
Dihydromyricetin Protects Intestinal Barrier Integrity by Promoting IL-22 Expression in ILC3s through the AMPK/SIRT3/STAT3 Signaling Pathway

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. FITC-Dextran Permeability Assays
2.3. Intestinal Lamina Propria Immune Cells Isolation
2.4. Cell Culture and Treatments
2.5. Flow Cytometry
2.6. Histological Assessment
2.7. Immunoblotting
2.8. Quantitative Real-Time PCR
2.9. Statistical Analysis
3. Results
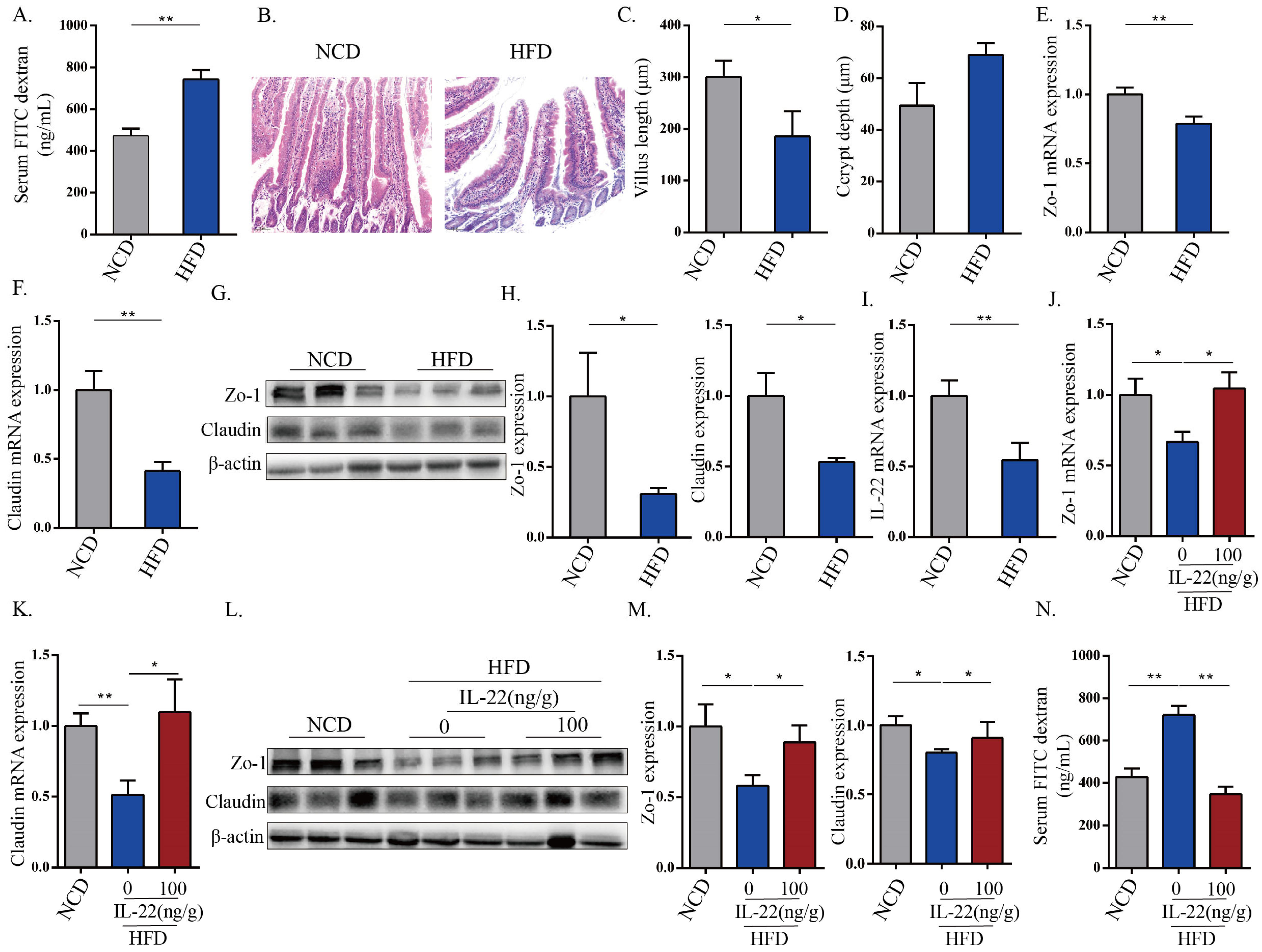
3.1. HFD-Induced Intestinal Epithelial Barrier Destruction Was Associated with the Down-Expression of IL-22
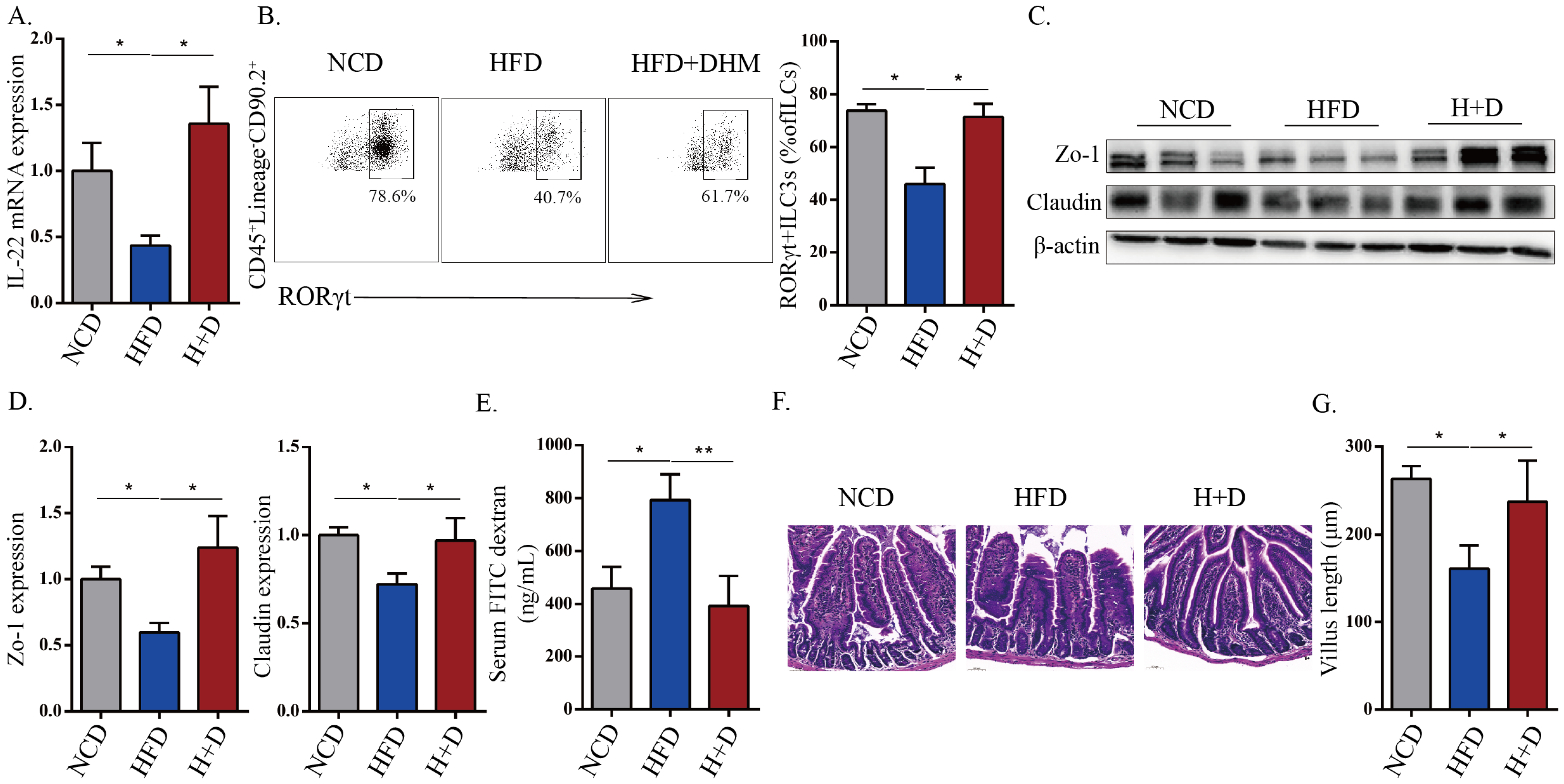
3.2. DHM Protected against HFD-Induced Intestinal Barrier Destruction That Was Associated with Increased IL-22 and ILC3 Frequency
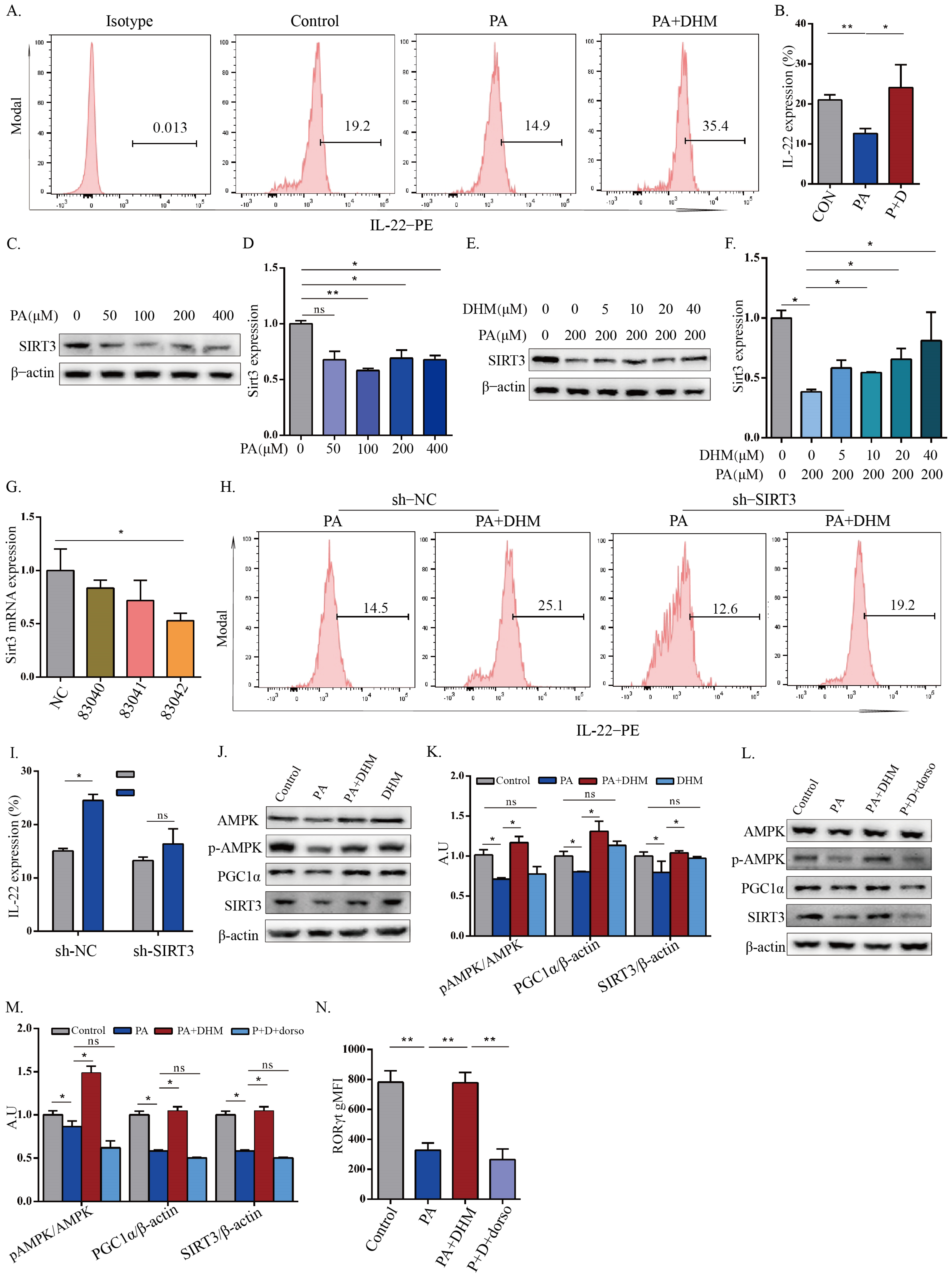
3.3. DHM Promoted IL-22 Expression in MNK3 Cells through SIRT3 via AMPK/PGC1α Signaling
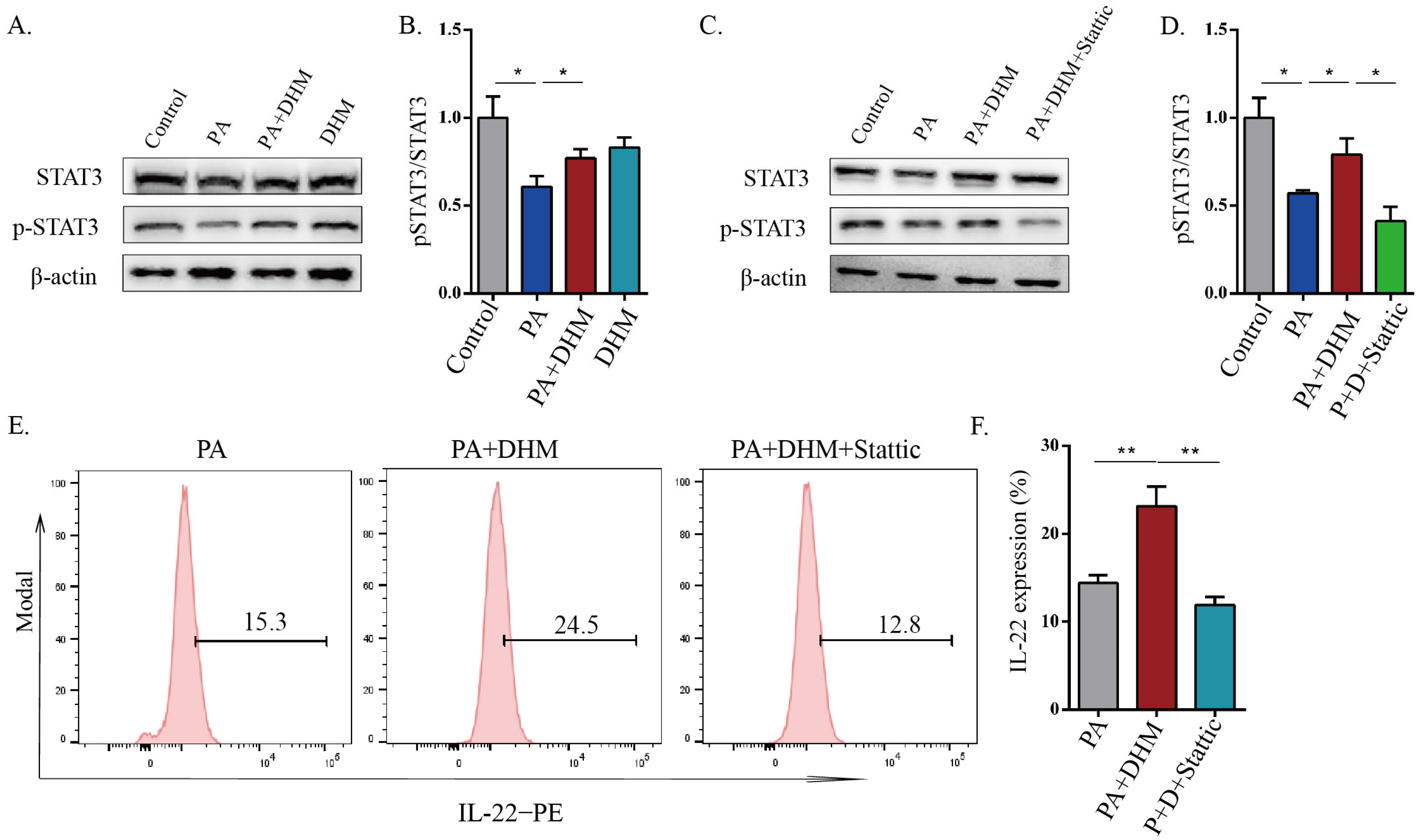
3.4. DHM Promoted IL-22 Expression in MNK3 Cells via STAT3 Phosphorylation
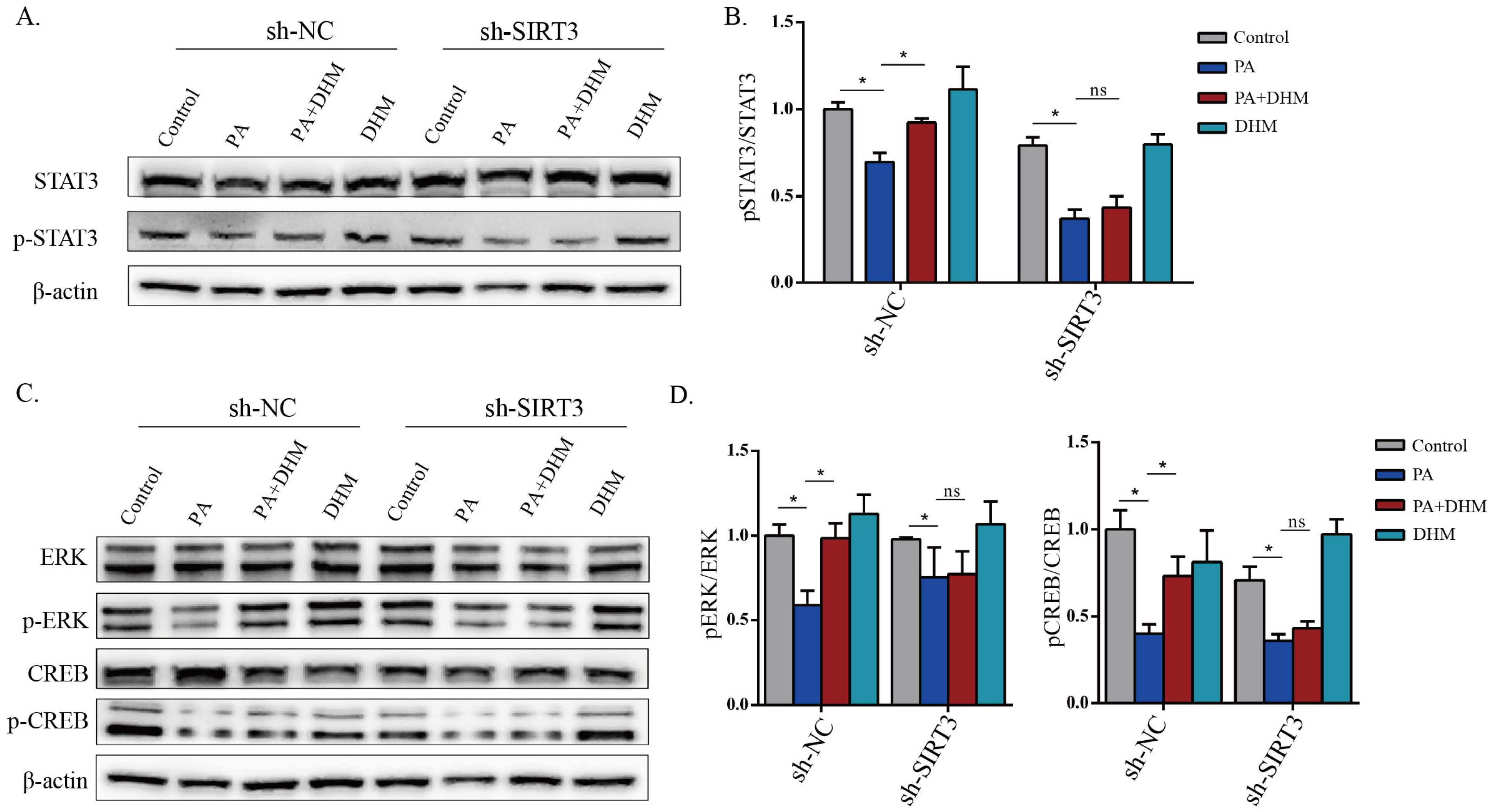
3.5. DHM Stimulated STAT3 Activation through SIRT3 in MNK3 Cells via ERK-CREB Signaling
4. Discussion
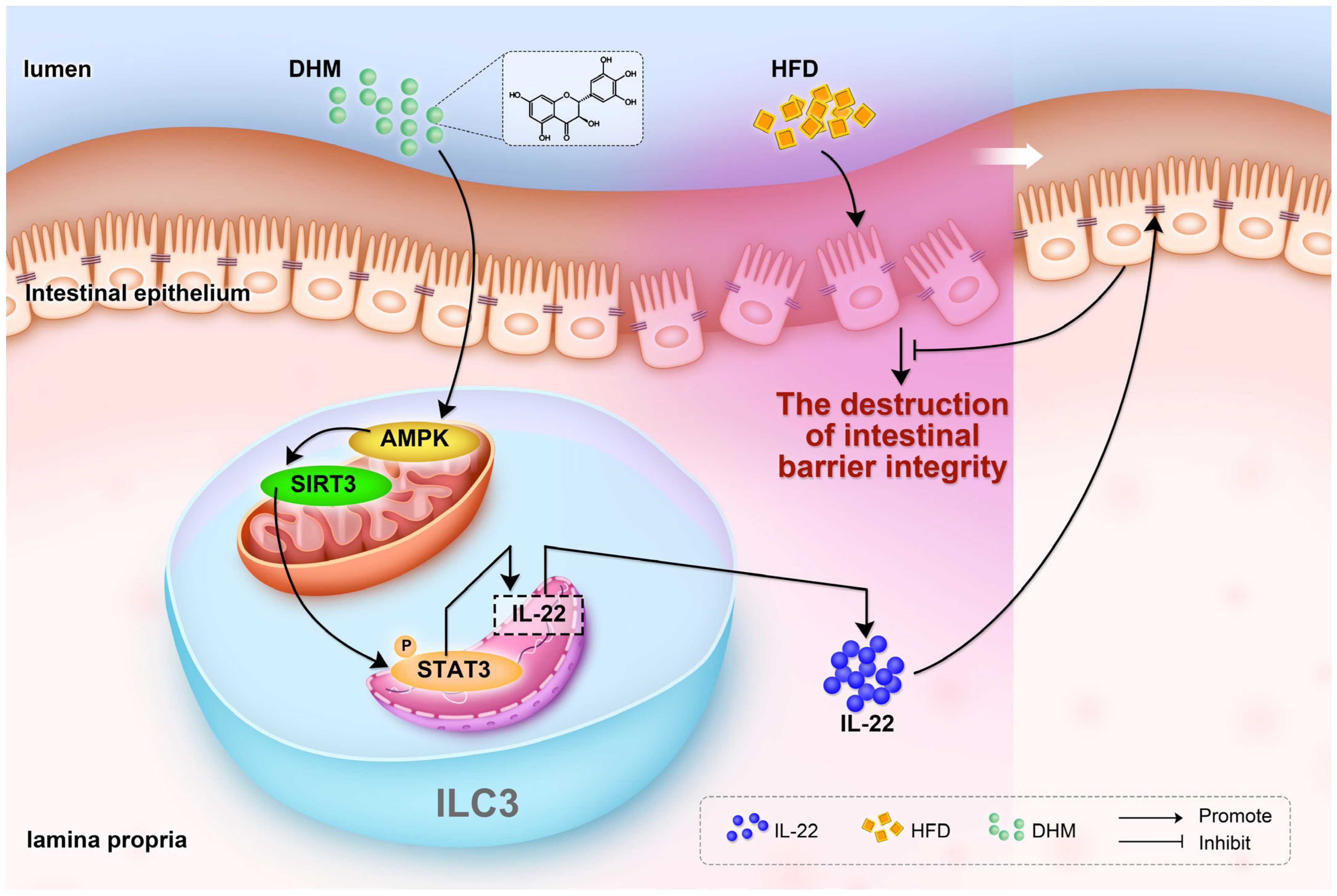
