
New Potentiality of Bioactive Substances: Regulating the NLRP3 Inflammasome in Autoimmune Diseases


Abstract
:1. Introduction
2. NLRP3 Inflammasome
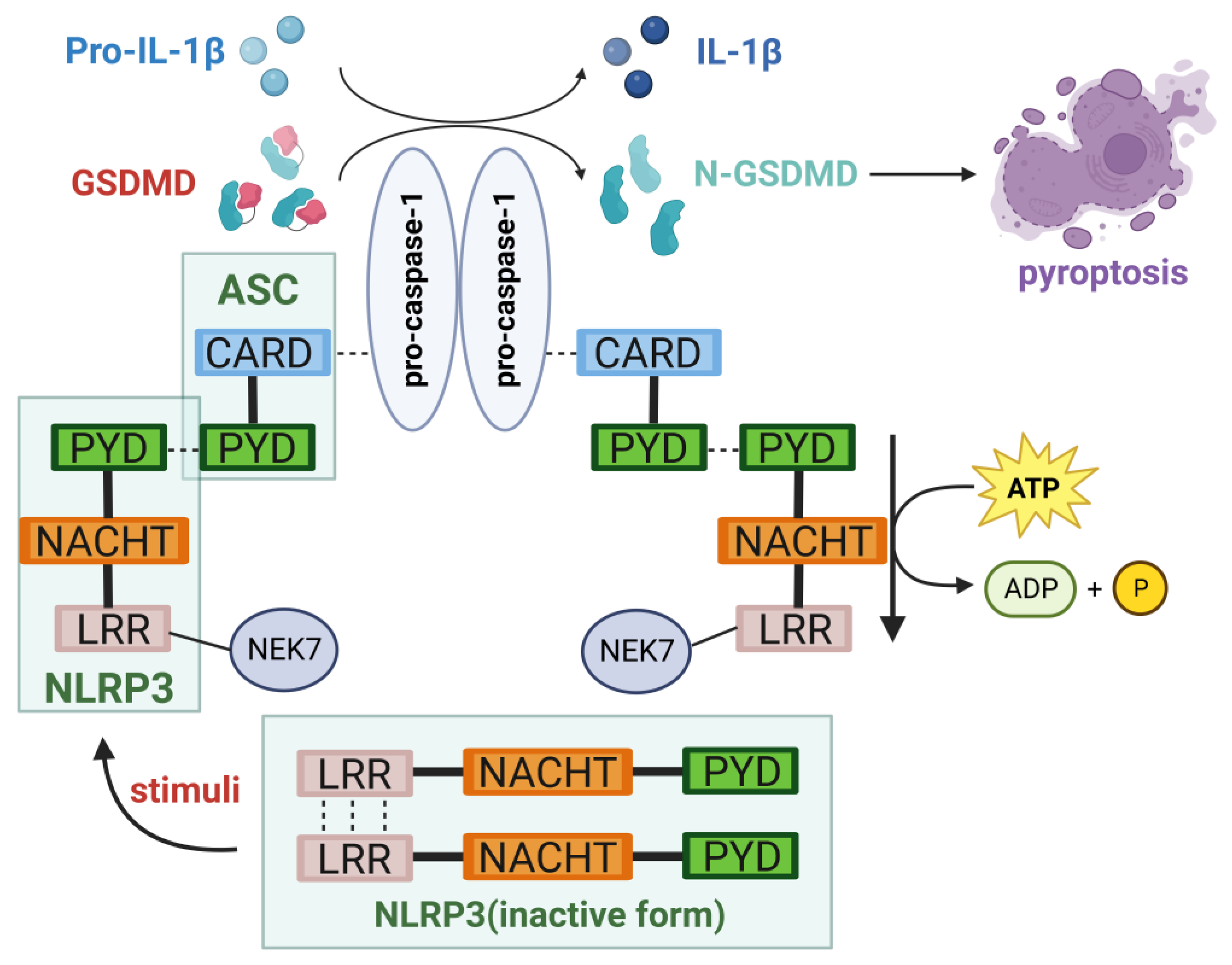
2.1. Structure of the NLRP3 Inflammasome
2.2. Activation of the NLRP3 Inflammasome
2.2.1. Canonical Pathway
2.2.2. Other Pathways
2.3. Inducing Pyroptosis
2.4. Regulation of the NLRP3 Inflammasome
3. The Biological Functions of NLRP3 Inflammasome
4. NLRP3 Inflammasome in Autoimmune Diseases
4.1. Rheumatoid Arthritis
4.2. Systemic Lupus Erythematosus
4.3. Systemic Sclerosis
5. Bioactive Substances Regulating the NLRP3 Inflammasome for the Treatment of Autoimmune Diseases
5.1. Inhibiting the Activation of the NLRP3 Inflammasome
5.2. Regulating the TLR4/NLRP3/NF-KB/GSDMD Signaling Pathway
5.3. Regulating Other Pathways Related to NLRP3
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, F.-S.; Gershwin, M.E. Human Autoimmune Diseases: A Comprehensive Update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Zhan, X.; Li, Q.; Xu, G.; Xiao, X.; Bai, Z. The Mechanism of NLRP3 Inflammasome Activation and Its Pharmacological Inhibitors. Front. Immunol. 2023, 13, 1109938. [Google Scholar] [CrossRef]
- de Alba, E. Structure and Interdomain Dynamics of Apoptosis-Associated Speck-like Protein Containing a CARD (ASC). J. Biol. Chem. 2009, 284, 32932–32941. [Google Scholar] [CrossRef]
- Xiao, L.; Magupalli, V.G.; Wu, H. Cryo-EM Structures of the Active NLRP3 Inflammasome Disc. Nature 2023, 613, 595–600. [Google Scholar] [CrossRef]
- Andreeva, L.; David, L.; Rawson, S.; Shen, C.; Pasricha, T.; Pelegrin, P.; Wu, H. NLRP3 Cages Revealed by Full-Length Mouse NLRP3 Structure Control Pathway Activation. Cell 2021, 184, 6299–6312.e22. [Google Scholar] [CrossRef]
- Fu, J.; Wu, H. Structural Mechanisms of NLRP3 Inflammasome Assembly and Activation. Annu. Rev. Immunol. 2023, 41, 301–316. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Yang, J.; Chen, Y.; Zhou, B.; Abbott, D.W.; Xiao, T.S. Caspase-1 Engages Full-Length Gasdermin D through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity 2020, 53, 106–114.e5. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhang, Z.; Magupalli, V.G.; Pablo, J.L.; Dong, Y.; Vora, S.M.; Wang, L.; Fu, T.-M.; Jacobson, M.P.; Greka, A.; et al. Gasdermin D Pore Structure Reveals Preferential Release of Mature Interleukin-1. Nature 2021, 593, 607–611. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Sharma, M.; de Alba, E. Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes. Int. J. Mol. Sci. 2021, 22, 872. [Google Scholar] [CrossRef]
