
Comprehensive Analysis of the Structure and Function of Peptide:N-Glycanase 1 and Relationship with Congenital Disorder of Deglycosylation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
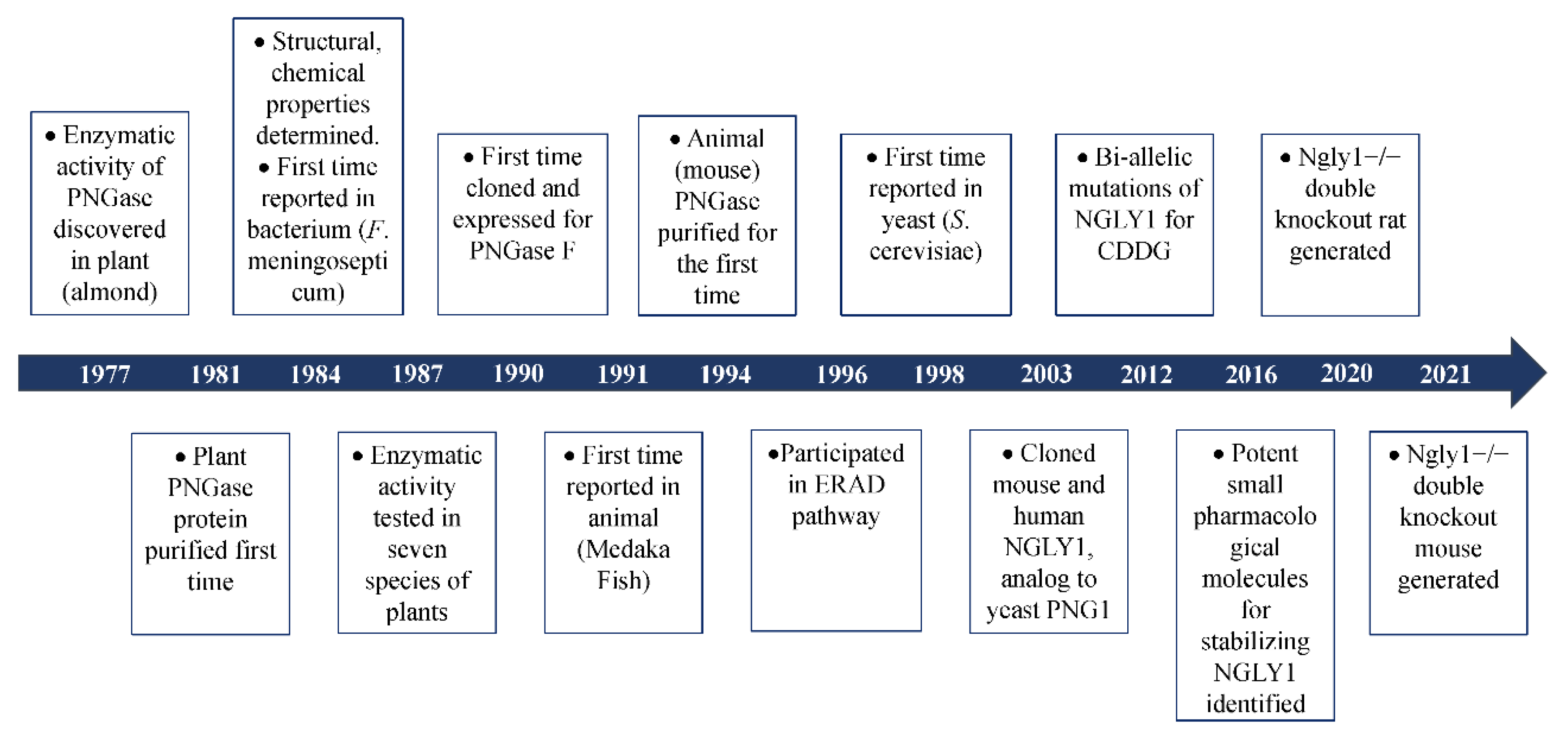
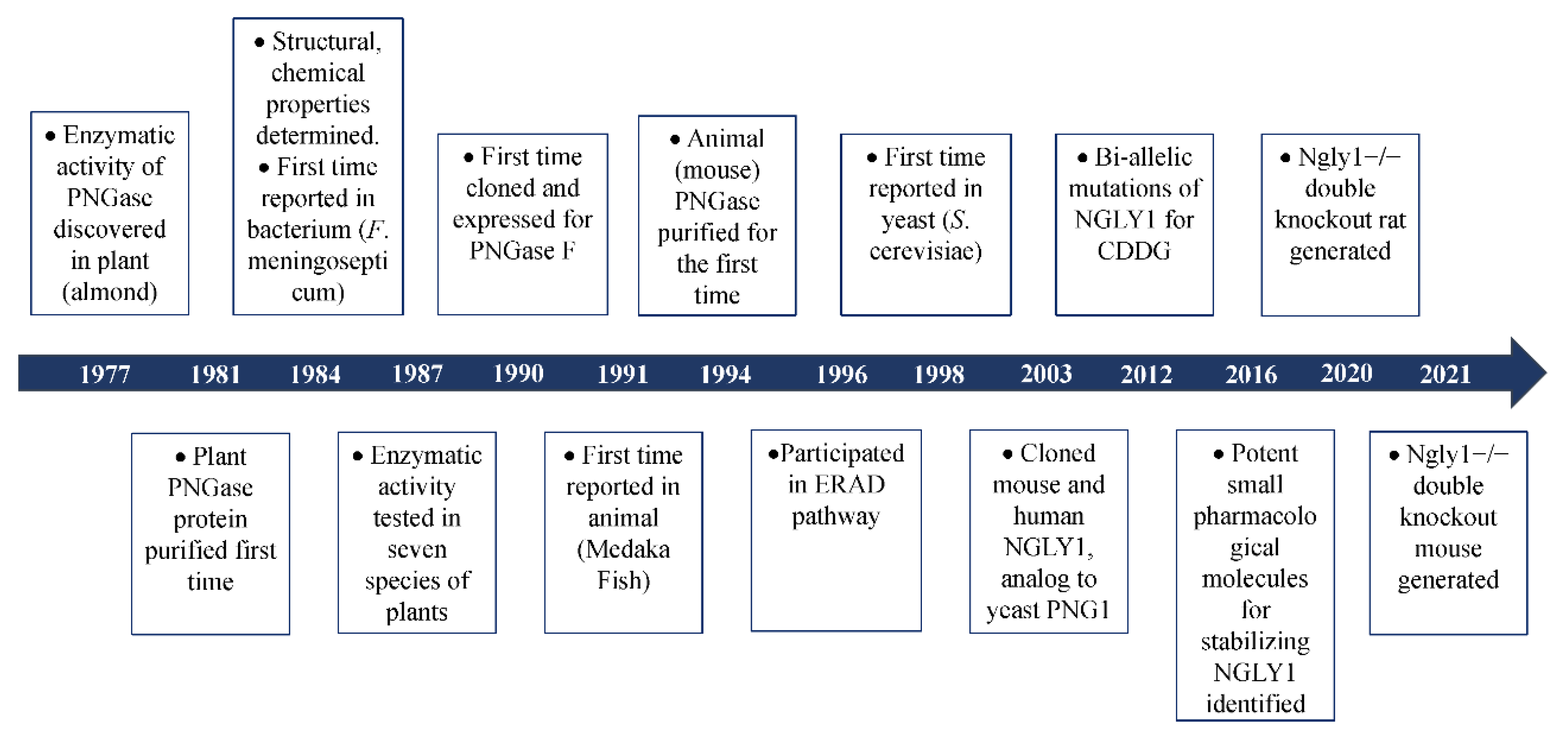
2. The Discovery of Peptide:N-Glycanase
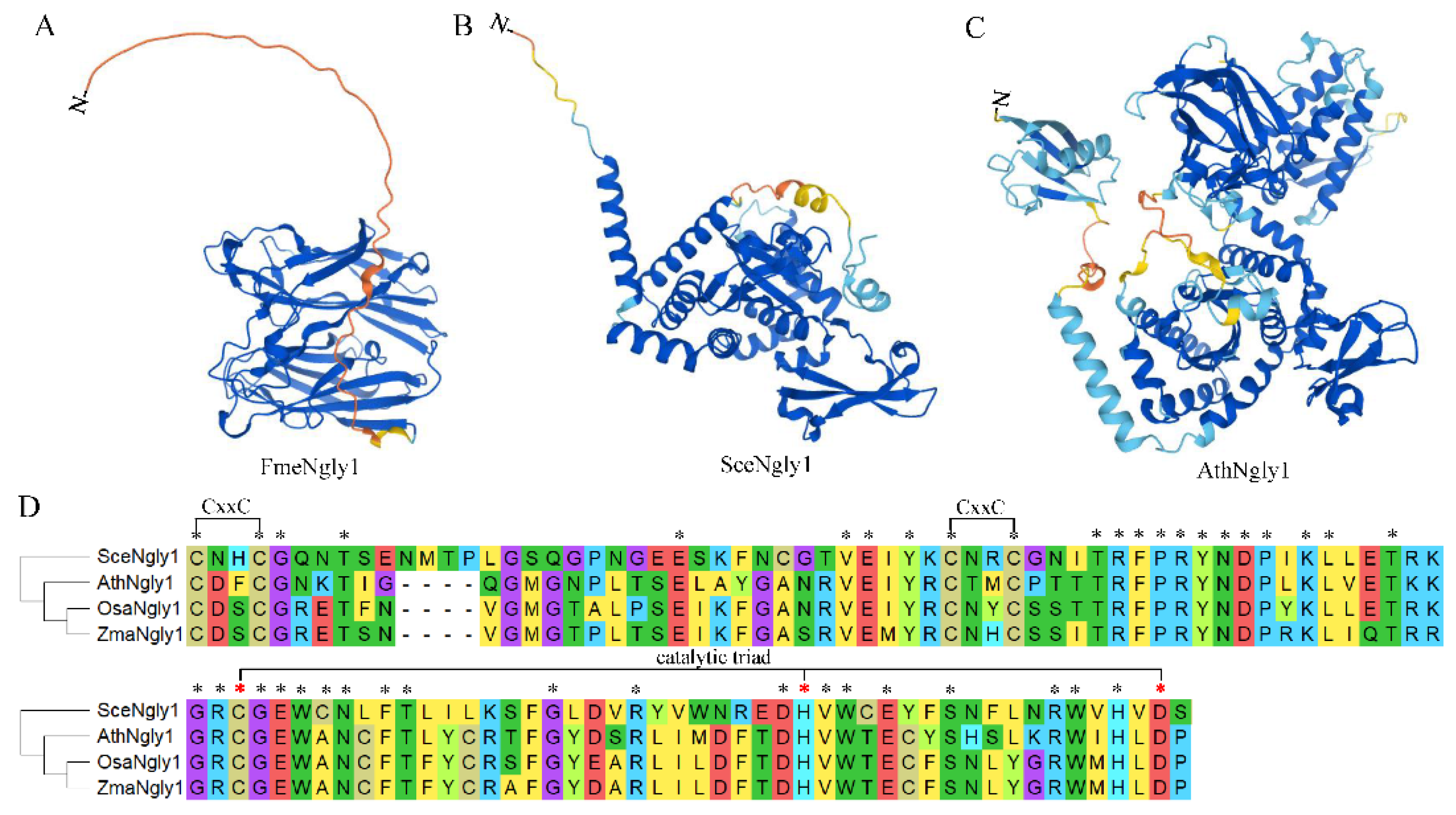
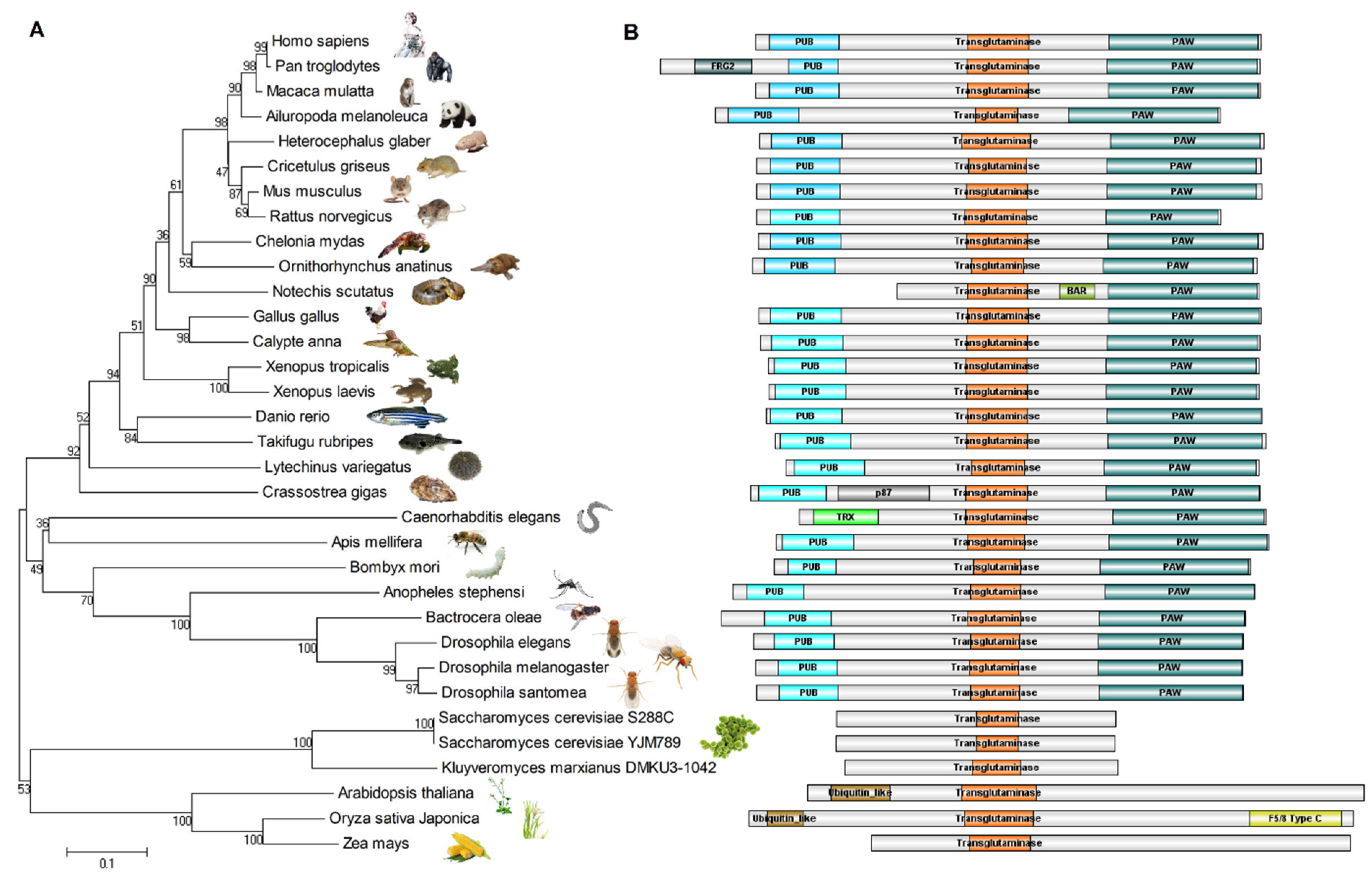
3. Protein Structure of Non-Animal Ngly1s
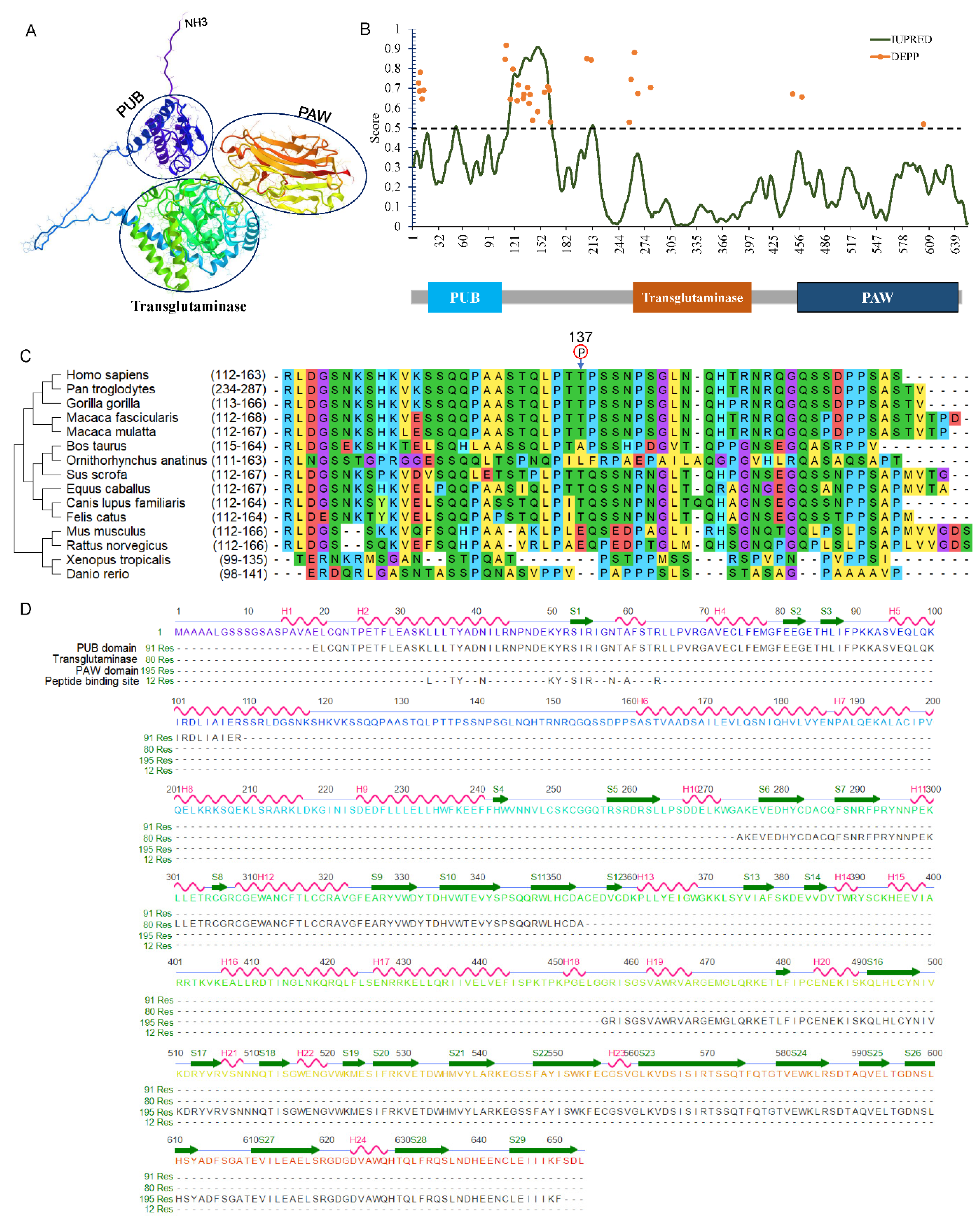
4. General Structures of Animal NGLY1
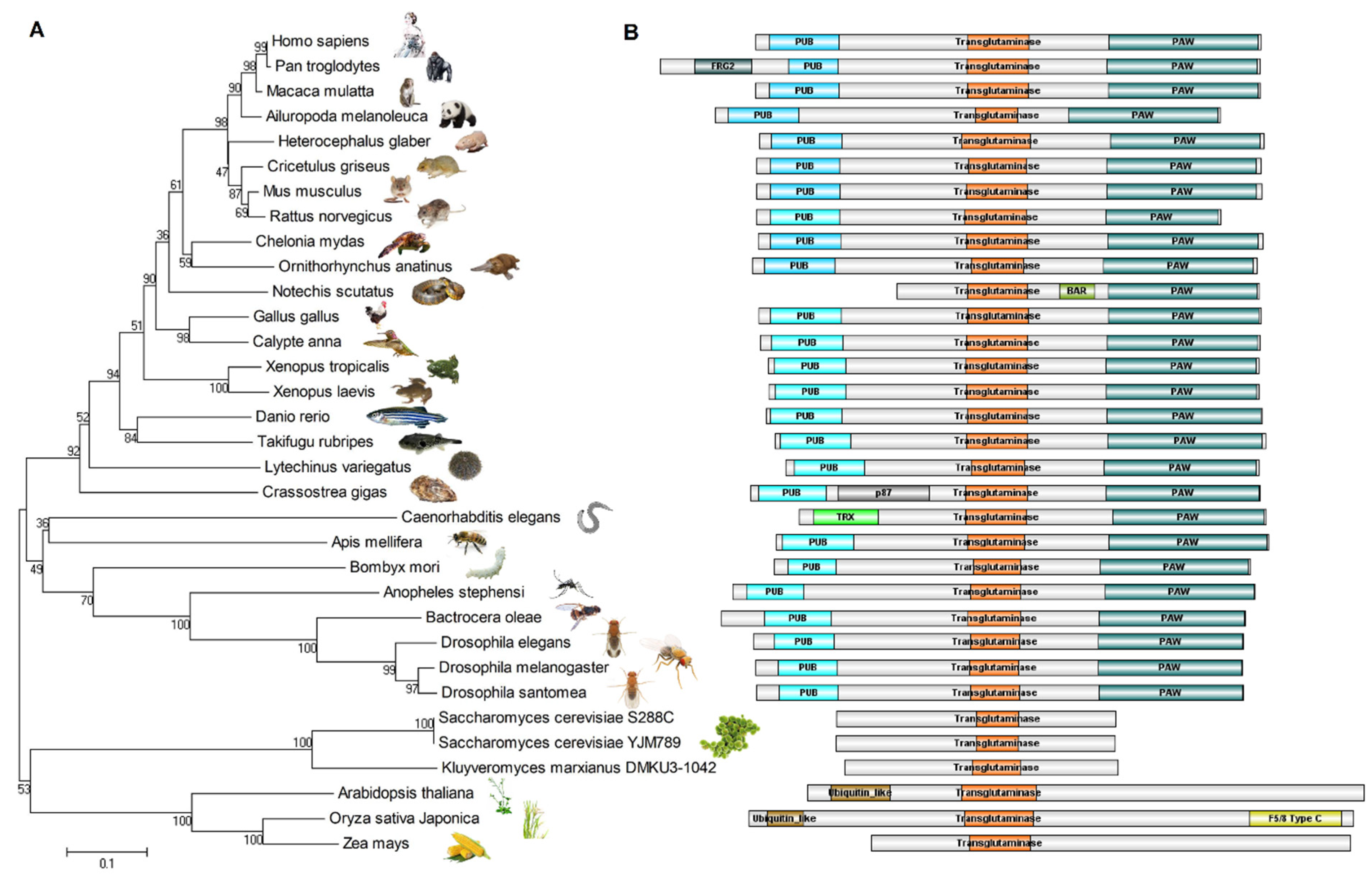
5. Evolutionary Relationships of NGLY1
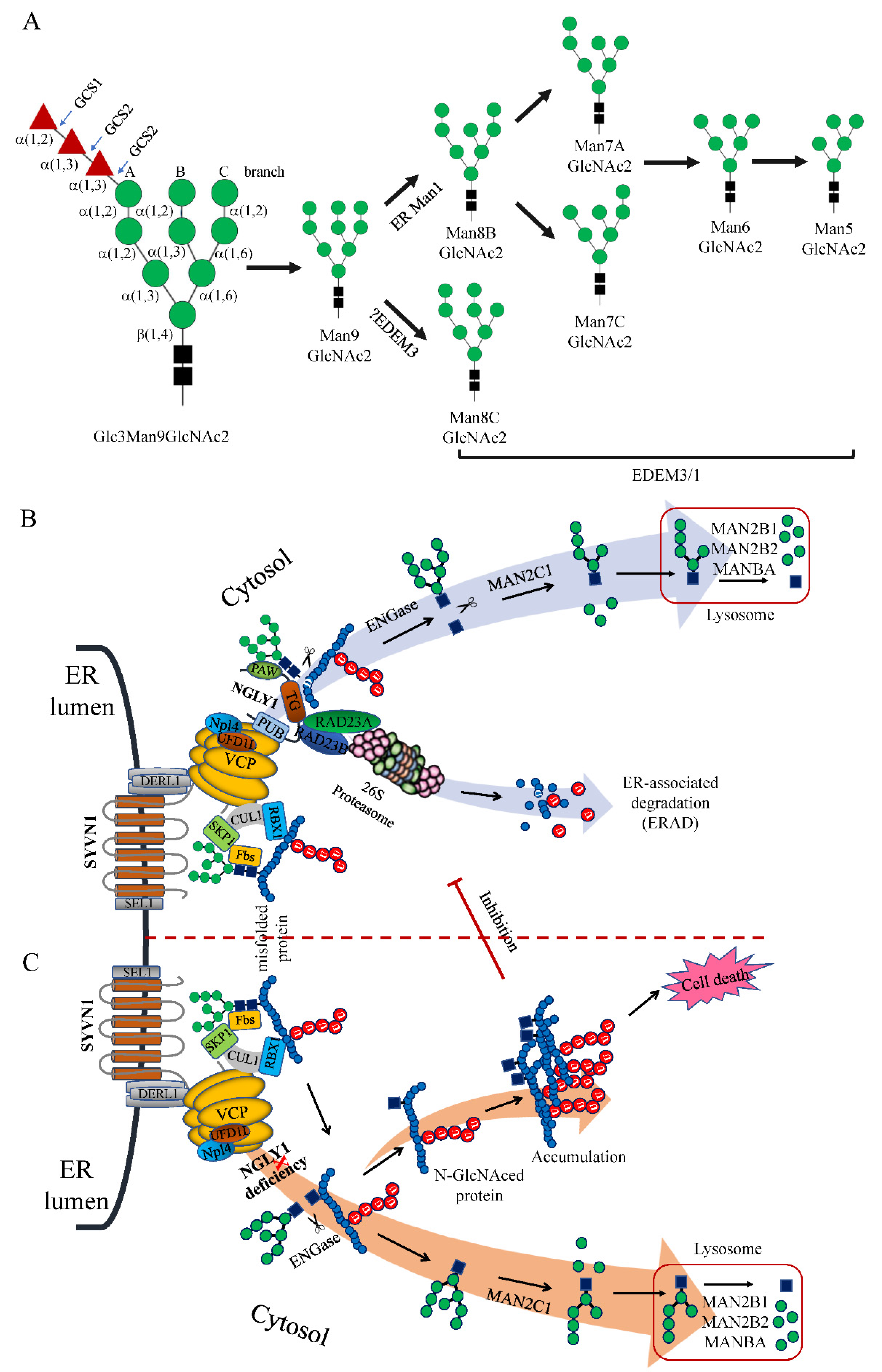
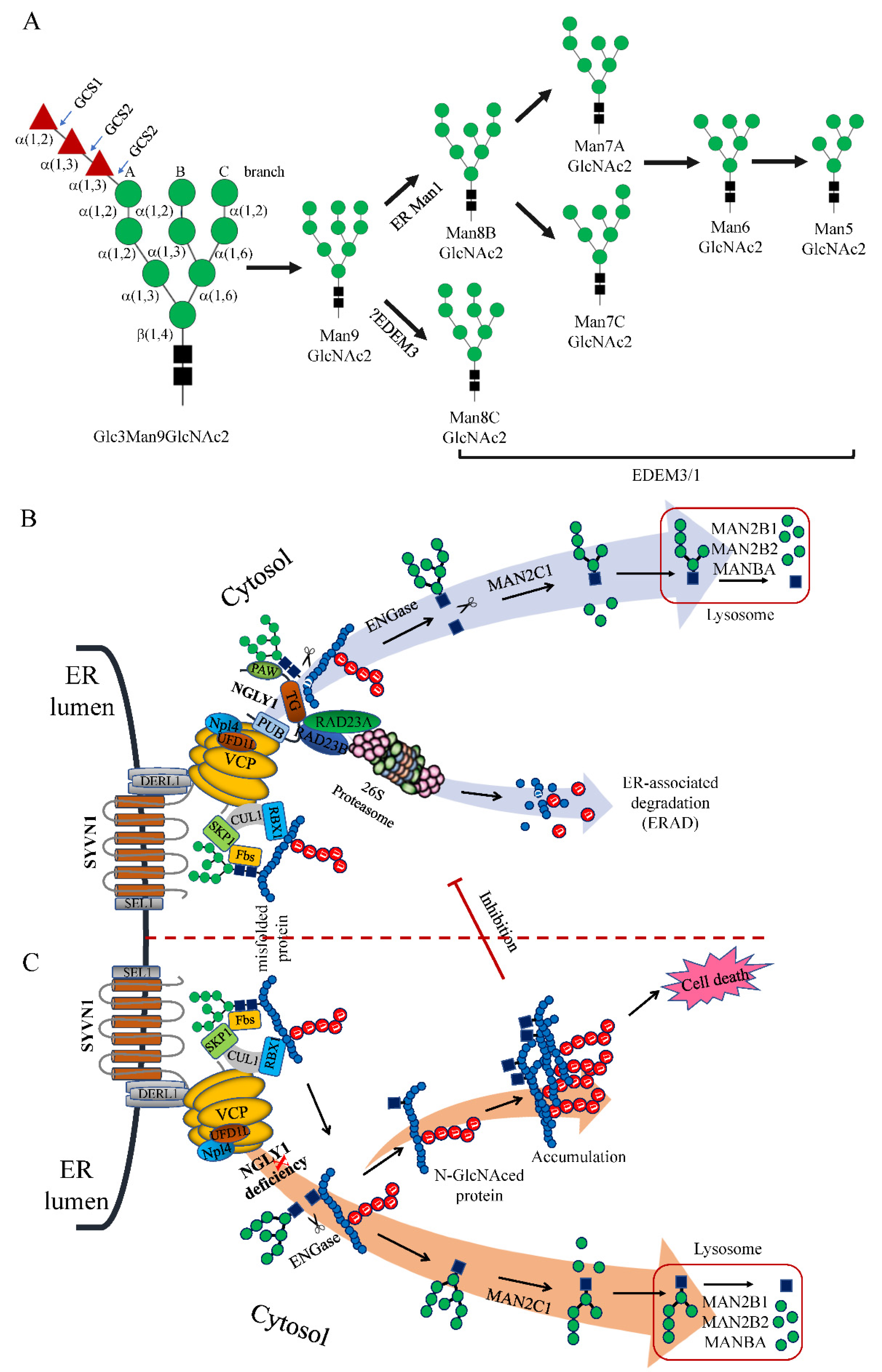
6. Roles of NGLY1 in the ERAD Pathway
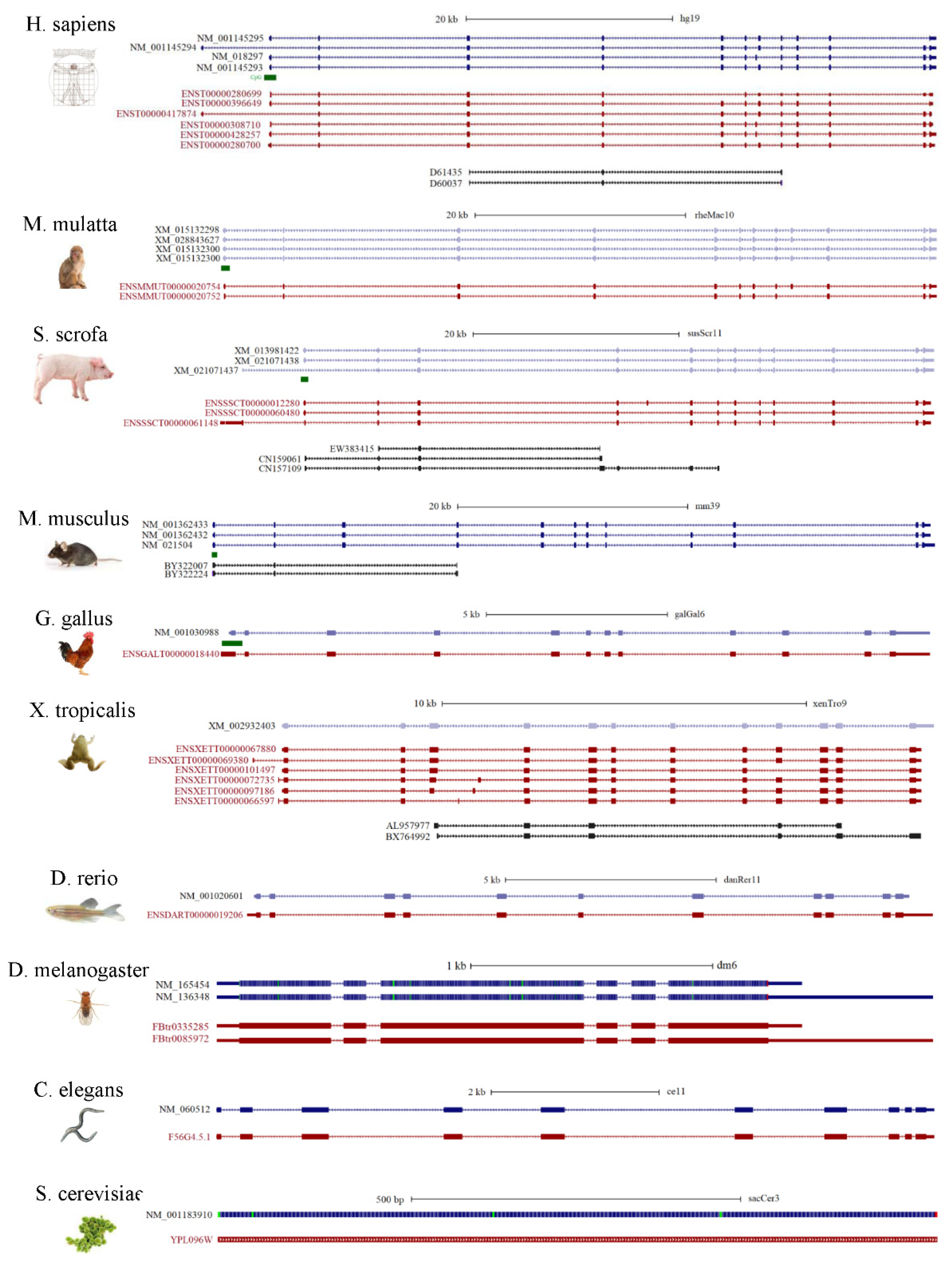
7. Alternative Splicing Patterns of NGLY1 in Human and Other Species
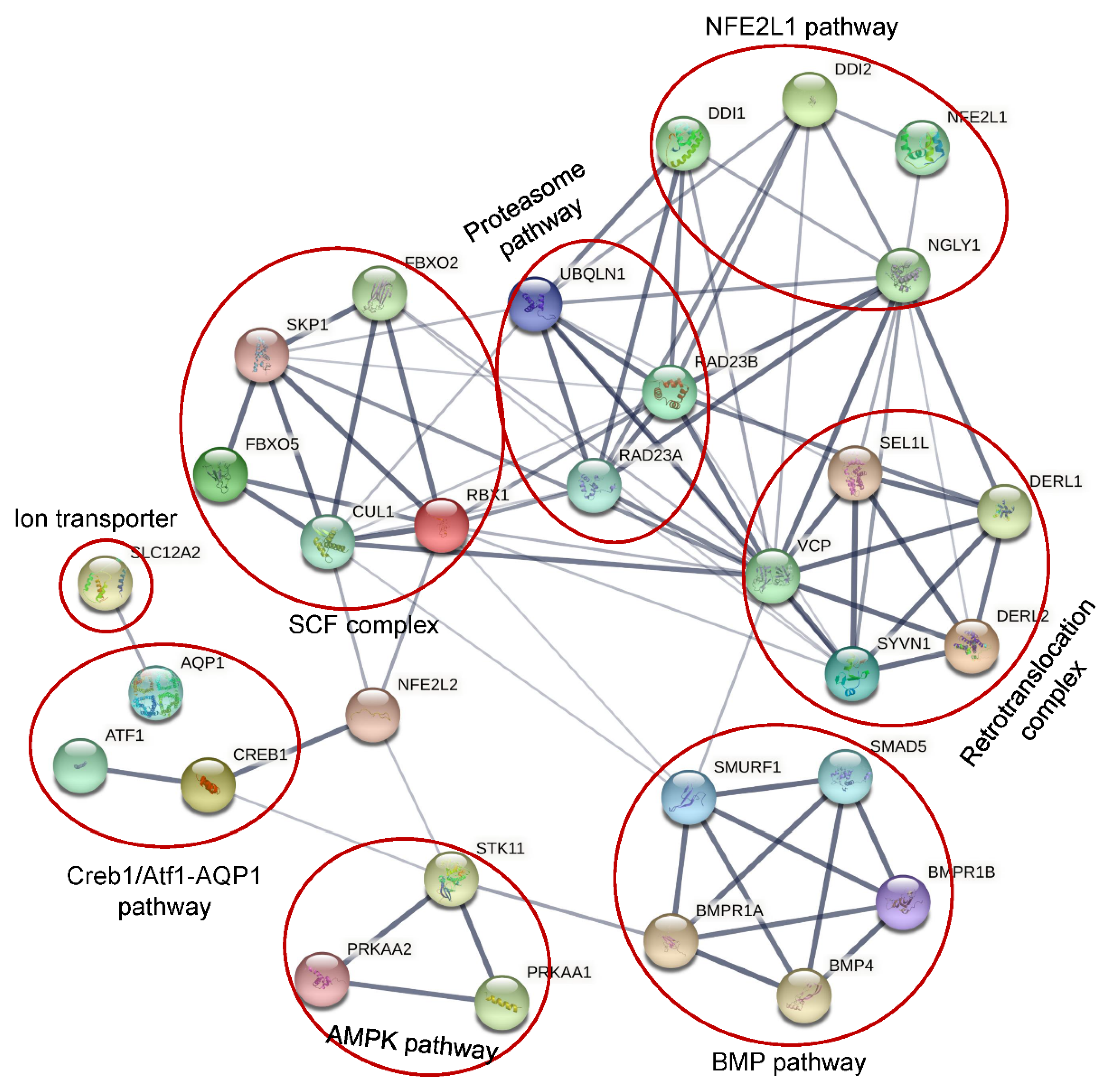
8. Different Functional Pathways Participated in by NGLY1
8.1. NFE2L1 Pathway
8.2. Creb1/Atf1-AQP Pathway
8.3. BMP Pathway
8.4. AMPK Pathway
8.5. SLC12A2
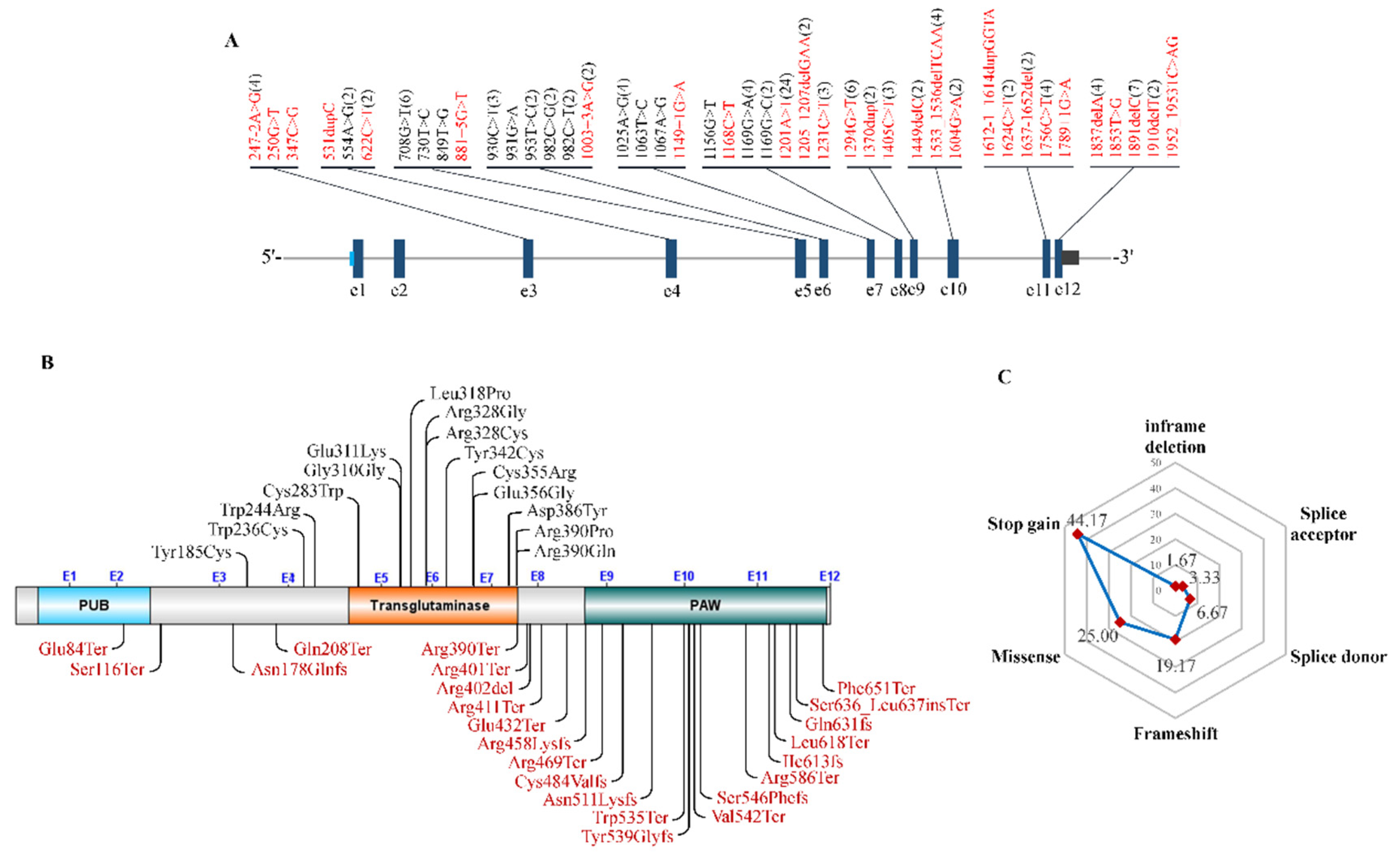
9. Mutations for NGLY1-Related Congenital Disorders
10. Potential Treatments for NGLY1-CDDG
10.1. Exogenous Restoration of NGLY1 Expression
10.2. Targeting ENGase with Small Inhibitors
10.3. Inhibition FOXB6 (Fbs2)
10.4. Activation of NFE2L2
11. Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Park, H.; Lennarz, W.J. Cytoplasmic peptide:N-glycanase (PNGase) in eukaryotic cells: Occurrence, primary structure, and potential functions. FASEB J. 2002, 16, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enns, G.M.; Shashi, V.; Bainbridge, M.; Gambello, M.J.; Zahir, F.R.; Bast, T.; Crimian, R.; Schoch, K.; Platt, J.; Cox, R.; et al. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med. 2014, 16, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N. Demonstration of a new amidase acting on glycopeptides. Biochem. Biophys. Res. Commun. 1977, 76, 1194–1201. [Google Scholar] [CrossRef]
- Sugiyama, K.; Ishihara, H.; Tejima, S.; Takahashi, N. Demonstration of a new glycopeptidase, from jack-bean meal, acting on aspartylglucosylamine linkages. Biochem. Biophys. Res. Commun. 1983, 112, 155–160. [Google Scholar] [CrossRef]
- Plummer, T.H., Jr.; Phelan, A.W.; Tarentino, A.L. Detection and quantification of peptide-N4-(N-acetyl-beta-glucosaminyl)asparagine amidases. Eur. J. Biochem. 1987, 163, 167–173. [Google Scholar] [CrossRef]
- Diepold, A.; Li, G.; Lennarz, W.J.; Nurnberger, T.; Brunner, F. The Arabidopsis AtPNG1 gene encodes a peptide:N-glycanase. Plant J. Cell Mol. Biol. 2007, 52, 94–104. [Google Scholar] [CrossRef]
- Takahashi, N.; Nishibe, H. Some characteristics of a new glycopeptidase acting on aspartylglycosylamine linkages. J. Biochem. 1978, 84, 1467–1473. [Google Scholar] [CrossRef]
- Plummer, T.H., Jr.; Tarentino, A.L. Facile cleavage of complex oligosaccharides from glycopeptides by almond emulsin peptide:N-glycosidase. J. Biol. Chem. 1981, 256, 10243–10246. [Google Scholar] [CrossRef]
- Nishibe, H.; Takahashi, N. The release of carbohydrate moieties from human fibrinogen by almond glycopeptidase without alteration in fibrinogen clottability. Biochim. Biophys. Acta 1981, 661, 274–279. [Google Scholar] [CrossRef]
- Tarentino, A.L.; Plummer, T.H., Jr. Oligosaccharide accessibility to peptide:N-glycosidase as promoted by protein-unfolding reagents. J. Biol. Chem. 1982, 257, 10776–10780. [Google Scholar] [CrossRef]
- Plummer, T.H., Jr.; Elder, J.H.; Alexander, S.; Phelan, A.W.; Tarentino, A.L. Demonstration of peptide:N-glycosidase F activity in endo-beta-N-acetylglucosaminidase F preparations. J. Biol. Chem. 1984, 259, 10700–10704. [Google Scholar] [CrossRef]
- Ishii, K.; Iwasaki, M.; Inoue, S.; Kenny, P.T.; Komura, H.; Inoue, Y. Free sialooligosaccharides found in the unfertilized eggs of a freshwater trout, Plecoglossus altivelis. A large storage pool of complex-type bi-, tri-, and tetraantennary sialooligosaccharides. J. Biol. Chem. 1989, 264, 1623–1630. [Google Scholar] [CrossRef]
