Abstract
Alzheimer’s disease (AD), an age-related neurodegenerative disorder, is currently incurable. Imbalanced amyloid-beta (Aβ) generation and clearance are thought to play a pivotal role in the pathogenesis of AD. Historically, strategies targeting Aβ clearance have typically focused on central clearance, but with limited clinical success. Recently, the contribution of peripheral systems, particularly the liver, to Aβ clearance has sparked an increased interest. In addition, AD presents pathological features similar to those of metabolic syndrome, and the critical involvement of brain energy metabolic disturbances in this disease has been recognized. More importantly, the liver may be a key regulator in these abnormalities, far beyond our past understanding. Here, we review recent animal and clinical findings indicating that liver dysfunction represents an early event in AD pathophysiology. We further propose that compromised peripheral Aβ clearance by the liver and aberrant hepatic physiological processes may contribute to AD neurodegeneration. The role of a hepatic synthesis product, fibroblast growth factor 21 (FGF21), in the management of AD is also discussed. A deeper understanding of the communication between the liver and brain may lead to new opportunities for the early diagnosis and treatment of AD.
1. Introduction
Alzheimer’s disease (AD), the most common type of dementia, is a growing global health concern with enormous implications for individuals and society. There are currently 40 million people living with AD worldwide; this number is estimated to rise more than threefold by 2050 [1]. According to the latest report, the annual total cost of AD treatment is predicted to reach $2.54 trillion in 2030 and $9.12 trillion in 2050 [2]. As a progressive neurodegenerative disease with complex pathobiology, AD is characterized clinically by progressive cognitive impairment and pathologically by extracellular accumulation of amyloid-beta (Aβ) plaques, intracellular neurofibrillary tangles (NFTs) composed of highly phosphorylated tau proteins, and loss of neurons and synapses [1,3]. Unfortunately, the efficient disease-modifying therapeutic approach for AD is unavailable and there is an urgent need to advance our understanding of this disease process.
It is believed that the imbalance between Aβ production and clearance plays a pivotal role in AD pathogenesis. Therefore, strategies to scavenge Aβ from the brain are considered a priority in the treatment of AD [4,5]. Unfortunately, immunotherapy targeting the central clearance of Aβ shows limited efficacy, and several adverse effects have been reported in clinical trials, such as neuroinflammation, neuronal hyperactivity, and enhanced neurotoxicity linked to Aβ oligomerization [6,7]. This fact prompted more investigators to focus on the development of peripheral Aβ clearance-based strategies, a safer therapy [6,8]. Indeed, most of the brain Aβ can be cleared via transportation to the periphery [9,10]. Although it is still unclear how Aβ is cleared peripherally, several pathways have been proposed to mediate the efflux of brain Aβ into the periphery, including the blood–brain barrier (BBB) and perivascular lymphatic drainage pathway [11,12]. After some Aβ peptides are eliminated through phagocytosis or proteolytic degradation in the central nervous system (CNS), other Aβ peptides can be transferred into the blood through the BBB, in interstitial or cerebrospinal fluid. Some of these Aβ peptides are degraded by phagocytosis or Aβ-degrading enzymes, while others are carried by lipoproteins, erythrocytes, and albumin to peripheral organs or tissues, where they are degraded by hepatocytes or macrophages, or excreted by the kidneys and liver [12,13]. Apart from the classical amyloid hypothesis, increasing evidence suggests that AD may be a systemic metabolic syndrome [14,15,16]. Interestingly, recent studies have highlighted that the liver, a principal organ responsible for system-wide metabolic control and metabolic detoxification, contributes significantly to Aβ clearance [10,11,17], and hepatic dysfunction may increase the risk of developing AD [18,19,20]. This review aimed to discuss recent advances in our understanding of the crosstalk between the liver and AD pathology and summarize the potential of relevant therapeutic strategies for disease prevention and treatment. We proposed that liver dysfunction may contribute to AD pathology through several pathophysiological pathways (Figure 1).
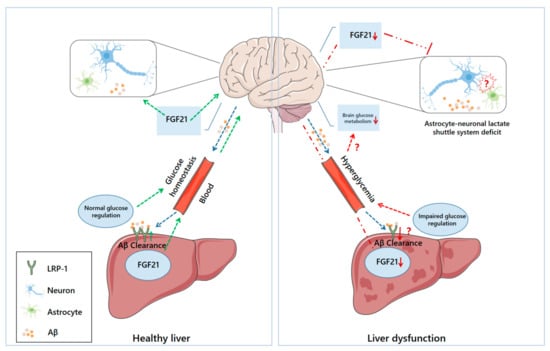
Figure 1.
The potential communication between liver dysfunction and AD neurodegeneration. Under normal physiological conditions (left), hepatocytes uptake circulating Aβ via LRP-1 and promote Aβ clearance. FGF21 can also be processed by the liver, and then delivered to the brain via the blood, where it can exert protective effects. Circulating FGF21 may contribute to the stability of the astrocyte–neuron–lactate shuttle system and cerebral energy homeostasis. Under pathological conditions (right), diminished LRP-1 hinders hepatic Aβ clearance, and the liver’s ability to synthesize FGF21 may also be compromised, leading to reduced supplementation of these substances to the brain. Although it is not yet clear what drives the impaired astrocyte–neuron–lactate shuttle system in AD and whether there is a causal relationship between that and liver dysfunction, lower FGF21 may weaken its protective effect on this communication system. Additionally, normal glucose regulation in the liver is essential for maintaining systemic glucose homeostasis. Conversely, compromised hepatic glucose regulation may induce hyperglycemia and disrupt glucose metabolism in the brain.
2. Background of Alzheimer’s Disease
AD is an age-related neurodegenerative disorder, with an insidious onset and deadly outcome. The clinical presentation of AD comprises memory loss and difficulties with thinking, speech, completing familiar tasks, and problem-solving skills [21,22]. The onset and progression of AD involves complex interactions of multiple factors. Largely, AD can be divided into early-onset familial forms and late-onset sporadic forms. Early-onset AD is strongly linked to mutations in the presenilin 1, presenilin 2, or amyloid precursor protein (APP) genes. In contrast, the large majority of AD cases are the sporadic form (>95%), which is thought to be induced by abnormal processing of Aβ [23,24]. Apart from aging, several risk factors contributing to sporadic AD have been proposed such as systemic inflammation, vascular disease, sleep disturbances, and gut microbiota dysbiosis [25,26,27,28]. The accumulation of Aβ and its oligomer in the brain is normally considered to be a key contributor to the pathogenesis of AD, but recently, several other mechanisms have also been proposed to explain its disease progression [22,29].
AD exhibits similar pathophysiological characteristics to type II diabetes mellitus, obesity, and other metabolic syndromes, including oxidative stress, abnormal fasting glucose, and insulin resistance [30,31]. Therefore, it has been suggested that AD should be considered in the context of metabolic disorders, and some even suggest terming AD as “Type 3 diabetes mellitus” [32,33,34]. Moreover, the role of several peripheral organs, including the liver and kidney, in facilitating circulating Aβ clearance and systemic metabolic regulation, has been increasingly recognized [11,35]. In this context, coupled with the limited success of strategies aimed to scavenge Aβ peptides centrally, therapeutic targets for AD are no longer solely based on past knowledge of this disease. With the emergence of novel treatment targets, treatment modalities for AD have become diversified. In the following sections, we discuss the contribution of peripheral organs in Aβ clearance, and the metabolic disruptions observed in AD.