- Yücel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.-H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides Induced Inflammatory Responses and Electrophysiological Dysfunctions in Human-Induced Pluripotent Stem Cell Derived Cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting Edge: TNF-α Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation1. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Gritsenko, A.; Green, J.P.; Brough, D.; Lopez-Castejon, G. Mechanisms of NLRP3 Priming in Inflammaging and Age Related Diseases. Cytokine Growth Factor Rev. 2020, 55, 15–25. [Google Scholar] [CrossRef]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 Inflammasome Is Triggered by Low Intracellular Potassium Concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Walev, I.; Reske, K.; Palmer, M.; Valeva, A.; Bhakdi, S. Potassium-Inhibited Processing of IL-1 Beta in Human Monocytes. EMBO J. 1995, 14, 1607–1614. [Google Scholar] [CrossRef]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.-D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e4. [Google Scholar] [CrossRef]
- Drinkall, S.; Lawrence, C.B.; Ossola, B.; Russell, S.; Bender, C.; Brice, N.B.; Dawson, L.A.; Harte, M.; Brough, D. The Two Pore Potassium Channel THIK-1 Regulates NLRP3 Inflammasome Activation. Glia 2022, 70, 1301–1316. [Google Scholar] [CrossRef]
- Dong, X.; Zheng, Z.; Lin, P.; Fu, X.; Li, F.; Jiang, J.; Zhu, P. ACPAs Promote IL-1β Production in Rheumatoid Arthritis by Activating the NLRP3 Inflammasome. Cell Mol. Immunol. 2020, 17, 261–271. [Google Scholar] [CrossRef]
- Zhao, N.; Li, C.; Di, B.; Xu, L. Recent Advances in the NEK7-Licensed NLRP3 Inflammasome Activation: Mechanisms, Role in Diseases and Related Inhibitors. J. Autoimmun. 2020, 113, 102515. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Y.; Wang, D.; Liu, C.; Qi, Z.; Tang, H.; Liu, Y.; Zhang, S.; Cui, Y.; Li, Y.; et al. ALK-JNK Signaling Promotes NLRP3 Inflammasome Activation and Pyroptosis via NEK7 during Streptococcus Pneumoniae Infection. Mol. Immunol. 2023, 157, 78–90. [Google Scholar] [CrossRef]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The Cytolytic P2Z Receptor for Extracellular ATP Identified as a P2X Receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Schorn, C.; Frey, B.; Lauber, K.; Janko, C.; Strysio, M.; Keppeler, H.; Gaipl, U.S.; Voll, R.E.; Springer, E.; Munoz, L.E.; et al. Sodium Overload and Water Influx Activate the NALP3 Inflammasome. J. Biol. Chem. 2011, 286, 35–41. [Google Scholar] [CrossRef]
- Green, J.P.; Yu, S.; Martín-Sánchez, F.; Pelegrin, P.; Lopez-Castejon, G.; Lawrence, C.B.; Brough, D. Chloride Regulates Dynamic NLRP3-Dependent ASC Oligomerization and Inflammasome Priming. Proc. Natl. Acad. Sci. USA 2018, 115, E9371–E9380. [Google Scholar] [CrossRef]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.-A.; Fromm, S.; et al. K+ Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Billingham, L.K.; Stoolman, J.S.; Vasan, K.; Rodriguez, A.E.; Poor, T.A.; Szibor, M.; Jacobs, H.T.; Reczek, C.R.; Rashidi, A.; Zhang, P.; et al. Mitochondrial Electron Transport Chain Is Necessary for NLRP3 Inflammasome Activation. Nat. Immunol. 2022, 23, 692–704. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Wang, L.; Sun, L.; Liu, W.; Li, Q.; Wang, J. IL-6 Promotes Collagen-Induced Arthritis by Activating the NLRP3 Inflammasome through the Cathepsin B/S100A9-Mediated Pathway. Int. Immunopharmacol. 2020, 88, 106985. [Google Scholar] [CrossRef]