- Seko, A.; Kitajima, K.; Iwasaki, M.; Inoue, S.; Inoue, Y. Structural studies of fertilization-associated carbohydrate-rich glycoproteins (hyosophorin) isolated from the fertilized and unfertilized eggs of flounder, Paralichthys olivaceus. Presence of a novel penta-antennary N-linked glycan chain in the tandem repeating glycopeptide unit of hyosophorin. J. Biol. Chem. 1989, 264, 15922–15929. [Google Scholar] [PubMed]
- Seko, A.; Kitajima, K.; Inoue, Y.; Inoue, S. Peptide:N-glycosidase activity found in the early embryos of Oryzias latipes (Medaka fish). The first demonstration of the occurrence of peptide:N-glycosidase in animal cells and its implication for the presence of a de-N-glycosylation system in living organisms. J. Biol. Chem. 1991, 266, 22110–22114. [Google Scholar]
- Suzuki, T.; Seko, A.; Kitajima, K.; Inoue, Y.; Inoue, S. Identification of peptide:N-glycanase activity in mammalian-derived cultured cells. Biochem. Biophys. Res. Commun. 1993, 194, 1124–1130. [Google Scholar] [CrossRef]
- Ftouhi-Paquin, N.; Hauer, C.R.; Stack, R.F.; Tarentino, A.L.; Plummer, T.H., Jr. Molecular cloning, primary structure, and properties of a new glycoamidase from the fungus Aspergillus tubigensis. J. Biol. Chem. 1997, 272, 22960–22965. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Park, H.; Kitajima, K.; Lennarz, W.J. Peptides glycosylated in the endoplasmic reticulum of yeast are subsequently deglycosylated by a soluble peptide:N-glycanase activity. J. Biol. Chem. 1998, 273, 21526–21530. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.; Ferreira, C.; Krasnewich, D.; Toro, C.; Latham, L.; Zein, W.M.; Lehky, T.; Brewer, C.; Baker, E.H.; Thurm, A.; et al. Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genet. Med. 2017, 19, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Haijes, H.A.; de Sain-van der Velden, M.G.M.; Prinsen, H.; Willems, A.P.; van der Ham, M.; Gerrits, J.; Couse, M.H.; Friedman, J.M.; van Karnebeek, C.D.M.; Selby, K.A.; et al. Aspartylglycosamine is a biomarker for NGLY1-CDDG, a congenital disorder of deglycosylation. Mol. Genet. Metab. 2019, 127, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Panneman, D.M.; Wortmann, S.B.; Haaxma, C.A.; van Hasselt, P.M.; Wolf, N.I.; Hendriks, Y.; Kusters, B.; van Emst-de Vries, S.; van de Westerlo, E.; Koopman, W.J.H.; et al. Variants in NGLY1 lead to intellectual disability, myoclonus epilepsy, sensorimotor axonal polyneuropathy and mitochondrial dysfunction. Clin. Genet. 2020, 97, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Tarentino, A.L.; Quinones, G.; Trumble, A.; Changchien, L.M.; Duceman, B.; Maley, F.; Plummer, T.H., Jr. Molecular cloning and amino acid sequence of peptide-N4-(N-acetyl-beta-D-glucosaminyl)asparagine amidase from flavobacterium meningosepticum. J. Biol. Chem. 1990, 265, 6961–6966. [Google Scholar] [CrossRef]
- Barsomian, G.D.; Johnson, T.L.; Borowski, M.; Denman, J.; Ollington, J.F.; Hirani, S.; McNeilly, D.S.; Rasmussen, J.R. Cloning and expression of peptide-N4-(N-acetyl-beta-D-glucosaminyl)asparagine amidase F in Escherichia coli. J. Biol. Chem. 1990, 265, 6967–6972. [Google Scholar] [CrossRef]
- Tarentino, A.L.; Gomez, C.M.; Plummer, J.T.H. Deglycosylation of asparagine-linked glycans by peptide:N-glycosidase F. Biochemistry 1985, 24, 4665–4671. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Tarentino, A.L.; Plummer, J.T.H.; Van Roey, P. Crystal Structure of Peptide-N4-(N-acetyl-.beta.-D-glucosaminyl)asparagine amidase F at 2.2-.ANG. Resolution. Biochemistry 1994, 33, 11699–11706. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Guan, C.; Cui, T.; Tarentino, A.L.; Plummer, T.H.; Van Roey, P. Active Site and Oligosaccharide Recognition Residues of Peptide-N4-(N-acetyl-β -D-glucosaminyl)asparagine Amidase F. J. Biol. Chem. 1995, 270, 29493–29497. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Park, H.; Hollingsworth, N.M.; Sternglanz, R.; Lennarz, W.J. PNG1, a Yeast Gene Encoding a Highly Conserved Peptide:N-Glycanase. J. Cell Biol. 2000, 149, 1039–1052. [Google Scholar] [CrossRef] [Green Version]