2.1. Central and Peripheral Aβ Catabolism
Various theories have been proposed to explain the pathogenesis of AD, including the amyloid, tau hyperphosphorylation, and mitochondrial dysfunction hypotheses [14,36,37,38]. Among them, the amyloid hypothesis remains the most accepted model to explain AD pathogenesis [39]. According to this theory, the imbalance between Aβ production and clearance could drive the sequential cleavage of APP by β and γ secretase enzymes in the brain, resulting in the accumulation of pathological forms of Aβ that subsequently form insoluble amyloidogenic fibers and aggregate into plaques [36,40]. Moreover, this aggregation will lead to kinase activation, which will result in microtubule-associated τ protein hyperphosphorylation and aggregation into insoluble NFTs [1]. Microglia infiltration in response to these aggregates can also mediate synapse loss by engulfing synapses, thereby exacerbating AD pathology, and ultimately contributing to dementia symptoms [41,42]. Unfortunately, immunotherapies designed to centrally scavenge Aβ have failed to modify disease symptoms, possibly due to the potential adverse effects of these treatments and the limited central clearance rate of Aβ. Recent studies found that peripheral organs contribute significantly to clearing Aβ accumulation in the brain. Experiments using radiation technology have shown that more than 60% radioactivity could be detected in peripheral organs, including the liver, kidney, gastrointestinal tract, and skin, after I125-labeled Aβ was administered intravenously or intracranially [9,10,43]. Additionally, peritoneal dialysis, a therapeutic method for removing metabolites and wastes from the blood, can significantly lower the plasma Aβ levels in patients and APP/PS1 mice, attenuate AD pathology (such as Tau hyperphosphorylation, glial activation, neuroinflammation, neuronal loss, and synaptic dysfunction), and increase Aβ phagocytosis in microglia, as well as rescue cognitive impairment in the animals [44]. It has also been reported that unilateral nephrectomy increased Aβ deposition in the brain and exacerbated AD pathologies, yet treatment with furosemide (a type of loop diuretic mainly acting on the kidney) exerted protective effects, as evidenced by the reduced Aβ levels in the blood and brain, with ameliorated AD symptoms and cognitive deficits [45]. These promising results have sparked an increased interest in developing strategies for the peripheral clearance of Aβ.
2.2. Metabolic Dysfunction in Alzheimer’s Disease
Remarkably, mounting evidence indicates that AD patients usually develop central and peripheral metabolic dysfunction [15,46,47,48]. The human brain consumes 20% of the body’s energy consumption at rest, although it represents only 2% of body weight; neurons expend 70–80% of total brain energy [49]. As a high energy-demand organ, the brain is sensitive to fuel supply and energy changes. Moreover, these metabolic alternations can significantly affect the onset and progression of multiple neurodegenerative diseases [49,50]. Glucose is the main energy substrate for the brain, and since brain neurons cannot synthesize or store glucose directly, their energy supply primarily depends on the continuous uptake of external glucose. Cerebral glucose metabolism involves two key processes: glucose transport and intracellular oxidative catabolism. The later involves complex pathways, the tricarboxylic acid (TCA) cycle and oxidative phosphorylation, which occur within the mitochondria, and the glycolysis and pentose phosphate pathway in the cytoplasm [14,51]. Since glucose is a polar hydrophilic molecule that cannot freely cross the BBB, normal physiological glucose transport in the brain is strongly dependent on the specific transport proteins, including sodium-dependent glucose transporters (SGLTs) and sodium-independent glucose transporters (GLUTs) [52,53]. Through intracellular catabolism, glucose transported to the brain is eventually converted into adenosine triphosphate (ATP) and its related metabolites to fuel neural activity and biosynthesis.
Consistent evidence from human studies and animal studies has revealed abnormal glucose metabolism in the brain that could occur prior to AD pathology and cognitive impairment, including reduced GLUT1 and GLUT3 [54,55], impaired insulin signaling [56,57], aberrant glucose metabolism [58,59,60], mitochondrial dysfunction and reduced ATP synthesis [61,62]. Furthermore, a significant reduction in cerebral glucose metabolism can be detected years or even decades before clinical symptoms appear in presenilin-1 gene mutation carriers [63]. In rat and Drosophila AD models, both recovery of the insulin signaling pathway, which is critical in regulating glucose transport across membranes, and overexpression of GLUT1 in neurons can effectively slow the progression of AD-related pathology [64,65]. These findings emphasize the role of aberrant brain energy metabolism in AD pathogenesis. Intriguingly, besides its role in facilitating Aβ clearance, the liver is the primary organ for glucose production and storage, and a normal hepatic function also markedly contributes to the maintenance of systemic glucose metabolism [66]. In fact, most AD patients exhibit fasting hyperglycemia, which may serve as a contributor to cerebral glucose dysregulation [33,67,68]. Furthermore, systemic administration of AD vectors directly impaired insulin signaling in the liver, which diminished the ability of insulin to inhibit hepatic glucose production, thus disrupting systemic glucose homeostasis [69]. Exposure to chronic hyperglycemia also exacerbates AD neurodegeneration and cognitive impairment in AD animals [70,71]. Notably, a large body of preclinical research and prospective cohort studies indicate that liver metabolic alterations or dysfunction could precede progressive cognitive decline and other typical symptoms of AD [20,72,73]. The liver is the first organ to exhibit metabolic dysfunction with AD progression [74]. Abnormal liver function enzymes were associated with a significantly increased AD risk [75]. The liver tissue from AD patients also contained less Aβ than that of healthy individuals, suggesting an impaired ability of the liver to uptake circulating Aβ [18]. These findings provide critical evidence that liver dysfunction as an early pathological event in AD, and will be discussed thoroughly later in this review.
3. Hepatic Metabolism in Health and Disease
The liver is an essential hub for numerous physiological processes, primarily responsible for system-wide metabolic regulation, metabolic detoxification, protein synthesis, immune system support, and lipid and cholesterol homeostasis [76]. Moreover, the liver’s ability to produce and store glycogen as a readily available glucose reserve for the body is vital. Upon feeding, the decline in glucagon and increase in insulin, followed by the increase of glycolysis and glycogen deposition, drive the liver to shift to the net glucose intake mode [16,76]. By contrast, as the body changes from the absorptive state to the fasting state, insulin diminishes, and glucagon increases, turning the liver into promoting net glucose output to meet the energy demands of the brain and other organs [76,77]. Liver-derived glucose production could represent 90% of endogenous glucose production and is of particular importance for cells that partially or completely rely on glucose as energy substrates, such as neurons, erythrocytes, and renal medullary cells [78,79]. These liver-associated physiological processes, however, are dysregulated in many metabolic disorders and AD [66,80]. During the fed state, insulin stimulates glucose absorption into muscle and adipose tissue and inhibits hepatic gluconeogenesis. In the case of hepatic insulin resistance, impaired suppression of hepatic glucose production can lead to hyperglycemia and compromises cerebral glucose transport [66,81,82]. Therefore, well-coordinated hepatic glucose metabolism is crucial for health.
Importantly, as suggested above, the liver is the principal organ responsible for Aβ peripheral clearance in normal physiological conditions. When effluxed from the brain, Aβ binds to other molecules. For instance, circulating Aβ can be transported to the liver via high-density lipoprotein (HDL) particles, and subsequently taken up into cells by hepatocyte low-density lipoprotein receptor-related protein-1 (LRP-1), where it is degraded or cleared through bile excretion [11,83,84]. Abnormalities in these processes have been observed in both AD patients and animal models. Notably, an earlier study reported that the liver tissue from AD patients contains fewer Aβ molecules than that of healthy individuals, implying that the liver’s Aβ clearance function may be impaired in AD pathology [18]. Indeed, it is increasingly recognized that hepatic dysfunction is an early event in AD, preceding the pathological characteristics. In the following section, we discuss recent findings supporting hepatic dysfunction as a risk factor in AD.