- Jäger, E.; Murthy, S.; Schmidt, C.; Hahn, M.; Strobel, S.; Peters, A.; Stäubert, C.; Sungur, P.; Venus, T.; Geisler, M.; et al. Calcium-Sensing Receptor-Mediated NLRP3 Inflammasome Response to Calciprotein Particles Drives Inflammation in Rheumatoid Arthritis. Nat. Commun. 2020, 11, 4243. [Google Scholar] [CrossRef]
- Wu, X.; Ren, G.; Zhou, R.; Ge, J.; Chen, F.-H. The Role of Ca2+ in Acid-Sensing Ion Channel 1a-Mediated Chondrocyte Pyroptosis in Rat Adjuvant Arthritis. Lab. Investig. 2019, 99, 499–513. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.; Huang, Z.; Zhang, N.; Zhang, L.; Xing, R.; Wang, P. Agnuside Alleviates Synovitis and Fibrosis in Knee Osteoarthritis through the Inhibition of HIF-1α and NLRP3 Inflammasome. Mediators Inflamm. 2021, 2021, e5534614. [Google Scholar] [CrossRef]
- Guo, X.; Chen, G. Hypoxia-Inducible Factor Is Critical for Pathogenesis and Regulation of Immune Cell Functions in Rheumatoid Arthritis. Front. Immunol. 2020, 11, 1668. [Google Scholar] [CrossRef]
- Shin, M.S.; Kang, Y.; Lee, N.; Wahl, E.R.; Kim, S.H.; Kang, K.S.; Lazova, R.; Kang, I. Self Double-Stranded (Ds)DNA Induces IL-1β Production from Human Monocytes by Activating NLRP3 Inflammasome in the Presence of Anti-dsDNA Antibodies. J. Immunol. Baltim. Md 1950 2013, 190, 1407–1415. [Google Scholar] [CrossRef]
- Bierschenk, D.; Papac-Milicevic, N.; Bresch, I.P.; Kovacic, V.; Bettoni, S.; Dziedzic, M.; Wetsel, R.A.; Eschenburg, S.; Binder, C.J.; Blom, A.M.; et al. C4b-Binding Protein Inhibits Particulate- and Crystalline-Induced NLRP3 Inflammasome Activation. Front. Immunol. 2023, 14, 1149822. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. PtdIns4P on Dispersed Trans-Golgi Network Mediates NLRP3 Inflammasome Activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Nanda, S.K.; Prescott, A.R.; Figueras-Vadillo, C.; Cohen, P. IKKβ Is Required for the Formation of the NLRP3 Inflammasome. EMBO Rep. 2021, 22, e50743. [Google Scholar] [CrossRef]
- Lee, B.; Hoyle, C.; Green, J.P.; Wellens, R.; Martin-Sanchez, F.; Williams, D.; Seoane, P.I.; Bennett, H.; Adamson, A.; Lopez-Castejon, G.; et al. NLRP3 Activation in Response to Disrupted Endocytic Traffic. bioRxiv 2021, 2021-09. [Google Scholar] [CrossRef]
- Zhu, F.; Ma, J.; Li, W.; Liu, Q.; Qin, X.; Qian, Y.; Wang, C.; Zhang, Y.; Li, Y.; Jiang, D.; et al. The Orphan Receptor Nur77 Binds Cytoplasmic LPS to Activate the Non-Canonical NLRP3 Inflammasome. Immunity 2023, 56, 753–767.e8. [Google Scholar] [CrossRef]
- Moretti, J.; Jia, B.; Hutchins, Z.; Roy, S.; Yip, H.; Wu, J.; Shan, M.; Jaffrey, S.R.; Coers, J.; Blander, J.M. Caspase-11 Interaction with NLRP3 Potentiates the Noncanonical Activation of the NLRP3 Inflammasome. Nat. Immunol. 2022, 23, 705–717. [Google Scholar] [CrossRef]