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.; Yi, Y.; Lu, T.; Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020, 217, e20192195. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Cheng, S.; Li, Y.; Wen, Z.; Ma, X.; Jiang, X.; Wang, Y.; Han, X. Faecal Microbiota Transplantation Reduces Susceptibility to Epithelial Injury and Modulates Tryptophan Metabolism of the Microbial Community in a Piglet Model. J. Crohn’s Colitis 2018, 12, 1359–1374. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Qiu, J.; Tu, T.; Yang, X.; Deng, L.; Anders, R.A.; Zhou, L.; Fu, Y.X. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 2014, 40, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Geremia, A.; Arancibia-Carcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Zhang, X.; Tan, L.; Jin, R.; Huang, S.; Li, X. Multitarget and promising role of dihydromyricetin in the treatment of metabolic diseases. Eur. J. Pharmacol. 2020, 870, 172888. [Google Scholar] [CrossRef]
- Dong, S.; Zhu, M.; Wang, K.; Zhao, X.; Hu, L.; Jing, W.; Lu, H.; Wang, S. Dihydromyricetin improves DSS-induced colitis in mice via modulation of fecal-bacteria-related bile acid metabolism. Pharmacol. Res. 2021, 171, 105767. [Google Scholar] [CrossRef]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q.; et al. Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxid. Redox Signal. 