4. Liver Dysfunction and AD Pathology
4.1. Animal Studies on the Crosstalk between Liver Dysfunction and AD Pathology
The role of liver dysfunction in aging and AD is beginning to receive attention. The contribution of liver dysfunction to the pathogenesis of AD is well-described in animal models (Table 1). LRP-1 is an endocytic and signaling receptor expressed in multiple tissues, including LRP-1 existing on the cell surface and soluble form of LRP1 (sLRP1) in plasma [59,85]. This receptor is required for the cerebral and systemic clearance of Aβ. In the brain, Aβ is combined with apolipoprotein E (ApoE) and transported to peripheral blood through LRP-1. Subsequently, most circulating Aβ binds to sLRP-1 to avoid excessive free Aβ concentrations in the blood [84,85]. In the liver, Aβ can be absorbed by hepatocytes via surface LRP-1 and then is eliminated. Nevertheless, both LRP-1 expression in the liver and hepatic Aβ uptake can be significantly reduced in aging and AD animals, suggesting impaired Aβ peripheral transport and clearance [86,87].

Table 1.
Animal studies on the crosstalk between liver dysfunction and AD pathology.
In light of the growing body of research suggesting that obesity and high-fat diets may increase the risk of AD, Kim et al. performed an important study to evaluate the relevance of non-alcoholic fatty liver disease (NAFLD) to AD pathology. They demonstrated that chronic NAFLD induced advanced pathological hallmarks of AD, including glial cell activation, neuroinflammation, neuronal apoptosis, and β-amyloid plaque load, in both wild-type and APP-Tg mice [88]. Intriguingly, decreased brain expression of LRP-1 was also observed in these animals. Additional evidence indicates that a Western diet feeding accelerated amyloid pathology in APPswe mice, which is paralleled by hypercholesterolemia and fatty liver [90]. Moreover, using an analytical method for in-depth tissue metabolome profiling, Wang et al. revealed remarkable differences in metabolome changes in the liver and brain in 5xFAD mice compared to wild-type mice [89]. In a similar manner, Zheng et al. investigated the metabolic changes in the liver, kidney, and heart of APP/PS1 mice with aging. They found that during the progression of AD, metabolic disorders existed in the peripheral organs of APP/PS1 mice, among which the liver was the first organ affected, mainly involving disturbances in energy metabolism, amino acid metabolism, nucleic acid metabolism, as well as ketone and fatty acid metabolism [74]. Furthermore, abnormal cholesterol levels can affect Aβ synthesis, clearance, and neurotoxicity. Elevated serum cholesterol levels have been shown to positively associate with an increased risk of dementia, while certain cholesterol-lowering therapies can reduce the production of Aβ [91,92]. Results from another study, which measured hepatic cholesterol and its metabolites in animals, revealed a fundamental defect of liver cholesterol in APP/PS1 and AppNL-G-F mice; in particular, impaired bile acid synthesis was observed [72]. Since bile acid synthesis plays a pivotal role in the excretion of cholesterol, these results collectively suggest that the overall hepatic metabolic profile is compromised in the progression of AD.
4.2. Clinical Data on the Crosstalk between Liver Dysfunction and AD or Dementia
Apart from animal studies, the interactions between liver dysfunction and AD/dementia have been extensively investigated in some human studies (Table 2). An early autopsy study observed that Aβ levels (Aβ40 and Aβ42) in the white and gray matter from AD patients are significantly higher than that from non-dementia subjects, and the liver tissue from AD patients contains less Aβ, suggesting Aβ clearance, particularly Aβ clearance by the liver, might be compromised [18]. Notably, this study also measured plasma Aβ42/40 ratios in AD patients, but they varied widely among individuals. This distinction could be due to age and/or gender differences of the subjects as well as other potential variables. Intriguingly, cirrhosis patients were found to have higher plasma A40 and A42 levels than healthy individuals, which were further elevated in cirrhosis patients with hepatitis B virus infection [20]. Furthermore, this study also examined blood biochemical parameters to assess hepatic function. Multiple linear regression results revealed a positive association between aberrant hepatic function and elevated plasma Aβ levels, suggesting liver dysfunction may lead to reduced peripheral Aβ clearance by the liver [20].
Table 2.
Clinical data on the crosstalk between liver dysfunction and AD or dementia.
Several studies examined the association between altered liver enzymes and AD dementia diagnosis. In a recent study, serum hepatic function markers, cognitive performance, and AD pathological profiles were measured in 1581 AD Neuroimaging Initiative participants, with an 8 year follow-up. The results of the regression analysis revealed a strong association between elevated aspartate aminotransferase (AST) to alanine aminotransferase (ALT) ratio (peripheral liver function markers), lower levels of ALT, and AD diagnosis [73]. In particular, lower levels of ALT were correlated with increased Aβ deposition and exacerbated brain atrophy, while higher AST to ALT ratios were significantly correlated with reduced glucose metabolism in some brain regions, including the bilateral frontal, parietal, and temporal lobes. Similarly, in a longitudinal study designed to establish the association of ALT and AST with the risk of dementia, low ALT and AST levels were found to be related to a higher risk of dementia in an average 18.3 year follow-up [75]. Notably, a population-based cross-sectional survey conducted by Chen et al. [93] pointed to cirrhosis as one of the co-morbidities significantly associated with mild cognitive impairment and dementia (OR 3.29, 95% CI 1.29–8.41). Given the progressive increase in liver fibrosis with aging, Solfrizzi et al. evaluated the link between NAFLD fibrosis score and risk of dementia. Their preliminary results suggest that older adults with high NAFLD fibrosis scores do not show a markedly increased risk of dementia [94]. Nevertheless, at the subsequent 8 year follow-up, frail older adults with high fibrosis scores were at an increased overall risk of developing dementia.
Taken together, although such studies remain quite limited, these findings support liver dysfunction as an early event related to AD and might be implicated in the pathogenesis of AD. Further prospective studies with a larger sample size are needed to investigate the correlation between liver dysfunction/disease and the risk of AD.
5. The Liver as a Potential Therapeutic Target for Alzheimer’s Disease
5.1. Strategies to Enhance Hepatic Aβ Degradation
A better understanding of the link between liver dysfunction in the pathogenesis of AD may present new opportunities for its early diagnosis and management. For this purpose, many attempts have been made to explore the therapeutic potential of improving liver function in the treatment of AD treatment. LRP-1, the main cell surface receptor involved in cerebral and systemic toxic Aβ clearance, is thought to be one of the therapeutic targets for AD [84]. Several measures aimed to restore or enhance LRP1-driven Aβ regulatory systems have shown great potential in mitigating AD pathological processes. For instance, fluvastatin treatment increased LRP-1 expression levels at the BBB, resulting in increased Aβ clearance and diminished accumulation in a transgenic mouse model of AD [95]. Low-level administration of recombinant LRP cluster IV has been proven to facilitate Aβ clearance, and alleviate pathological alternations and functional disorders, but this therapy did not affect phosphorylated LRP-1 levels in the brain and liver [96]. Importantly, portal insulin injection facilitated LRP-1 transport to the hepatic plasma membrane and enhanced LRP-1-dependent Aβ uptake by the liver [97]. Intriguingly, orally administered extracts of Withania somnifera (WS), a drug plant widely used for the treatment of neurological disorders, have been reported to reverse Aβ plaque burden and behavioral deficits in middle-aged transgenic AD mice [87]. The positive effects of WS largely depended on the modulation of hepatic LRP. Although upregulation of plasma slurp and hepatic LRP was observed after WS administration, the extract did not induce changes in brain LRP. Significantly, selective downregulation of hepatic LRP eliminated these therapeutic benefits [87]. These lines of evidence appear to corroborate the efficacy of enhanced hepatic Aβ clearance in slowing AD progression.