- Davidson, G.S.; Joe, R.M.; Roy, S.; Meirelles, O.; Allen, C.P.; Wilson, M.R.; Tapia, P.H.; Manzanilla, E.E.; Dodson, A.E.; Chakraborty, S.; et al. The proteomics of quiescent and nonquiescent cell differentiation in yeast stationary-phase cultures. Mol. Biol. Cell 2011, 22, 988–998. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; Huh, W.-K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef]
- Zhao, G.; Li, G.; Zhou, X.; Matsuo, I.; Ito, Y.; Suzuki, T.; Lennarz, W.J.; Schindelin, H. Structural and mutational studies on the importance of oligosaccharide binding for the activity of yeast PNGase. Glycobiology 2008, 19, 118–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Choi, J.M.; Lee, C.; Yi, K.J.; Cho, Y. Structure of a peptide:N -glycanase-Rad23 complex: Insight into the deglycosylation for denatured glycoproteins. Proc. Natl. Acad. Sci. USA 2005, 102, 9144–9149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katiyar, S.; Li, G.; Lennarz, W.J. A complex between peptide:N-glycanase and two proteasome-linked proteins suggests a mechanism for the degradation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 13774–13779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Katiyar, S.; Li, G.; Zhou, X.; Lennarz, W.J.; Schindelin, H. The N-terminus of yeast peptide:N-glycanase interacts with the DNA repair protein Rad23. Biochem. Biophys. Res. Commun. 2004, 323, 149–155. [Google Scholar] [CrossRef]
- Suzuki, T.; Hara, I.; Nakano, M.; Zhao, G.; Lennarz, W.J.; Schindelin, H.; Taniguchi, N.; Totani, K.; Matsuo, I.; Ito, Y. Site-specific Labeling of Cytoplasmic Peptide:N-Glycanase by N,N′-Diacetylchitobiose-related Compounds. J. Biol. Chem. 2006, 281, 22152–22160. [Google Scholar] [CrossRef] [Green Version]
- Chantret, I.; Kodali, V.P.; Lahmouich, C.; Harvey, D.J.; Moore, S.E. Endoplasmic Reticulum-associated Degradation (ERAD) and Free Oligosaccharide Generation in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 41786–41800. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Park, H.; Till, E.A.; Lennarz, W.J. The PUB Domain: A Putative Protein–Protein Interaction Domain Implicated in the Ubiquitin-Proteasome Pathway. Biochem. Biophys. Res. Commun. 2001, 287, 1083–1087. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; I Hurwitz, D.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Della Mea, M.; Caparrós-Ruiz, D.; Claparols, I.; Serafini-Fracassini, D.; Rigau, J.; Kirst, M.; Myburg, A.A.; De León, J.P.; Kirst, M.E.; Scott, J.; et al. AtPng1p. The First Plant Transglutaminase. Plant Physiol. 2004, 135, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Masahara-Negishi, Y.; Hosomi, A.; Della Mea, M.; Serafini-Fracassini, D.; Suzuki, T. A plant peptide:N-glycanase orthologue facilitates glycoprotein ER-associated degradation in yeast. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 1457–1462. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.R.; Mohammed, S. Toward a Comprehensive Characterization of a Human Cancer Cell Phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerks, T.; Copley, R.R.; Schultz, J.; Ponting, C.P.; Bork, P. Systematic identification of novel protein domain families associated with nuclear functions. Genome. Res. 2002, 12, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Bucher, P. The UBA domain: A sequence motif present in multiple enzyme classes of the ubiquitination pathway. Trends Biochem. Sci. 1996, 21, 172–173. [Google Scholar] [CrossRef]