2019, 30, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallows, W.C.; Yu, W.; Smith, B.C.; Devries, M.K.; Ellinger, J.J.; Someya, S.; Shortreed, M.R.; Prolla, T.; Markley, J.L.; Smith, L.M.; et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 2011, 41, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Luccia, B.; Gilfillan, S.; Cella, M.; Colonna, M.; Huang, S.C. ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J. Exp. Med. 2019, 216, 2231–2241. [Google Scholar] [CrossRef]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Wang, B.; Kaliannan, K.; Wang, X.; Lang, H.; Hui, S.; Huang, L.; Zhang, Y.; Zhou, M.; Chen, M.; et al. Gut Microbiota Mediates the Protective Effects of Dietary Capsaicin against Chronic Low-Grade Inflammation and Associated Obesity Induced by High-Fat Diet. MBio 2017, 8, e00470-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Hou, P.; Yao, Y.; Yue, J.; Zhang, Q.; Yi, L.; Mi, M. Dihydromyricetin Improves High-Fat Diet-Induced Hyperglycemia through ILC3 Activation via a SIRT3-Dependent Mechanism. Mol. Nutr. Food Res. 2022, 66, e2101093. [Google Scholar] [CrossRef]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Longman, R.S.; Diehl, G.E.; Victorio, D.A.; Huh, J.R.; Galan, C.; Miraldi, E.R.; Swaminath, A.; Bonneau, R.; Scherl, E.J.; Littman, D.R. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 2014, 211, 1571–1583. [Google Scholar] [CrossRef]