Moreover, regular exercise is an effective intervention in the management of diabetes mellitus and AD. Previous studies by our group and others have reported the positive effects of exercise interventions in preventing the progression of AD [98,99,100]; exercise also shows the potential to improve liver metabolic function [101,102]. To be noted, in an experimental model of AD, treadmill exercise reversed diminished plasma sLRP-1, as well as compromised LRP-1 mRNA and protein content in the liver, aside from a reduction in brain Aβ plaque load and up-regulation of hippocampal LRP-1 [103]. In this regard, exercise could be a powerful means to facilitate both central and peripheral Aβ clearance, with it also being the most cost-effective.
5.2. Strategies to Regulate Hepatic Synthesis Products
Some liver-derived synthesis products have also been reported to alleviate AD disease progression [104,105]. Fibroblast growth factor 21 (FGF21) is a peptide hormone highly synthesized in the liver and functions to maintain energy homeostasis in multiple tissues [106,107]. The physiology of FGF21 is complicated, as it can be synthesized in multiple organs and act on multiple target tissues, including the liver, brain, heart, and adipose tissue [107]. Under most physiological conditions, circulating FGF21 is predominantly derived from the liver. The specific ablation of hepatocytes using albumin Cre leads to a dramatic decrease in circulating levels of FGF21 [108]. Furthermore, systemically administered FGF21 can cross the BBB in a non-saturable manner and maintain integrity [109]. Since FGF21 receptors (FGFRs), including FGFR1, FGFR2, and FGFR3, as well as an obligate coreceptor, β-klotho, are expressed in the brain, it has been suggested that circulating FGF21 may exert many positive effects on the brain [107]. Additionally steatosis, inflammation, hepatocyte damage, and fibrosis in the liver can develop from FGF21 deficiency [110,111,112], whereas FGF21 analogue administration or upregulation of its endogenous level attenuates these processes [113,114]. FGF21 has attracted growing interest as a therapeutic agent for numerous metabolic syndromes including obesity and diabetes [106,115]. More importantly, compared with healthy subjects, the plasma FGF21 levels were significantly lower in AD patients, suggesting impaired hepatic ability to synthesize FGF21 in AD pathology [116].
Given the large body of research suggesting a strong link between AD/dementia and metabolic disease, a seminal study by Chen et al. investigated the effects of FGF21 on AD pathology [104]. After an intracranial injection of Aβ25-35, the rats were injected subcutaneously with FGF21 (twice daily for 8 days). The results showed that FGF21 exerted numerous protective effects, including alleviation in Aβ25-35-induced neuronal apoptosis, tau pathology, mitochondrial damage, and oxidative stress, paralleled by a significant improvement in the animals’ cognitive deficits [104]. Furthermore, previous studies demonstrated that the astrocyte–neuronal–lactate shuttle (ANLS) is critical for memory formation and consolidation [117,118]. Abnormalities in this communication have been identified in AD, including reduced lactate content, down-regulation of GLUTs and monocarboxylate transport proteins (MCTs), and lower lactate dehydrogenase (LDHA) in the cortex and hippocampus [119,120,121]. Based on these findings, the latest study from the same group revealed that FGF21 ameliorates neurodegeneration in transgenic AD animals through modulation of the ANLS system [105]. Consistent with previous reports, in both in vivo and in vitro models of AD, a compromised ANLS was found, and FGF21 delivered centrally or peripherally alleviated AD-related degeneration in mice; blocking FGFR1, however, prevented the beneficial effects induced by FGF21, including the alleviation of tau pathology and the upregulation of ATP, MCT2, and MCT4 in the brain [105]. Interestingly, ablation of MCT2 and MCT4 also eliminated the beneficial effect of FGF21 on cortical and hippocampal ATP levels, suggesting that ANLS regulation could be a target for the neuroprotection of FGF21. Intriguingly, in a rodent model of neurodegeneration, caloric restriction, a commonly adopted dietary intervention in preclinical research, significantly increased the mRNA expression of FGF21 in the liver [122]. This is in line with elevated circulating FGF21 and its expression in the brain, as well as alleviated synaptic loss and cognitive deficits. Similarly, FGF21 treatment improved metabolic dysfunction-related cognitive impairment and AD-like degeneration in obese rats [123,124]. These findings emphasized the potential contribution of FGF21, a hormone primarily derived from the liver, in preventing AD-like neurodegeneration.
6. Limitations and Future Perspectives
More recently, the role of liver dysfunction in AD pathology has emerged as an exciting field of investigation. Encouraging findings from animal studies also reflect the translational value of developing strategies to restore hepatic physiological function for AD management. Nevertheless, given the limited literature available, many questions remain to be addressed in this nascent field. Current evidence points to a reduction in hepatic LRP-1 under AD pathology, which results in a compromised peripheral clearance of Aβ, but whether liver dysfunction directly exacerbates brain Aβ load and accompanying neurodegeneration is not yet well-defined. As one of the peripheral organs responsible for Aβ clearance, the resection of the kidney has been proven to increase Aβ burden in the brain and exacerbated associated pathologies [45]. Along these lines, as the liver is in charge of eliminating the majority of the circulating Aβ, inhibition of its ability to uptake and clear circulating Aβ, or hepatectomy removal, may shed light on these issues. In addition, the confirmation that peripheral FGF21 delivery protects against AD via cerebral FGF1 and MCTs emphasizes the essential role of circulating FGF21 for cerebral energy homeostasis [105]. Given that liver accounts for more than 80% of the FGF21 product in circulation, a novel liver-specific knockout of FGF21 mouse model has been established to identify the contribution of liver-derived FGF21 in metabolic regulation [125,126]. This transgenic model might serve as a powerful tool for understanding the potential role of liver FGF21 deficiency in AD-like pathologies. Intriguingly, FGF21 has been shown to be an exercise-responsive factor. Circulating FGF21 elevates after different forms of exercise, and the synthesis of FGF21 in response to exercise may mediate the multiple health benefits conferred by exercise [127,128]. A recent study revealed that the protective effect of exercise on cardiac function was eliminated in liver-specific FGF21 knockout mice [129]. Similar to exercise, FGF21 is protective against the development of AD; whether the induction of liver FGF21 is required for the beneficial effects of exercise for AD would be an issue worthy of further investigation.
Remarkably, although studies have revealed a connection between hepatic pathological alternations and AD, little is known about how different forms and degrees of liver pathologies are involved in AD progression. Further studies are required to elucidate these critical issues. In this sense, targeted metabolomics analyses on liver tissue from experimental animal models and the wider employment of liver disease-related animal models would be useful to identify potential pathogenic factors, as well as to determine potential differences among these pathological processes, and earlier AD markers. Since AD normally triggers irreversible tissue damage after onset, a better understanding of these physiological processes will undoubtedly facilitate the early diagnosis and management of AD.