- Allen, M.D.; Buchberger, A.; Bycroft, M. The PUB domain functions as a p97 binding module in human peptide N-glycanase. J. Biol. Chem. 2006, 281, 25502–25508. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, Y.; Uekusa, Y.; Sumiyoshi, A.; Sasakawa, H.; Hirao, T.; Suzuki, T.; Kato, K. NMR characterization of the interaction between the PUB domain of peptide:N-glycanase and ubiquitin-like domain of HR23. FEBS Lett. 2012, 586, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, V.; Akutsu, M.; Olma, M.H.; Gomes, L.C.; Kawasaki, M.; Dikic, I. Binding of OTULIN to the PUB domain of HOIP controls NF-kappaB signaling. Mol. Cell 2014, 54, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Olzmann, J.A.; Shaler, T.A.; Sowa, M.E.; Bennett, E.J.; Richter, C.M.; Tyler, R.E.; Greenblatt, E.J.; Harper, J.W.; Kopito, R.R. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2011, 14, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Kitajima, K.; Emori, Y.; Inoue, Y.; Inoue, S. Site-specific de-N-glycosylation of diglycosylated ovalbumin in hen oviduct by endogenous peptide:N-glycanase as a quality control system for newly synthesized proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 6244–6249. [Google Scholar] [CrossRef] [Green Version]
- Katiyar, S.; Joshi, S.; Lennarz, W.J. The retrotranslocation protein Derlin-1 binds peptide:N-glycanase to the endoplasmic reticulum. Mol. Biol. Cell 2005, 16, 4584–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Zhou, X.; Wang, L.; Li, G.; Kisker, C.; Lennarz, W.J.; Schindelin, H. Structure of the mouse peptide N-glycanase-HR23 complex suggests co-evolution of the endoplasmic reticulum-associated degradation and DNA repair pathways. J. Biol. Chem. 2006, 281, 13751–13761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taga, E.M.; Waheed, A.; Van Etten, R.L. Structural and chemical characterization of a homogeneous peptide N-glycosidase from almond. Biochemistry 1984, 23, 815–822. [Google Scholar] [CrossRef]
- Maerz, S.; Funakoshi, Y.; Negishi, Y.; Suzuki, T.; Seiler, S. The Neurospora peptide:N-glycanase ortholog PNG1 is essential for cell polarity despite its lack of enzymatic activity. J. Biol. Chem. 2010, 285, 2326–2332. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Madura, K. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell Biol. 2002, 22, 4902–4913. [Google Scholar] [CrossRef] [Green Version]
- Lambertson, D.; Chen, L.; Madura, K. Investigating the importance of proteasome-interaction for Rad23 function. Curr. Genet. 2003, 42, 199–208. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Heinen, C.; Hoogstraten, D. The ubiquitin receptor Rad23: At the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair 2009, 8, 449–460. [Google Scholar] [CrossRef]
- van Eeuwen, T.; Shim, Y.; Kim, H.J.; Zhao, T.; Basu, S.; Garcia, B.A.; Kaplan, C.D.; Min, J.H.; Murakami, K. Cryo-EM structure of TFIIH/Rad4-Rad23-Rad33 in damaged DNA opening in nucleotide excision repair. Nat. Commun. 2021, 12, 3338. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, G.; Truglio, J.J.; Wang, L.; Li, G.; Lennarz, W.J.; Schindelin, H. Structural and biochemical studies of the C-terminal domain of mouse peptide-N-glycanase identify it as a mannose-binding module. Proc. Natl. Acad. Sci. USA 2006, 103, 17214–17219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Nei, M. Efficiencies of fast algorithms of phylogenetic inference under the criteria of maximum parsimony, minimum evolution, and maximum likelihood when a large number of sequences are used. Mol. Biol. Evol. 2000, 17, 1251–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, Y.; Haucke, V. Membrane shaping by the Bin/amphiphysin/Rvs (BAR) domain protein superfamily. Cell. Mol. Life Sci. 2011, 68, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Mochizuki, N. Structural characteristics of BAR domain superfamily to sculpt the membrane. Semin. Cell. Dev. Biol. 2010, 21, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Michelot, A.; Koskela, E.V.; Tkach, V.; Stamou, D.; Drubin, D.G.; Lappalainen, P. Membrane-sculpting BAR domains generate stable lipid microdomains. Cell Rep. 2013, 4, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Morone, N.; Suetsugu, S. Membrane re-modelling by BAR domain superfamily proteins via molecular and non-molecular factors. Biochem. Soc. Trans. 2018, 46, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tanabe, K.; Hara, I.; Taniguchi, N.; Colavita, A. Dual enzymatic properties of the cytoplasmic peptide:N-glycanase in C. elegans. Biochem. Biophys. Res. Commun. 2007, 358, 837–841. [Google Scholar] [CrossRef]