- Fachi, J.L.; Pral, L.P.; Dos, S.J.; Codo, A.C.; de Oliveira, S.; Felipe, J.S.; Zambom, F.; Camara, N.; Vieira, P.; Colonna, M.; et al. Hypoxia enhances ILC3 responses through HIF-1alpha-dependent mechanism. Mucosal Immunol. 2021, 14, 828–841. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutz, S.; Wang, X.; Ouyang, W. The IL-20 subfamily of cytokines—From host defence to tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Ibiza, S.; Garcia-Cassani, B.; Ribeiro, H.; Carvalho, T.; Almeida, L.; Marques, R.; Misic, A.M.; Bartow-McKenney, C.; Larson, D.M.; Pavan, W.J.; et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 2016, 535, 440–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, E.; Lavoie, S.; Fonseca-Pereira, D.; Bae, S.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Gallini, C.C.; Glickman, J.N.; Fuller, M.H.; et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity 2019, 51, 871–884. [Google Scholar] [CrossRef]
- Martel, J.; Chang, S.H.; Ko, Y.F.; Hwang, T.L.; Young, J.D.; Ojcius, D.M. Gut barrier disruption and chronic disease. Trends Endocrinol. Metab. 2022, 33, 247–265. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Robles, H.; Castro-Ochoa, K.F.; Citalan-Madrid, A.F.; Schnoor, M. Beneficial effects of nutritional supplements on intestinal epithelial barrier functions in experimental colitis models in vivo. World J. Gastroenterol. 2019, 25, 4181–4198. [Google Scholar] [CrossRef]