Moreover, developing treatments targeted at liver protection will not only help us understand the peripheral pathological mechanisms of AD, but will also guide our efforts to develop innovative treatment modalities for this disease. To be noted, some liver-targeted mitochondrial uncoupling agents, such as derivatives of 2,4-dinitrophenol-methyl ether, have shown the potential to attenuate hepatic gluconeogenesis, insulin resistance, hepatic lipid peroxidation, protein carbonylation, and systemic inflammation [130,131]. In addition, J147, a novel AD candidate used to alleviate neurological dysfunction, has been reported to reduce free fatty acid levels in the liver via modulation of AMPK/ACC1 signaling [132]. Given that accumulated free fatty acid in the liver is the major driver of NAFLD and the potential link between NAFLD and AD pathology, the liver is probably an attractive target for the neuroprotection of J147. In this regard, the treatment efficacy and molecular mechanisms of these agents in AD pathobiology will deserve further investigation.
In light of the limited effect of monotherapy, combined therapies for AD are increasingly being advocated to enhance the treatment efficacy of traditional therapy. Whether the combination of traditional treatment strategies with interventions to enhance peripheral Aβ clearance or supplementation with FGF21 will have additive effects remains to be elucidated. If successfully applied in clinical practice, these combined approaches will greatly benefit patients with AD and reduce associated social and economic burdens.
7. Conclusions
Peripheral organs, especially the liver, are of importance for Aβ clearance, and disturbances in peripheral Aβ metabolism may contribute to AD progression. Furthermore, aberrant liver function might also undermine the communication between the liver and brain, including normal glucose metabolism, and the supply of certain key hepatic synthesis products, resulting in metabolic disorders. Improvements in our understanding of the interaction between hepatic disorders and the brain will help identify the peripheral mechanisms of AD pathogenesis and contribute to its early diagnosis and treatment. In addition, the failure of monotherapies has prompted increasing agreement that combination therapies directed to multiple targets may accumulate their beneficial effects in AD treatment. Thus, combinatorial approaches targeting the liver with other pathologies may potentially represent new AD therapeutic strategies.
Author Contributions
Z.H., Q.Z. and X.Z. reviewed the literature and drafted the manuscript. Z.H., H.W.L. and X.Z. drew the figure. H.W.L. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a grant from the National Institute of Aging, National Institutes of Health (1RF1AG058603 to Q.Z.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
AD: Alzheimer’s disease; Aβ, Amyloid-beta; APP, Amyloid precursor protein; ATP, Adenosine triphosphate; ApoE, Apolipoprotein E; AST, Aspartate aminotransferase; ALT, Alanine aminotransferase; ANLS, Astrocyte-neuronal lactate shuttle; BBB, Blood-brain barrier; CNS, Central nerves system; FGF21, Fibroblast growth factor 21; GLUTs, Glucose transporters; HDL, High-density lipoprotein; LDHA, Lactate dehydrogenase; LRP-1, Low-density lipoprotein receptor-related protein-1; MCTs, Monocarboxylate transport proteins; NAFLD, Non-alcoholic fatty liver disease; NFTs, Neurofibrillary tangles; RBC, red blood cell; WS, Withania somnifera.
References
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Wei, C.; Chen, S.; Li, F.; Tang, Y.; Qin, W.; Zhao, L.; Jin, H.; Xu, H.; Wang, F.; et al. The cost of Alzheimer’s disease in China and re-estimation of costs worldwide. Alzheimer’s Dement. 2018, 14, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Giunta, B.; Zhou, H.-D.; Tan, J.; Wang, Y.-J. Immunotherapy for Alzheimer disease—The challenge of adverse effects. Nat. Rev. Neurol. 2012, 8, 465–469. [Google Scholar] [CrossRef]
- Busche, M.A.; Grienberger, C.; Keskin, A.D.; Song, B.; Neumann, U.; Staufenbiel, M.; Förstl, H.; Konnerth, A. Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer’s models. Nat. Neurosci. 2015, 18, 1725–1727. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Wang, Y.-R.; Xiang, Y.; Zhou, H.-D.; Giunta, B.; Mañucat-Tan, N.B.; Tan, J.; Zhou, X.-F.; Wang, Y.-J. Clearance of Amyloid-Beta in Alzheimer’s Disease: Shifting the Action Site from Center to Periphery. Mol. Neurobiol. 2015, 51, 1–7. [Google Scholar] [CrossRef]
- Qosa, H.; Abuasal, B.S.; Romero, I.A.; Weksler, B.; Couraud, P.-O.; Keller, J.N.; Kaddoumi, A. Differences in amyloid-β clearance across mouse and human blood–brain barrier models: Kinetic analysis and mechanistic modeling. Neuropharmacology 2014, 79, 668–678. [Google Scholar] [CrossRef]
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, D.-Y.; Wang, Y.-J. Peripheral clearance of brain-derived Aβ in Alzheimer’s disease: Pathophysiology and therapeutic perspectives. Transl. Neurodegener. 2020, 9, 16. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A systemic view of Alzheimer disease—insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Xin, S.-H.; Tan, L.; Cao, X.; Yu, J.-T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef]
- Clarke, J.R.; Ribeiro, F.C.; Frozza, R.L.; De Felice, F.G.; Lourenco, M.V. Metabolic Dysfunction in Alzheimer’s Disease: From Basic Neurobiology to Clinical Approaches. J. Alzheimer’s Dis. 2018, 64, S405–S426. [Google Scholar] [CrossRef]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin resistance in Alzheimer’s disease. Transl. Res. 2017, 183, 26–40. [Google Scholar] [CrossRef]
- Sutcliffe, J.G.; Hedlund, P.B.; Thomas, E.A.; Bloom, F.E.; Hilbush, B.S. Peripheral reduction of β-amyloid is sufficient to reduce brain β-amyloid: Implications for Alzheimer’s disease. J. Neurosci. Res. 2011, 89, 808–814. [Google Scholar] [CrossRef]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef]
- Astarita, G.; Jung, K.-M.; Berchtold, N.C.; Nguyen, V.Q.; Gillen, D.L.; Head, E.; Cotman, C.W.; Piomelli, D. Deficient Liver Biosynthesis of Docosahexaenoic Acid Correlates with Cognitive Impairment in Alzheimer’s Disease. PLoS ONE 2010, 5, e12538. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Wang, Q.-H.; Zhang, T.; Liu, Y.-H.; Yao, X.-Q.; Zeng, F.; Li, J.; Zhou, F.-Y.; Wang, L.; Yan, J.-C.; et al. Associations Between Hepatic Functions and Plasma Amyloid-Beta Levels—Implications for the Capacity of Liver in Peripheral Amyloid-Beta Clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef]
- Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 2018, 15, 276. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s Disease and Vascular Aging. J. Am. Coll. Cardiol. 2020, 75, 942–951. [Google Scholar] [CrossRef]
- Irwin, M.R.; Vitiello, M.V. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 2019, 18, 296–306. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Lee, B.Y.; Leonenko, Z. Recent Progress in Alzheimer’s Disease Research, Part 1: Pathology. J. Alzheimer’s Dis. 2017, 57, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 3306. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Van Giau, V. Type 3 diabetes and its role implications in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef]
- Leszek, J.; Trypka, E.; Tarasov, V.V.; Ashraf, G.; Aliev, G. Type 3 Diabetes Mellitus: A Novel Implication of Alzheimers Disease. Curr. Top. Med. Chem. 2017, 17, 1331–1335. [Google Scholar] [CrossRef]
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Front. Aging Neurosci. 2019, 11, 174. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Small, S.A.; Duff, K. Linking Aβ and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef]
- Ortiz, J.M.P.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the Amyloid Hypothesis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 493–510. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2017, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Colonna, M. Microglia in Alzheimer’s disease: A target for immunotherapy. J. Leukoc. Biol. 2019, 106, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.F.; Elbert, D.L.; Kasten, T.P.; Patterson, B.W.; Sigurdson, W.C.; Connors, R.E.; Ovod, V.; Munsell, L.Y.; Mawuenyega, K.G.; Miller-Thomas, M.M.; et al. Amyloid-β efflux from the central nervous system into the plasma. Ann. Neurol. 2014, 76, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.-S.; Shen, L.-L.; Bu, X.-L.; Zhang, W.-W.; Chen, S.-H.; Huang, Z.-L.; Xiong, J.-X.; Gao, C.-Y.; Dong, Z.; He, Y.-N.; et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017, 134, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.-Y.; Cheng, Y.; Zhuang, Z.-Q.; He, C.-Y.; Pan, Q.-G.; Tang, M.-Z.; Hu, X.-L.; Shen, Y.-Y.; Wang, Y.-R.; Chen, S.-H.; et al. Physiological clearance of amyloid-beta by the kidney and its therapeutic potential for Alzheimer’s disease. Mol. Psychiatry 2021, 26, 6074–6082. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Deng, D.; Yan, N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. [Google Scholar] [CrossRef]
- Szablewski, L. Glucose Transporters in Brain: In Health and in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 1307–1320. [Google Scholar] [CrossRef]
- Biswas, J.; Gupta, S.; Verma, D.K.; Gupta, P.; Singh, A.; Tiwari, S.; Goswami, P.; Sharma, S.; Singh, S. Involvement of glucose related energy crisis and endoplasmic reticulum stress: Insinuation of streptozotocin induced Alzheimer’s like pathology. Cell Signal. 2018, 42, 211–226. [Google Scholar] [CrossRef]
- Dineley, K.T.; Jahrling, J.B.; Denner, L. Insulin resistance in Alzheimer’s disease. Neurobiol. Dis. 2014, 72, 92–103. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, F.; Head, E.; Butterfield, D.A.; Perluigi, M.; Barone, E. Brain insulin resistance triggers early onset Alzheimer disease in Down syndrome. Neurobiol. Dis. 2020, 137, 104772. [Google Scholar] [CrossRef]
- Adams, J.N.; Lockhart, S.N.; Li, L.; Jagust, W.J. Relationships Between Tau and Glucose Metabolism Reflect Alzheimer’s Disease Pathology in Cognitively Normal Older Adults. Cereb. Cortex 2019, 29, 1997–2009. [Google Scholar] [CrossRef]
- Dato, V.A.; Chiabrando, G.A. The Role of Low-Density Lipoprotein Receptor-Related Protein 1 in Lipid Metabolism, Glucose Homeostasis and Inflammation. Int. J. Mol. Sci. 2018, 19, 1780. [Google Scholar] [CrossRef]
- Carter, S.F.; Chiotis, K.; Nordberg, A.; Rodriguez-Vieitez, E. Longitudinal association between astrocyte function and glucose metabolism in autosomal dominant Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 348–356. [Google Scholar] [CrossRef]
- Ebanks, B.; Ingram, T.L.; Chakrabarti, L. ATP synthase and Alzheimer’s disease: Putting a spin on the mitochondrial hypothesis. Aging 2020, 12, 16647–16662. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef]
- Lu, X.-Y.; Huang, S.; Chen, Q.-B.; Zhang, D.; Li, W.; Ao, R.; Leung, F.C.-Y.; Zhang, Z.; Huang, J.; Tang, Y.; et al. Metformin Ameliorates Aβ Pathology by Insulin-Degrading Enzyme in a Transgenic Mouse Model of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2020, 2020, 2315106. [Google Scholar] [CrossRef]
- Niccoli, T.; Cabecinha, M.; Tillmann, A.; Kerr, F.; Wong, C.T.; Cardenes, D.; Vincent, A.J.; Bettedi, L.; Li, L.; Grönke, S.; et al. Increased Glucose Transport into Neurons Rescues Aβ Toxicity in Drosophila. Curr. Biol. 2016, 26, 2291–2300. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Bangen, K.J.; Himali, J.J.; Beiser, A.S.; Nation, D.A.; Libon, D.J.; Fox, C.S.; Seshadri, S.; Wolf, P.A.; McKee, A.C.; Au, R.; et al. Interaction Between Midlife Blood Glucose and APOE Genotype Predicts Later Alzheimer’s Disease Pathology. J. Alzheimer’s Dis. 2016, 53, 1553–1562. [Google Scholar] [CrossRef]
- Zangerolamo, L.; Vettorazzi, J.F.; Solon, C.; Bronczek, G.A.; Engel, D.F.; Kurauti, M.A.; Soares, G.M.; Rodrigues, K.S.; Velloso, L.A.; Boschero, A.C.; et al. The bile acid TUDCA improves glucose metabolism in streptozotocin-induced Alzheimer’s disease mice model. Mol. Cell. Endocrinol. 2021, 521, 111116. [Google Scholar] [CrossRef]
- Jiang, S.; Gavrikova, T.A.; Pereboev, A.; Messina, J.L. Adenovirus infection results in alterations of insulin signaling and glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1295–E1304. [Google Scholar] [CrossRef]
- Ferreiro, E.; Lanzillo, M.; Canhoto, D.; da Silva, A.M.C.; Mota, S.I.; Dias, I.S.; Ferreira, I.L.; Fontes, A.R.; Mastrella, G.; Pinheiro, P.; et al. Chronic hyperglycemia impairs hippocampal neurogenesis and memory in an Alzheimer’s disease mouse model. Neurobiol. Aging 2020, 92, 98–113. [Google Scholar] [CrossRef]
- Huang, R.; Tian, S.; Zhang, H.; Zhu, W.; Wang, S. Chronic hyperglycemia induces tau hyperphosphorylation by downregulating OGT-involved O-GlcNAcylation in vivo and in vitro. Brain Res. Bull. 2020, 156, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Seeger, D.; Golovko, S.; Golovko, M.; Combs, C. Liver Bile Acid Changes in Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7451. [Google Scholar] [CrossRef] [PubMed]