- Kato, T.; Kawahara, A.; Ashida, H.; Yamamoto, K. Unique Peptide:N-glycanase of Caenorhabditis elegans has Activity of Protein Disulphide Reductase as well as of Deglycosylation. J. Biochem. 2007, 142, 175–181. [Google Scholar] [CrossRef]
- Chung, C.-Y.; Majewska, N.; Wang, Q.; Paul, J.T.; Betenbaugh, M.J. SnapShot: N-Glycosylation Processing Pathways across Kingdoms. Cell 2017, 171. [Google Scholar] [CrossRef]
- Bai, L.; Wang, T.; Zhao, G.; Kovach, A.; Li, H. The atomic structure of a eukaryotic oligosaccharyltransferase complex. Nature 2018, 555, 328–333. [Google Scholar] [CrossRef]
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.J. Role of N-oligosaccharide endoplasmic reticulum processing reactions in glycoprotein folding and degradation. Biochem. J. 2000, 348, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Caramelo, J.J.; Parodi, A.J. Getting In and Out from Calnexin/Calreticulin Cycles. J. Biol. Chem. 2008, 283, 10221–10225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U. Protein Misfolding Diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Harada, Y.; Hosomi, A.; Masahara-Negishi, Y.; Seino, J.; Fujihira, H.; Funakoshi, Y.; Suzuki, T.; Dohmae, N.; Suzuki, T. Endo-β-N-acetylglucosaminidase forms N -GlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1398–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Phillips, B.P.; Gomez-Navarro, N.; Miller, E.A. Protein quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 2020, 65, 96–102. [Google Scholar] [CrossRef]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a Lectin Required for Glycoprotein Endoplasmic Reticulum-associated Degradation, Recognizes Mannose-trimmed N-Glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.; Zuber, C. Quality control of glycoprotein folding and ERAD: The role of N-glycan handling, EDEM1 and OS-9. Histochem. Cell Biol. 2016, 147, 269–284. [Google Scholar] [CrossRef]
- George, G.; Ninagawa, S.; Yagi, H.; Furukawa, J.-I.; Hashii, N.; Ishii-Watabe, A.; Deng, Y.; Matsushita, K.; Ishikawa, T.; Mamahit, Y.P.; et al. Purified EDEM3 or EDEM1 alone produces determinant oligosaccharide structures from M8B in mammalian glycoprotein ERAD. eLife 2021, 10, e70357. [Google Scholar] [CrossRef]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1–SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinari, M.; Calanca, V.; Galli, C.; Lucca, P.; Paganetti, P. Role of EDEM in the Release of Misfolded Glycoproteins from the Calnexin Cycle. Science 2003, 299, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Cesaratto, F.; Sasset, L.; Myers, M.P.; Re, A.; Petris, G.; Burrone, O.R. BiP/GRP78 Mediates ERAD Targeting of Proteins Produced by Membrane-Bound Ribosomes Stalled at the STOP-Codon. J. Mol. Biol. 2018, 431, 123–141. [Google Scholar] [CrossRef]
- Bernasconi, R.; Galli, C.; Calanca, V.; Nakajima, T.; Molinari, M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol. 2010, 188, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Hosokawa, N.; Wada, I.; Nagata, K. EDEM As an Acceptor of Terminally Misfolded Glycoproteins Released from Calnexin. Science 2003, 299, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, C.; Buchberger, A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 999–1006. [Google Scholar] [CrossRef]
- Jarosch, E.; Taxis, C.; Volkwein, C.; Bordallo, J.; Finley, D.; Wolf, D.H.; Sommer, T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 2002, 4, 134–139. [Google Scholar] [CrossRef]
- Yoshida, Y.; Chiba, T.; Tokunaga, F.; Kawasaki, H.; Iwai, K.; Suzuki, T.; Ito, Y.; Matsuoka, K.; Yoshida, M.; Tanaka, K.; et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002, 418, 438–442. [Google Scholar] [CrossRef]