- Suzuki, T.; Hara, H. Role of flavonoids in intestinal tight junction regulation. J. Nutr. Biochem. 2011, 22, 401–408. [Google Scholar] [CrossRef]
- Gil-Cardoso, K.; Gines, I.; Pinent, M.; Ardevol, A.; Blay, M.; Terra, X. Effects of flavonoids on intestinal inflammation, barrier integrity and changes in gut microbiota during diet-induced obesity. Nutr. Res. Rev. 2016, 29, 234–248. [Google Scholar] [CrossRef]
- Oteiza, P.I.; Fraga, C.G.; Mills, D.A.; Taft, D.H. Flavonoids and the gastrointestinal tract: Local and systemic effects. Mol. Asp. Med. 2018, 61, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Yuan, L.; Liu, J.; Muhammad, I.; Cao, C.; Shi, C.; Zhang, Y.; Li, R.; Li, C.; Liu, F. Dihydromyricetin attenuates Escherichia coli lipopolysaccharide-induced ileum injury in chickens by inhibiting NLRP3 inflammasome and TLR4/NF-kappaB signalling pathway. Vet. Res. 2020, 51, 72. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio-Domingo, P.; Romera-Hernandez, M.; Karrich, J.J.; Cornelissen, F.; Papazian, N.; Lindenbergh-Kortleve, D.J.; Butler, J.A.; Boon, L.; Coles, M.C.; Samsom, J.N.; et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J. Exp. Med. 2015, 212, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Sonnenberg, G.F.; Monticelli, L.A.; Elloso, M.M.; Fouser, L.A.; Artis, D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 