- Matikainen, S.; Nyman, T.A.; Cypryk, W. Function and Regulation of Noncanonical Caspase-4/5/11 Inflammasome. J. Immunol. Baltim. Md 1950 2020, 204, 3063–3069. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Hagar, J.; Powell, D.; Aachoui, Y.; Ernst, R.; Miao, E. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.B.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 Inflammasome Activation and Cell Death. Cell Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Biby, S.; Kaur, B.; Zhang, S. A Patent Review of NLRP3 Inhibitors to Treat Autoimmune Diseases. Expert Opin. Ther. Pat. 2023, 32, 401–421. [Google Scholar] [CrossRef]
- Stutz, A.; Kolbe, C.-C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 Inflammasome Assembly Is Regulated by Phosphorylation of the Pyrin Domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, H.; Hao, Y.; Lin, H.; Dong, M.; Ye, J.; Song, L.; Wang, Y.; Li, Q.; Shan, B.; et al. Myeloid PTEN Promotes Chemotherapy-Induced NLRP3-Inflammasome Activation and Antitumour Immunity. Nat. Cell Biol. 2020, 22, 716–727. [Google Scholar] [CrossRef]
- Niu, T.; De Rosny, C.; Chautard, S.; Rey, A.; Patoli, D.; Groslambert, M.; Cosson, C.; Lagrange, B.; Zhang, Z.; Visvikis, O.; et al. NLRP3 Phosphorylation in Its LRR Domain Critically Regulates Inflammasome Assembly. Nat. Commun. 2021, 12, 5862. [Google Scholar] [CrossRef]
- Zhang, A.; Xing, J.; Xia, T.; Zhang, H.; Fang, M.; Li, S.; Du, Y.; Li, X.C.; Zhang, Z.; Zeng, M.-S. EphA2 Phosphorylates NLRP3 and Inhibits Inflammasomes in Airway Epithelial Cells. EMBO Rep. 2020, 21, e49666. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, C.-S.; Harrison, K.; Hwang, I.-Y.; Nabar, N.R.; Wang, M.; Kehrl, J.H. AKT Regulates NLRP3 Inflammasome Activation by Phosphorylating NLRP3 Serine 5. J. Immunol. Baltim. Md 1950 2020, 205, 2255–2264. [Google Scholar] [CrossRef]
- Taatjes, D.J. The Continuing SAGA of TFIID and RNA Polymerase II Transcription. Mol. Cell 2017, 68, 1–2. [Google Scholar] [CrossRef]
- Bittner, Z.A.; Liu, X.; Mateo Tortola, M.; Tapia-Abellán, A.; Shankar, S.; Andreeva, L.; Mangan, M.; Spalinger, M.; Kalbacher, H.; Düwell, P.; et al. BTK Operates a Phospho-Tyrosine Switch to Regulate NLRP3 Inflammasome Activity. J. Exp. Med. 2021, 218, e20201656. [Google Scholar] [CrossRef]
- Dufies, O.; Doye, A.; Courjon, J.; Torre, C.; Michel, G.; Loubatier, C.; Jacquel, A.; Chaintreuil, P.; Majoor, A.; Guinamard, R.R.; et al. Escherichia Coli Rho GTPase-Activating Toxin CNF1 Mediates NLRP3 Inflammasome Activation via P21-Activated Kinases-1/2 during Bacteraemia in Mice. Nat. Microbiol. 2021, 6, 401–412. [Google Scholar] [CrossRef]