- Yoshida, Y.; Tokunaga, F.; Chiba, T.; Iwai, K.; Tanaka, K.; Tai, T. Fbs2 Is a New Member of the E3 Ubiquitin Ligase Family That Recognizes Sugar Chains. J. Biol. Chem. 2003, 278, 43877–43884. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Adachi, E.; Fukiya, K.; Iwai, K.; Tanaka, K. Glycoprotein-specific ubiquitin ligases recognize N -glycans in unfolded substrates. EMBO Rep. 2005, 6, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, T.; Yoshida, Y.; Kumanomidou, T.; Hasegawa, Y.; Suzuki, A.; Yamane, T.; Tanaka, K. Structural basis for the selection of glycosylated substrates by SCF Fbs1 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2007, 104, 5777–5781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, Y.; Mizushima, T.; Tanaka, K. Sugar-Recognizing Ubiquitin Ligases: Action Mechanisms and Physiology. Front. Physiol. 2019, 10, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Bhattacharya, A.; Qi, L. Endoplasmic reticulum quality control in cancer: Friend or foe. Semin. Cancer Biol. 2015, 33, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantret, I.; Moore, S.E.H. Free oligosaccharide regulation during mammalian protein N-glycosylation. Glycobiology 2007, 18, 210–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.E. Oligosaccharide transport: Pumping waste from the ER into lysosomes. Trends Cell Biol. 1999, 9, 441–446. [Google Scholar] [CrossRef]
- Suzuki, T.; Funakoshi, Y. Free N-linked oligosaccharide chains: Formation and degradation. Glycoconj. J. 2006, 23, 291–302. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Chantret, I.; Fasseu, M.; Zaoui, K.; Le Bizec, C.; Yayé, H.S.; Dupré, T.; Moore, S.E.H. Identification of Roles for Peptide:N-Glycanase and Endo-β-N-Acetylglucosaminidase (Engase1p) during Protein N-Glycosylation in Human HepG2 Cells. PLoS ONE 2010, 5, e11734. [Google Scholar] [CrossRef] [Green Version]
- Karousis, E.; Mühlemann, O. Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb. Perspect. Biol. 2018, 11, a032862. [Google Scholar] [CrossRef]
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—Mechanisms of ER stress-induced cell death. Biol. Chem. 2013, 395, 1–13. [Google Scholar] [CrossRef]
- Kario, E.; Tirosh, B.; Ploegh, H.L.; Navon, A. N-Linked Glycosylation Does Not Impair Proteasomal Degradation but Affects Class I Major Histocompatibility Complex Presentation. J. Biol. Chem. 2008, 283, 244–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, D.; Hirsch, C.; Stern, P.; Tortorella, M.; Ploegh, H.L. A glycosylated type I membrane protein becomes cytosolic when peptide:N-glycanase is compromised. EMBO J. 2004, 23, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owings, K.G.; Lowry, J.B.; Bi, Y.; Might, M.; Chow, C.Y. Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet. 2018, 27, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Asahina, M.; Fujinawa, R.; Nakamura, S.; Yokoyama, K.; Tozawa, R.; Suzuki, T. Ngly1−/− rats develop neurodegenerative phenotypes and pathological abnormalities in their peripheral and central nervous systems. Hum. Mol. Genet. 2020, 29, 1635–1647. [Google Scholar] [CrossRef] [Green Version]
- Tambe, M.A.; Ng, B.G.; Freeze, H.H. N-Glycanase 1 Transcriptionally Regulates Aquaporins Independent of Its Enzymatic Activity. Cell Rep. 2019, 29, 4620–4631.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, W.F.; Jakob, P.; Sun, H.; Clauder-Münster, S.; Ghidelli-Disse, S.; Ordonez, D.; Boesche, M.; Bantscheff, M.; Collier, P.; Haase, B.; et al. Loss of N-Glycanase 1 Alters Transcriptional and Translational Regulation in K562 Cell Lines. G3 Genes Genomes Genet. 2020, 10, 1585–1597. [Google Scholar] [CrossRef] [Green Version]
- Misaghi, S.; Pacold, M.E.; Blom, D.; Ploegh, H.L.; Korbel, G.A. Using a Small Molecule Inhibitor of Peptide:N-Glycanase to Probe Its Role in Glycoprotein Turnover. Chem. Biol. 2004, 11, 1677–1687. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Han, J.W.; Chan, J.Y. Nuclear Factor Erythroid-2 Like 1 (NFE2L1): Structure, function and regulation. Gene 2016, 584, 17–25. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal Degradation Is Transcriptionally Controlled by TCF11 via an ERAD-Dependent Feedback Loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Lehrbach, N.; Breen, P.C.; Ruvkun, G. Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 2019, 177, 737–750.e15. [Google Scholar] [CrossRef] [Green Version]