2011, 34, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Gnafakis, S.; Shomrat, O. Innate Lymphoid Cell-Epithelial Cell Modules Sustain Intestinal Homeostasis. Immunity 2020, 52, 452–463. [Google Scholar] [CrossRef]
- Neill, D.R.; Flynn, R.J. Origins and evolution of innate lymphoid cells: Wardens of barrier immunity. Parasite Immunol. 2018, 40, e12436. [Google Scholar] [CrossRef]
- Robinette, M.L.; Fuchs, A.; Cortez, V.S.; Lee, J.S.; Wang, Y.; Durum, S.K.; Gilfillan, S.; Colonna, M. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 2015, 16, 306–317. [Google Scholar] [CrossRef]
- Domingues, R.G.; Hepworth, M.R. Immunoregulatory Sensory Circuits in Group 3 Innate Lymphoid Cell (ILC3) Function and Tissue Homeostasis. Front. Immunol. 2020, 11, 116. [Google Scholar] [CrossRef]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Goverse, G.; Labao-Almeida, C.; Ferreira, M.; Molenaar, R.; Wahlen, S.; Konijn, T.; Koning, J.; Veiga-Fernandes, H.; Mebius, R.E. Vitamin A Controls the Presence of RORgamma+ Innate Lymphoid Cells and Lymphoid Tissue in the Small Intestine. J. Immunol. 2016, 196, 5148–5155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepahi, A.; Liu, Q.; Friesen, L.; Kim, C.H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 2021, 14, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Bostikova, Z.; Moserova, M.; Pavek, P.; Stiborova, M.; Hodek, P. Role of dihydromyricetin in cytochrome P450-mediated metabolism and carcinogen activation. Neuro Enocrinology Lett. 2015, 36 (Suppl. 