- Zhang, Z.; Meszaros, G.; He, W.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gámez, M.; Goginashvili, A.; et al. Protein Kinase D at the Golgi Controls NLRP3 Inflammasome Activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Palazón-Riquelme, P.; Worboys, J.D.; Green, J.; Valera, A.; Martín-Sánchez, F.; Pellegrini, C.; Brough, D.; López-Castejón, G. USP7 and USP47 Deubiquitinases Regulate NLRP3 Inflammasome Activation. EMBO Rep. 2018, 19, e44766. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, L.; Xu, T.; Guo, X.; Wang, C.; Li, Y.; Yang, Y.; Yang, D.; Sun, B.; Zhao, X.; et al. HUWE1 Mediates Inflammasome Activation and Promotes Host Defense against Bacterial Infection. J. Clin. Investig. 2020, 130, 6301–6316. [Google Scholar] [CrossRef]
- Ni, J.; Guan, C.; Liu, H.; Huang, X.; Yue, J.; Xiang, H.; Jiang, Z.; Tao, Y.; Cao, W.; Liu, J.; et al. Ubc13 Promotes K63-Linked Polyubiquitination of NLRP3 to Activate Inflammasome. J. Immunol. Baltim. Md 1950 2021, 206, 2376–2385. [Google Scholar] [CrossRef]
- Kawashima, A.; Karasawa, T.; Tago, K.; Kimura, H.; Kamata, R.; Usui-Kawanishi, F.; Watanabe, S.; Ohta, S.; Funakoshi-Tago, M.; Yanagisawa, K.; et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J. Immunol. 2017, 199, 3614–3622. [Google Scholar] [CrossRef]
- Xu, T.; Yu, W.; Fang, H.; Wang, Z.; Chi, Z.; Guo, X.; Jiang, D.; Zhang, K.; Chen, S.; Li, M.; et al. Ubiquitination of NLRP3 by Gp78/Insig-1 Restrains NLRP3 Inflammasome Activation. Cell Death Differ. 2022, 29, 1582–1595. [Google Scholar] [CrossRef]
- Tang, J.; Tu, S.; Lin, G.; Guo, H.; Yan, C.; Liu, Q.; Huang, L.; Tang, N.; Xiao, Y.; Pope, R.M.; et al. Sequential Ubiquitination of NLRP3 by RNF125 and Cbl-b Limits Inflammasome Activation and Endotoxemia. J. Exp. Med. 2020, 217, e20182091. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Xu, X.; Wu, J.; Peng, Y.; Li, J.; Luo, R.; Huang, L.; Liu, L.; Yu, S.; et al. YAP Promotes the Activation of NLRP3 Inflammasome via Blocking K27-Linked Polyubiquitination of NLRP3. Nat. Commun. 2021, 12, 2674. [Google Scholar] [CrossRef]
- Wan, P.; Zhang, Q.; Liu, W.; Jia, Y.; Ai, S.; Wang, T.; Wang, W.; Pan, P.; Yang, G.; Xiang, Q.; et al. Cullin1 Binds and Promotes NLRP3 Ubiquitination to Repress Systematic Inflammasome Activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 5793–5807. [Google Scholar] [CrossRef] [PubMed]
- Michelini, S.; Sarajlic, M.; Duschl, A.; Horejs-Hoeck, J. IL-1β Induces Expression of Costimulatory Molecules and Cytokines but Not Immune Feedback Regulators in Dendritic Cells. Hum. Immunol. 2018, 79, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Lalor, S.J.; Dungan, L.S.; Sutton, C.E.; Basdeo, S.A.; Fletcher, J.M.; Mills, K.H.G. Caspase-1–Processed Cytokines IL-1β and IL-18 Promote IL-17 Production by Γδ and CD4 T Cells That Mediate Autoimmunity. J. Immunol. 2011, 186, 5738–5748. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhang, Y.; Han, C.; Hu, X.; Zhang, H.; Xu, X.; Tian, J.; Liu, Y.; Ding, Y.; Liu, J.; et al. Blockade of CD47 Ameliorates Autoimmune Inflammation in CNS by Suppressing IL-1-Triggered Infiltration of Pathogenic Th17 Cells. J. Autoimmun. 2016, 69, 74–85. [Google Scholar] [CrossRef]