- Lehrbach, N.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 2016, 5, e17721. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 2016, 5, e18357. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.; Kolstø, A.-B. Expression of the bZIP transcription factor TCF11 and its potential dimerization partners during development. Mech. Dev. 2000, 97, 141–148. [Google Scholar] [CrossRef]
- Delporte, C.; Bryla, A.; Perret, J. Aquaporins in Salivary Glands: From Basic Research to Clinical Applications. Int. J. Mol. Sci. 2016, 17, 166. [Google Scholar] [CrossRef] [Green Version]
- Zhi, H.; Yuan, W.-T. Expression of aquaporin 3, 4, and 8 in colonic mucosa of rat models with slow transit constipation. Zhonghua Wei Chang Wai Ke Za Zhi = Chin. J. Gastrointest. Surg. 2011, 14, 1124–1130. [Google Scholar]
- Galeone, A.; Han, S.Y.; Huang, C.; Hosomi, A.; Suzuki, T.; Jafar-Nejad, H. Tissue-specific regulation of BMP signaling by Drosophila N-glycanase 1. eLife 2017, 6, e27612. [Google Scholar] [CrossRef] [Green Version]
- Galeone, A.; Adams, J.; Matsuda, S.; Presa, M.F.; Pandey, A.; Han, S.Y.; Tachida, Y.; Hirayama, H.; Vaccari, T.; Suzuki, T.; et al. Regulation of BMP4/Dpp retrotranslocation and signaling by deglycosylation. eLife 2020, 9, e55596. [Google Scholar] [CrossRef]
- Bakrania, P.; Efthymiou, M.; Klein, J.C.; Salt, A.; Bunyan, D.J.; Wyatt, A.; Ponting, C.P.; Martin, A.; Williams, S.; Lindley, V.; et al. Mutations in BMP4 Cause Eye, Brain, and Digit Developmental Anomalies: Overlap between the BMP4 and Hedgehog Signaling Pathways. Am. J. Hum. Genet. 2008, 82, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Obara-Ishihara, T.; Kuhlman, J.; Niswander, L.; Herzlinger, D. The surface ectoderm is essential for nephric duct formation in intermediate mesoderm. Development 1999, 126, 1103–1108. [Google Scholar] [CrossRef]
- Tirosh-Finkel, L.; Zeisel, A.; Brodt-Ivenshitz, M.; Shamai, A.; Yao, Z.; Seger, R.; Domany, E.; Tzahor, E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development 2010, 137, 2989–3000. [Google Scholar] [CrossRef] [Green Version]
- Reis, L.M.; Tyler, R.C.; Schilter, K.F.; Abdul-Rahman, O.; Innis, J.W.; Kozel, B.A.; Schneider, A.S.; Bardakjian, T.M.; Lose, E.J.; Martin, D.M.; et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Qual. Life Res. 2011, 130, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, J.; Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Oretsky, O.; McCormick, E.M.; He, M.; Argon, Y.; Falk, M.J. Mitochondrial function requires NGLY1. Mitochondrion 2017, 38, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Han, S.Y.; Pandey, A.; Moore, T.; Galeone, A.; Duraine, L.; Cowan, T.M.; Jafar-Nejad, H. A conserved role for AMP-activated protein kinase in NGLY1 deficiency. PLoS Genet. 2020, 16, e1009258. [Google Scholar] [CrossRef] [PubMed]
- Talsness, D.M.; Owings, K.G.; Coelho, E.; Mercenne, G.; Pleinis, J.M.; Partha, R.; A Hope, K.; Zuberi, A.R.; Clark, N.L.; Lutz, C.M.; et al. A Drosophila screen identifies NKCC1 as a modifier of NGLY1 deficiency. eLife 2020, 9, e27831. [Google Scholar] [CrossRef]
- Rusan, Z.M.; Kingsford, O.A.; Tanouye, M.A. Modeling Glial Contributions to Seizures and Epileptogenesis: Cation-Chloride Cotransporters in Drosophila melanogaster. PLoS ONE 2014, 9, e101117. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Almutairi, M.M.; Pacheco-Andrade, R.; Almiahuob, M.Y.M.; Di Fulvio, M. Impact of Hybrid and Complex N-Glycans on Cell Surface Targeting of the Endogenous Chloride Cotransporter Slc12a2. Int. J. Cell Biol. 2015, 2015, 505294. [Google Scholar] [CrossRef] [Green Version]
- Delpire, E.; Gagnon, K.B. Na+-K+-2Cl− Cotransporter (NKCC) Physiological Function in Nonpolarized Cells and Transporting Epithelia. Compr. Physiol. 2018, 8, 871–901. [Google Scholar] [CrossRef]