1), 46–52. [Google Scholar]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell. 2011, 44, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Hui, S.; Lang, H.; Zhou, M.; Zhang, Y.; Kang, C.; Zeng, X.; Zhang, Q.; Yi, L.; Mi, M. SIRT3 Deficiency Promotes High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease in Correlation with Impaired Intestinal Permeability through Gut Microbial Dysbiosis. Mol. Nutr. Food Res. 2019, 63, e1800612. [Google Scholar] [CrossRef]
- Sun, W.; Liu, C.; Chen, Q.; Liu, N.; Yan, Y.; Liu, B. SIRT3: A New Regulator of Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 7293861. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Shangguan, X.; Zhou, W.; Cao, Y.; Zheng, Q.; Tu, J.; Hu, G.; Liang, Z.; Jiang, C.; Deng, L.; et al. Glucose limitation activates AMPK coupled SENP1-Sirt3 signalling in mitochondria for T cell memory development. Nat. Commun. 2021, 12, 4371. [Google Scholar] [CrossRef]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward Primers (5′→3′) | Reverse Primers (5′→3′) | |
|---|---|---|
| zo-1 | AGGACACCAAAGCATGTGAG | GGCATTCCTGCTGGTTACA |
| occludin | TTGAAAGTCCACCTCCTTACAGA | CCGGATAAAAAGAGTACGCTGG |