- Ben-Sasson, S.Z.; Hogg, A.; Hu-Li, J.; Wingfield, P.; Chen, X.; Crank, M.; Caucheteux, S.; Ratner-Hurevich, M.; Berzofsky, J.A.; Nir-Paz, R.; et al. IL-1 Enhances Expansion, Effector Function, Tissue Localization, and Memory Response of Antigen-Specific CD8 T Cells. J. Exp. Med. 2013, 210, 491–502. [Google Scholar] [CrossRef]
- Galozzi, P.; Bindoli, S.; Doria, A.; Sfriso, P. The Revisited Role of Interleukin-1 Alpha and Beta in Autoimmune and Inflammatory Disorders and in Comorbidities. Autoimmun. Rev. 2021, 20, 102785. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, J.; Tang, Y.; Jin, T.; Tao, J. Inflammasomes Cross-Talk with Lymphocytes to Connect the Innate and Adaptive Immune Response. J. Adv. Res. 2023. [Google Scholar] [CrossRef]
- Puren, A.J.; Fantuzzi, G.; Dinarello, C.A. Gene Expression, Synthesis, and Secretion of Interleukin 18 and Interleukin 1beta Are Differentially Regulated in Human Blood Mononuclear Cells and Mouse Spleen Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2256–2261. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Lee, A.J.; Nham, T.; Lusty, E.; Larché, M.J.; Lee, D.A.; Ashkar, A.A. Combined Stimulation with Interleukin-18 and Interleukin-12 Potently Induces Interleukin-8 Production by Natural Killer Cells. J. Innate Immun. 2017, 9, 511–525. [Google Scholar] [CrossRef]
- Yu, S.; Chen, Z.; Mix, E.; Zhu, S.-W.; Winblad, B.; Ljunggren, H.-G.; Zhu, J. Neutralizing Antibodies to IL-18 Ameliorate Experimental Autoimmune Neuritis by Counter-Regulation of Autoreactive Th1 Responses to Peripheral Myelin Antigen. J. Neuropathol. Exp. Neurol. 2002, 61, 614–622. [Google Scholar] [CrossRef]
- Kanai, T.; Watanabe, M.; Okazawa, A.; Sato, T.; Yamazaki, M.; Okamoto, S.; Ishii, H.; Totsuka, T.; Iiyama, R.; Okamoto, R.; et al. Macrophage-Derived IL-18-Mediated Intestinal Inflammation in the Murine Model of Crohn’s Disease. Gastroenterology 2001, 121, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid Arthritis. Lancet Lond. Engl. 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid Arthritis: Pathological Mechanisms and Modern Pharmacologic Therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Liu, N.; Sigdel, K.R.; Duan, L. Role of NLRP3 Inflammasome in Rheumatoid Arthritis. Front. Immunol. 2022, 13, 931690. [Google Scholar] [CrossRef]
- Miyoshi, M.; Liu, S. Collagen-Induced Arthritis Models. Methods Mol. Biol. Clifton N.J. 2018, 1868, 3–7. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Li, H. NLRP3 Inflammasome Plays an Important Role in the Pathogenesis of Collagen-Induced Arthritis. Mediators Inflamm. 2016, 2016, e9656270. [Google Scholar] [CrossRef]
- Choulaki, C.; Papadaki, G.; Repa, A.; Kampouraki, E.; Kambas, K.; Ritis, K.; Bertsias, G.; Boumpas, D.T.; Sidiropoulos, P. Enhanced Activity of NLRP3 Inflammasome in Peripheral Blood Cells of Patients with Active Rheumatoid Arthritis. Arthritis Res. Ther. 2015, 17, 257. [Google Scholar] [CrossRef]
