroselcholic (selenium-75) acid] for the investigation of bile acid malabsorption and measurement of bile acid pool loss: A systematic review and cost-effectiveness analysis. Health Technol. Assess. 2013, 17, 1–236. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, O.; Hojgaard, L.; Moller, E.H.; O Wielandt, T.; Thale, M.; Jarnum, S.; Krag, E. Effect of enterocoated cholestyramine on bowel habit after ileal resection: A double blind crossover study. BMJ 1985, 290, 1315–1318. [Google Scholar] [CrossRef] [Green Version]
- Barkun, A.N.; Love, J.; Gould, M.; Pluta, H.; Steinhart, A.H. Bile Acid Malabsorption in Chronic Diarrhea: Pathophysiology and Treatment. Can. J. Gastroenterol. 2013, 27, 653–659. [Google Scholar] [CrossRef]
- Mottacki, N.; Simrén, M.; Bajor, A. Review article: Bile acid diarrhoea—Pathogenesis, diagnosis and management. Aliment. Pharmacol. Ther. 2016, 43, 884–898. [Google Scholar] [CrossRef] [Green Version]
- Keely, S.J.; Walters, J. The Farnesoid X Receptor: Good for BAD. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 725–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Appleby, R.; Moghul, I.; Khan, S.; Yee, M.; Manousou, P.; Neal, T.D.; Walters, J.R.F. Non-alcoholic fatty liver disease is associated with dysregulated bile acid synthesis and diarrhea: A prospective observational study. PLoS ONE 2019, 14, e0211348. [Google Scholar] [CrossRef] [Green Version]
- Mroz, M.S.; Keating, N.; Ward, J.B.; Sarker, R.; Amu, S.; Aviello, G.; Donowitz, M.; Fallon, P.; Keely, S. Farnesoid X receptor agonists attenuate colonic epithelial secretory function and prevent experimental diarrhoea in vivo. Gut 2013, 63, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Kelly, O.B.; Mroz, M.S.; Ward, J.B.J.; Colliva, C.; Scharl, M.; Pellicciari, R.; Gilmer, J.F.; Fallon, P.G.; Hofmann, A.F.; Roda, A.; et al. Ursodeoxycholic acid attenuates colonic epithelial secretory function. J. Physiol. 2013, 591, 2307–2318. [Google Scholar] [CrossRef]
- Hvas, C.L.; Ott, P.; Paine, P.; Lal, S.; Jørgensen, S.P.; Dahlerup, J.F. Obeticholic acid for severe bile acid diarrhea with intestinal failure: A case report and review of the literature. World J. Gastroenterol. 2018, 24, 2320–2326. [Google Scholar] [CrossRef]
- Walters, J.R.F.; Johnston, I.M.; Nolan, J.D.; Vassie, C.; Pruzanski, M.E.; Shapiro, D.A. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment. Pharmacol. Ther. 2014, 41, 54–64. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, D.; Li, Y.; Dai, S.; Qu, L.; Chen, X.; Guo, M.; Wei, H.; Chen, Y. Structural basis of tropifexor as a potent and selective agonist of farnesoid X receptor. Biochem. Biophys. Res. Commun. 2020, 534, 1047–1052. [Google Scholar] [CrossRef]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non–Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 9, 395–410. [Google Scholar] [CrossRef]
- Camilleri, M.; Nord, S.L.; Burton, D.; Oduyebo, I.; Zhang, Y.; Chen, J.; Im, K.; Bhad, P.; Badman, M.K.; Sanders, D.S.; et al. Randomised clinical trial: Significant biochemical and colonic transit effects of the farnesoid X receptor agonist tropifexor in patients with primary bile acid diarrhoea. Aliment. Pharmacol. Ther. 2020, 52, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, N.; Stankov, K.; Mikov, M. Probiotics—Interactions with Bile Acids and Impact on Cholesterol Metabolism. Appl. Biochem. Biotechnol. 2012, 168, 1880–1895. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Rainaldi, S.; Bovenga, F.; Murzilli, S.; Moschetta, A. Microbiota Modification with Probiotics Induces Hepatic Bile Acid Synthesis via Downregulation of the Fxr-Fgf15 Axis in Mice. Cell Rep. 2014, 7, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Joyce, S.A.; Gahan, C.G. Bile Acid Modifications at the Microbe-Host Interface: Potential for Nutraceutical and Pharmaceutical Interventions in Host Health. Annu. Rev. Food Sci. Technol. 2016, 7, 313–333. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Jang, Y.S.; Kang, D.; Chang, D.K.; Min, Y.W. Efficacy and Safety of New Lactobacilli Probiotics for Unconstipated Irritable Bowel Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients 2019, 11, 2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Seekatz, A.M.; Rao, K.; Santhosh, K.; Young, V.B. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med. 2016, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Weingarden, A.R.; Chen, C.; Bobr, A.; Yao, D.; Lu, Y.; Nelson, V.M.; Sadowsky, M.J.; Khoruts, A. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Liver Physiol. 2014, 306, G310–G319. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Vincenzo, F.; Puca, P.; Lopetuso, L.R.; Petito, V.; Masi, L.; Bartocci, B.; Murgiano, M.; De Felice, M.; Petronio, L.; Gasbarrini, A.; et al. Bile Acid-Related Regulation of Mucosal Inflammation and Intestinal Motility: From Pathogenesis to Therapeutic Application in IBD and Microscopic Colitis. Nutrients 2022, 14, 2664. https://doi.org/10.3390/nu14132664
Di Vincenzo F, Puca P, Lopetuso LR, Petito V, Masi L, Bartocci B, Murgiano M, De Felice M, Petronio L, Gasbarrini A, et al. Bile Acid-Related Regulation of Mucosal Inflammation and Intestinal Motility: From Pathogenesis to Therapeutic Application in IBD and Microscopic Colitis. Nutrients. 2022; 14(13):2664. https://doi.org/10.3390/nu14132664
Chicago/Turabian StyleDi Vincenzo, Federica, Pierluigi Puca, Loris Riccardo Lopetuso, Valentina Petito, Letizia Masi, Bianca Bartocci, Marco Murgiano, Margherita De Felice, Lorenzo Petronio, Antonio Gasbarrini, and et al. 2022. "Bile Acid-Related Regulation of Mucosal Inflammation and Intestinal Motility: From Pathogenesis to Therapeutic Application in IBD and Microscopic Colitis" Nutrients 14, no. 13: 2664. https://doi.org/10.3390/nu14132664
APA StyleDi Vincenzo, F., Puca, P., Lopetuso, L. R., Petito, V., Masi, L., Bartocci, B., Murgiano, M., De Felice, M., Petronio, L., Gasbarrini, A., & Scaldaferri, F. (2022). Bile Acid-Related Regulation of Mucosal Inflammation and Intestinal Motility: From Pathogenesis to Therapeutic Application in IBD and Microscopic Colitis. Nutrients, 14(13), 2664. https://doi.org/10.3390/nu14132664