- Nho, K.; Kueider-Paisley, A.; Ahmad, S.; Mahmoudiandehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; et al. Association of Altered Liver Enzymes With Alzheimer Disease Diagnosis, Cognition, Neuroimaging Measures, and Cerebrospinal Fluid Biomarkers. JAMA Netw. Open 2019, 2, e197978. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Cai, A.; Shu, Q.; Niu, Y.; Xu, P.; Li, C.; Lin, L.; Gao, H. Tissue-Specific Metabolomics Analysis Identifies the Liver as a Major Organ of Metabolic Disorders in Amyloid Precursor Protein/Presenilin 1 Mice of Alzheimer’s Disease. J. Proteome Res. 2019, 18, 1218–1227. [Google Scholar] [CrossRef]
- Lu, Y.; Pike, J.R.; Selvin, E.; Mosley, T.; Palta, P.; Sharrett, A.R.; Thomas, A.; Loehr, L.; Barritt, A.S.; Hoogeveen, R.C.; et al. Low Liver Enzymes and Risk of Dementia: The Atherosclerosis Risk in Communities (ARIC) Study. J. Alzheimer’s Dis. 2021, 79, 1775–1784. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin Resistance in Nonalcoholic Fatty Liver Disease. Curr. Pharm. Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef]
- Ekberg, K.; Landau, B.R.; Wajngot, A.; Chandramouli, V.; Efendic, S.; Brunengraber, H.; Wahren, J. Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes 1999, 48, 292–298. [Google Scholar] [CrossRef]
- Oosterveer, M.H.; Schoonjans, K. Hepatic glucose sensing and integrative pathways in the liver. Cell. Mol. Life Sci. 2014, 71, 1453–1467. [Google Scholar] [CrossRef]
- Wijesekara, N.; Gonçalves, R.A.; De Felice, F.G.; Fraser, P.E. Impaired peripheral glucose homeostasis and Alzheimer’s disease. Neuropharmacology 2018, 136, 172–181. [Google Scholar] [CrossRef]
- Leão, L.L.; Tangen, G.; Barca, M.L.; Engedal, K.; Santos, S.H.S.; Machado, F.S.M.; de Paula, A.M.B.; Monteiro-Junior, R.S. Does hyperglycemia downregulate glucose transporters in the brain? Med. Hypotheses 2020, 139, 109614. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J. Diabetes Res. 2020, 2020, 4981814. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. eLife 2017, 6, e29595. [Google Scholar] [CrossRef]
- Sagare, A.P.; Deane, R.; Zlokovic, B.V. Low-density lipoprotein receptor-related protein 1: A physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacol. Ther. 2012, 136, 94–105. [Google Scholar] [CrossRef]
- Cai, Z.; Qiao, P.-F.; Wan, C.-Q.; Cai, M.; Zhou, N.-K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Tamaki, C.; Ohtsuki, S.; Iwatsubo, T.; Hashimoto, T.; Yamada, K.; Yabuki, C.; Terasaki, T. Major Involvement of Low-Density Lipoprotein Receptor-Related Protein 1 in the Clearance of Plasma Free Amyloid β-Peptide by the Liver. Pharm. Res. 2006, 23, 1407–1416. [Google Scholar] [CrossRef]
- Sehgal, N.; Gupta, A.; Valli, R.K.; Joshi, S.D.; Mills, J.T.; Hamel, E.; Khanna, P.; Jain, S.C.; Thakur, S.S.; Ravindranath, V. Withania somnifera reverses Alzheimer’s disease pathology by enhancing low-density lipoprotein receptor-related protein in liver. Proc. Natl. Acad. Sci. USA 2012, 109, 3510–3515. [Google Scholar] [CrossRef]
- Kim, D.-G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.-A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflammation 2016, 13, 1. [Google Scholar] [CrossRef]
- Wang, X.; Han, W.; Yang, J.; Westaway, D.; Li, L. Development of chemical isotope labeling LC-MS for tissue metabolomics and its application for brain and liver metabolome profiling in Alzheimer’s disease mouse model. Anal. Chim. Acta 2019, 1050, 95–104. [Google Scholar] [CrossRef]
- Wiȩckowska-Gacek, A.; Mietelska-Porowska, A.; Chutorański, D.; Wydrych, M.; Długosz, J.; Wojda, U. Western Diet Induces Impairment of Liver-Brain Axis Accelerating Neuroinflammation and Amyloid Pathology in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 654509. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef]
- Wanamaker, B.L.; Swiger, K.J.; Blumenthal, R.S.; Martin, S.S. Cholesterol, Statins, and Dementia: What the Cardiologist Should Know. Clin. Cardiol. 2015, 38, 243–250. [Google Scholar] [CrossRef]
- Chen, T.-B.; Yiao, S.-Y.; Sun, Y.; Lee, H.-J.; Yang, S.-C.; Chiu, M.-J.; Chen, T.-F.; Lin, K.-N.; Tang, L.-Y.; Lin, C.-C.; et al. Comorbidity and dementia: A nationwide survey in Taiwan. PLoS ONE 2017, 12, e0175475. [Google Scholar] [CrossRef]
- Solfrizzi, V.; Scafato, E.; Custodero, C.; Loparco, F.; Ciavarella, A.; Panza, F.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Napoli, N.; et al. Liver fibrosis score, physical frailty, and the risk of dementia in older adults: The Italian Longitudinal Study on Aging. Alzheimer’s Dementia Transl. Res. Clin. Interv. 2020, 6, e12065. [Google Scholar] [CrossRef]
- Shinohara, M.; Sato, N.; Kurinami, H.; Takeuchi, D.; Takeda, S.; Shimamura, M.; Yamashita, T.; Uchiyama, Y.; Rakugi, H.; Morishita, R. Reduction of Brain β-Amyloid (Aβ) by Fluvastatin, a Hydroxymethylglutaryl-CoA Reductase Inhibitor, through Increase in Degradation of Amyloid Precursor Protein C-terminal Fragments (APP-CTFs) and Aβ Clearance. J. Biol. Chem. 2010, 285, 22091–22102. [Google Scholar] [CrossRef]
- Sagare, A.; Deane, R.; Bell, R.D.; Johnson, B.; Hamm, K.; Pendu, R.; Marky, A.; Lenting, P.; Wu, Z.; Zarcone, T.; et al. Clearance of amyloid-β by circulating lipoprotein receptors. Nat. Med. 2007, 13, 1029–1031. [Google Scholar] [CrossRef]
- Tamaki, C.; Ohtsuki, S.; Terasaki, T. Insulin Facilitates the Hepatic Clearance of Plasma Amyloid β-Peptide (1–40) by Intracellular Translocation of Low-Density Lipoprotein Receptor-Related Protein 1 (LRP-1) to the Plasma Membrane in Hepatocytes. Mol. Pharmacol. 2007, 72, 850–855. [Google Scholar] [CrossRef]
- Wu, C.; Yang, L.; Tucker, D.; Dong, Y.; Zhu, L.; Duan, R.; Liu, T.C.-Y.; Zhang, Q. Beneficial Effects of Exercise Pretreatment in a Sporadic Alzheimer’s Rat Model. Med. Sci. Sports Exerc. 2018, 50, 945–956. [Google Scholar] [CrossRef]
- Yang, L.; Wu, C.; Li, Y.; Dong, Y.; Wu, C.Y.; Lee, R.H.; Brann, D.W.; Lin, H.W.; Zhang, Q. Long-term exercise pre-training attenuates Alzheimer’s disease-related pathology in a transgenic rat model of Alzheimer’s disease. Geroscience 2022, 44, 1457–1477. [Google Scholar] [CrossRef]
- Yu, F.; Vock, D.M.; Zhang, L.; Salisbury, D.; Nelson, N.W.; Chow, L.S.; Smith, G.; Barclay, T.R.; Dysken, M.; Wyman, J.F. Cognitive Effects of Aerobic Exercise in Alzheimer’s Disease: A Pilot Randomized Controlled Trial. J. Alzheimer’s Dis. 2021, 80, 233–244. [Google Scholar] [CrossRef]