- Abuduxikuer, K.; Zou, L.; Wang, L.; Chen, L.; Wang, J.-S. Novel NGLY1 gene variants in Chinese children with global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty. J. Hum. Genet. 2020, 65, 387–396. [Google Scholar] [CrossRef]
- Kariminejad, A.; Shakiba, M.; Shams, M.; Namiranian, P.; Eghbali, M.; Talebi, S.; Makvand, M.; Jaeken, J.; Najmabadi, H.; Hennekam, R.C. NGLY1 deficiency: Novel variants and literature review. Eur. J. Med Genet. 2021, 64, 104146. [Google Scholar] [CrossRef]
- Pandey, A.; Jafar-Nejad, H. Tracing the NGLY1 footprints: Insights from Drosophila. J. Biochem. 2021, 171, 153–160. [Google Scholar] [CrossRef]
- He, P.; E Grotzke, J.; Ng, B.G.; Gunel, M.; Jafar-Nejad, H.; Cresswell, P.; Enns, G.M.; Freeze, H.H. A congenital disorder of deglycosylation: Biochemical characterization of N-glycanase 1 deficiency in patient fibroblasts. Glycobiology 2015, 25, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.; Bogdańska, A.; Różdżyńska-Świątkowska, A.; Wierzbicka-Rucińska, A.; Tylki-Szymańska, A. NGLY1 deficiency: Novel patient, review of the literature and diagnostic algorithm. JIMD Rep. 2020, 51, 82–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; A Kwofie, M.; Lennarz, W.J. Ngly1, a mouse gene encoding a deglycosylating enzyme implicated in proteasomal degradation: Expression, genomic organization, and chromosomal mapping. Biochem. Biophys. Res. Commun. 2003, 304, 326–332. [Google Scholar] [CrossRef]
- Asahina, M.; Fujinawa, R.; Hirayama, H.; Tozawa, R.; Kajii, Y.; Suzuki, T. Reversibility of motor dysfunction in the rat model of NGLY1 deficiency. Mol. Brain 2021, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Fujihira, H.; Masahara-Negishi, Y.; Tamura, M.; Huang, C.; Harada, Y.; Wakana, S.; Takakura, D.; Kawasaki, N.; Taniguchi, N.; Kondoh, G.; et al. Lethality of mice bearing a knockout of the Ngly1-gene is partially rescued by the additional deletion of the Engase gene. PLoS Genet. 2017, 13, e1006696. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Might, M.; Vankayalapati, H.; Kuberan, B. Repurposing of Proton Pump Inhibitors as first identified small molecule inhibitors of endo -β- N -acetylglucosaminidase (ENGase) for the treatment of NGLY1 deficiency, a rare genetic disease. Bioorganic Med. Chem. Lett. 2017, 27, 2962–2966. [Google Scholar] [CrossRef]
- Yoshida, Y.; Asahina, M.; Murakami, A.; Kawawaki, J.; Yoshida, M.; Fujinawa, R.; Iwai, K.; Tozawa, R.; Matsuda, N.; Tanaka, K.; et al. Loss of peptide:N-glycanase causes proteasome dysfunction mediated by a sugar-recognizing ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2021, 118, e2102922118. [Google Scholar] [CrossRef]
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 2018, 215, 2600–2616. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [Green Version]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A ΔΨm Independent Pharmacological Regulator of Mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, X.; Wu, J.; Chen, H.; Lu, G. Comprehensive Analysis of the Structure and Function of Peptide:N-Glycanase 1 and Relationship with Congenital Disorder of Deglycosylation. Nutrients 2022, 14, 1690. https://doi.org/10.3390/nu14091690
Miao X, Wu J, Chen H, Lu G. Comprehensive Analysis of the Structure and Function of Peptide:N-Glycanase 1 and Relationship with Congenital Disorder of Deglycosylation. Nutrients. 2022; 14(9):1690. https://doi.org/10.3390/nu14091690
Chicago/Turabian StyleMiao, Xiangguang, Jin Wu, Hongping Chen, and Guanting Lu. 2022. "Comprehensive Analysis of the Structure and Function of Peptide:N-Glycanase 1 and Relationship with Congenital Disorder of Deglycosylation" Nutrients 14, no. 9: 1690. https://doi.org/10.3390/nu14091690
APA StyleMiao, X., Wu, J., Chen, H., & Lu, G. (2022). Comprehensive Analysis of the Structure and Function of Peptide:N-Glycanase 1 and Relationship with Congenital Disorder of Deglycosylation. Nutrients, 14(9), 1690. https://doi.org/10.3390/nu14091690