| sirt3 | CGCTAAACTTCTCCCGGGTT | ACACTAGTCCTCGCCAAACG |
| Il22 | CATGCAGGAGGTGGTACCTT | CAGACGCAAGCATTTCTCAG |
| 18sRNA | ACGGACCAGAGCGAAAGCAT | TGTCAATCCTGTCCGTGTCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Yue, J.; Yao, Y.; Hou, P.; Zhang, T.; Zhang, Q.; Yi, L.; Mi, M. Dihydromyricetin Protects Intestinal Barrier Integrity by Promoting IL-22 Expression in ILC3s through the AMPK/SIRT3/STAT3 Signaling Pathway. Nutrients 2023, 15, 355. https://doi.org/10.3390/nu15020355
Zhou J, Yue J, Yao Y, Hou P, Zhang T, Zhang Q, Yi L, Mi M. Dihydromyricetin Protects Intestinal Barrier Integrity by Promoting IL-22 Expression in ILC3s through the AMPK/SIRT3/STAT3 Signaling Pathway. Nutrients. 2023; 15(2):355. https://doi.org/10.3390/nu15020355
Chicago/Turabian StyleZhou, Jie, Jing Yue, Yu Yao, Pengfei Hou, Ting Zhang, Qianyong Zhang, Long Yi, and Mantian Mi. 2023. "Dihydromyricetin Protects Intestinal Barrier Integrity by Promoting IL-22 Expression in ILC3s through the AMPK/SIRT3/STAT3 Signaling Pathway" Nutrients 15, no. 2: 355. https://doi.org/10.3390/nu15020355
APA StyleZhou, J., Yue, J., Yao, Y., Hou, P., Zhang, T., Zhang, Q., Yi, L., & Mi, M. (2023). Dihydromyricetin Protects Intestinal Barrier Integrity by Promoting IL-22 Expression in ILC3s through the AMPK/SIRT3/STAT3 Signaling Pathway. Nutrients, 15(2), 355. https://doi.org/10.3390/nu15020355