- Smart, N.A.; King, N.; McFarlane, J.R.; Graham, P.L.; Dieberg, G. Effect of exercise training on liver function in adults who are overweight or exhibit fatty liver disease: A systematic review and meta-analysis. Br. J. Sports Med. 2018, 52, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, D.; Gharakhanlou, R.; Naghdi, N.; Salimi, M.; Azimi, M.; Shahed, A.; Heysieattalab, S. Treadmill Exercise Ameliorates Spatial Learning and Memory Deficits Through Improving the Clearance of Peripheral and Central Amyloid-Beta Levels. Neurochem. Res. 2018, 43, 1561–1574. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, S.-T.; Sun, Y.; Xu, Z.; Wang, Y.; Yao, S.-Y.; Yao, W.-B.; Gao, X.-D. Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer’s disease. Redox Biol. 2019, 22, 101133. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Y.; Chen, S.-T.; Chen, Y.-J.; Shen, J.; Yao, W.-B.; Gao, X.-D.; Chen, S. Modulation of the Astrocyte-Neuron Lactate Shuttle System contributes to Neuroprotective action of Fibroblast Growth Factor 21. Theranostics 2020, 10, 8430–8445. [Google Scholar] [CrossRef]
- Lewis, J.E.; Ebling, F.J.; Samms, R.J.; Tsintzas, K. Going Back to the Biology of FGF21: New Insights. Trends Endocrinol. Metab. 2019, 30, 491–504. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 Is Liver Derived and Enhances Glucose Uptake During Refeeding and Overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef]
- Tan, B.K.; Hallschmid, M.; Adya, R.; Kern, W.; Lehnert, H.; Randeva, H.S. Fibroblast Growth Factor 21 (FGF21) in Human Cerebrospinal Fluid: Relationship with plasma FGF21 and body adiposity. Diabetes 2011, 60, 2758–2762. [Google Scholar] [CrossRef]
- Zarei, M.; Pizarro-Delgado, J.; Barroso, E.; Palomer, X.; Vázquez-Carrera, M. Targeting FGF21 for the Treatment of Nonalcoholic Steatohepatitis. Trends Pharmacol. Sci. 2020, 41, 199–208. [Google Scholar] [CrossRef]
- Singhal, G.; Kumar, G.; Chan, S.; Fisher, F.M.; Ma, Y.; Vardeh, H.G.; Nasser, I.A.; Flier, J.S.; Maratos-Flier, E. Deficiency of fibroblast growth factor 21 (FGF21) promotes hepatocellular carcinoma (HCC) in mice on a long term obesogenic diet. Mol. Metab. 2018, 13, 56–66. [Google Scholar] [CrossRef]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef]
- Tucker, B.; Li, H.; Long, X.; Rye, K.-A.; Ong, K.L. Fibroblast growth factor 21 in non-alcoholic fatty liver disease. Metabolism 2019, 101, 153994. [Google Scholar] [CrossRef]
- Tillman, E.J.; Rolph, T. FGF21: An Emerging Therapeutic Target for Non-Alcoholic Steatohepatitis and Related Metabolic Diseases. Front. Endocrinol. 2020, 11, 601290. [Google Scholar] [CrossRef]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef]
- Conte, M.; Sabbatinelli, J.; Chiariello, A.; Martucci, M.; Santoro, A.; Monti, D.; Arcaro, M.; Galimberti, D.; Scarpini, E.; Bonfigli, A.R.; et al. Disease-specific plasma levels of mitokines FGF21, GDF15, and Humanin in type II diabetes and Alzheimer’s disease in comparison with healthy aging. GeroScience 2021, 43, 985–1001. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate Produced by Glycogenolysis in Astrocytes Regulates Memory Processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, X.; Dang, R.; Zhang, W.; Zhang, J.; Yao, Z. Lactate Deficit in an Alzheimer Disease Mouse Model: The Relationship With Neuronal Damage. J. Neuropathol. Exp. Neurol. 2018, 77, 1163–1176. [Google Scholar] [CrossRef]
- Lu, W.; Huang, J.; Sun, S.; Huang, S.; Gan, S.; Xu, J.; Yang, M.; Xu, S.; Jiang, X. Changes in lactate content and monocarboxylate transporter 2 expression in Abeta(2)(5)(-)(3)(5)-treated rat model of Alzheimer’s disease. Neurol. Sci. 2015, 36, 871–876. [Google Scholar] [CrossRef]
- Lu, W.; Sun, S.; Li, Y.; Xu, S.; Gan, S.; Xu, J.; Qiu, G.; Zhuo, F.; Huang, S.; Jiang, X.; et al. Curcumin Ameliorates Memory Deficits by Enhancing Lactate Content and MCT2 Expression in APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease. Anat. Rec. 2019, 302, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Rühlmann, C.; Wölk, T.; Blümel, T.; Stahn, L.; Vollmar, B.; Kuhla, A. Long-term caloric restriction in ApoE-deficient mice results in neuroprotection via Fgf21-induced AMPK/mTOR pathway. Aging 2016, 8, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yuan, J.; Yu, Z.; Lin, L.; Jiang, Y.; Cao, Z.; Zhuang, P.; Whalen, M.J.; Song, B.; Wang, X.-J.; et al. FGF21 Attenuates High-Fat Diet-Induced Cognitive Impairment via Metabolic Regulation and Anti-inflammation of Obese Mice. Mol. Neurobiol. 2018, 55, 4702–4717. [Google Scholar] [CrossRef] [PubMed]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Wang, X.; Liang, G.; Li, X.; Jiang, C.; Pratchayasakul, W.; Chattipakorn, N.; et al. FGF21 and DPP-4 inhibitor equally prevents cognitive decline in obese rats. Biomed. Pharmacother. 2018, 97, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Feng, B.; Huang, L.; Li, J.; Luo, T.; Jiang, X.; Han, X.; Che, L.; Xu, S.; Lin, Y.; et al. Time-restricted feeding improves the reproductive function of female mice via liver fibroblast growth factor 21. Clin. Transl. Med. 2020, 10, e195. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Barrett, T.; Tournier, C.; Davis, R.J. Fibroblast Growth Factor 21 Mediates Glycemic Regulation by Hepatic JNK. Cell Rep. 2016, 14, 2273–2280. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, S.H.; Min, Y.-K.; Yang, H.-M.; Lee, J.-B.; Lee, M.-S. Acute Exercise Induces FGF21 Expression in Mice and in Healthy Humans. PLoS ONE 2013, 8, e63517. [Google Scholar] [CrossRef]
- Cuevas-Ramos, D.; Almeda-Valdes, P.; Meza-Arana, C.E.; Brito-Córdova, G.; Gómez-Pérez, F.J.; Mehta, R.; Oseguera-Moguel, J.; Aguilar-Salinas, C.A. Exercise Increases Serum Fibroblast Growth Factor 21 (FGF21) Levels. PLoS ONE 2012, 7, e38022. [Google Scholar] [CrossRef]
- Jin, L.; Geng, L.; Ying, L.; Shu, L.; Ye, K.; Yang, R.; Liu, Y.; Wang, Y.; Cai, Y.; Jiang, X.; et al. FGF21-Sirtuin 3 Axis Confers the Protective Effects of Exercise Against Diabetic Cardiomyopathy by Governing Mitochondrial Integrity. Circulation 2022. [Google Scholar] [CrossRef]
- Perry, R.J.; Kim, T.; Zhang, X.-M.; Lee, H.-Y.; Pesta, D.; Popov, V.B.; Zhang, D.; Rahimi, Y.; Jurczak, M.J.; Cline, G.W.; et al. Reversal of Hypertriglyceridemia, Fatty Liver Disease, and Insulin Resistance by a Liver-Targeted Mitochondrial Uncoupler. Cell Metab. 2013, 18, 740–748. [Google Scholar] [CrossRef]
- Goedeke, L.; Murt, K.N.; Di Francesco, A.; Camporez, J.P.; Nasiri, A.R.; Wang, Y.; Zhang, X.; Cline, G.W.; de Cabo, R.; Shulman, G.I. Sex- and strain-specific effects of mitochondrial uncoupling on age-related metabolic diseases in high-fat diet-fed mice. Aging Cell 2022, 21, e13539. [Google Scholar] [CrossRef] [PubMed]
- Kepchia, D.; Huang, L.; Currais, A.; Liang, Z.; Fischer, W.; Maher, P. The Alzheimer’s disease drug candidate J147 decreases blood plasma fatty acid levels via modulation of AMPK/ACC1 signaling in the liver. Biomed. Pharmacother. 2022, 147, 112648. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).