- Ruscitti, P.; Cipriani, P.; Di Benedetto, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Ciccia, F.; Alvaro, S.; Triolo, G.; Giacomelli, R. Monocytes from Patients with Rheumatoid Arthritis and Type 2 Diabetes Mellitus Display an Increased Production of Interleukin (IL)-1β via the Nucleotide-Binding Domain and Leucine-Rich Repeat Containing Family Pyrin 3(NLRP3)-Inflammasome Activation: A Possible Implication for Therapeutic Decision in These Patients. Clin. Exp. Immunol. 2015, 182, 35–44. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, C.-L.; Wang, S.; Mo, L.-W.; Yang, G.-D.; Hu, J.; Zhang, F. Role of NLRP3 and NLRP1 Inflammasomes Signaling Pathways in Pathogenesis of Rheumatoid Arthritis. Asian Pac. J. Trop. Med. 2014, 7, 827–831. [Google Scholar] [CrossRef]
- Lasithiotaki, I.; Giannarakis, I.; Tsitoura, E.; Samara, K.D.; Margaritopoulos, G.A.; Choulaki, C.; Vasarmidi, E.; Tzanakis, N.; Voloudaki, A.; Sidiropoulos, P.; et al. NLRP3 Inflammasome Expression in Idiopathic Pulmonary Fibrosis and Rheumatoid Lung. Eur. Respir. J. 2016, 47, 910–918. [Google Scholar] [CrossRef]
- Kolly, L.; Busso, N.; Palmer, G.; Talabot-Ayer, D.; Chobaz, V.; So, A. Expression and Function of the NALP3 Inflammasome in Rheumatoid Synovium. Immunology 2010, 129, 178–185. [Google Scholar] [CrossRef]
- Bokarewa, M.; Hultgren, O. Is Interleukin-18 Useful for Monitoring Rheumatoid Arthritis? Scand. J. Rheumatol. 2005, 34, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Sack, U.; Stiehl, P.; Geiler, G. Distribution of Macrophages in Rheumatoid Synovial Membrane and Its Association with Basic Activity. Rheumatol. Int. 1994, 13, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Q.; Cao, G.; Luo, M.; Hou, H.; Yue, C. Pyroptosis by NLRP3/Caspase-1/Gasdermin-D Pathway in Synovial Tissues of Rheumatoid Arthritis Patients. J. Cell Mol. Med. 2023, 27, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Wu, W.; Nan, Y.; Sun, W.; Wang, Y. SMAD2 Inhibits Pyroptosis of Fibroblast-like Synoviocytes and Secretion of Inflammatory Factors via the TGF-β Pathway in Rheumatoid Arthritis. Arthritis Res. Ther. 2023, 25, 144. [Google Scholar] [CrossRef]
- Li, G.; Xiu, L.; Li, X.; Ma, L.; Zhou, J. miR-155 Inhibits Chondrocyte Pyroptosis in Knee Osteoarthritis by Targeting SMAD2 and Inhibiting the NLRP3/Caspase-1 Pathway. J. Orthop. Surg. 2022, 17, 48. [Google Scholar] [CrossRef]
- Li, C.; Yin, W.; Yu, N.; Zhang, D.; Zhao, H.; Liu, J.; Liu, J.; Pan, Y.; Lin, L. miR-155 Promotes Macrophage Pyroptosis Induced by Porphyromonas Gingivalis through Regulating the NLRP3 Inflammasome. ORAL Dis. 2019, 25, 2030–2039. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Alivernini, S.; Ballantine, L.E.; Asquith, D.L.; Millar, N.L.; Gilchrist, D.S.; Reilly, J.; Ierna, M.; Fraser, A.R.; Stolarski, B.; et al. MicroRNA-155 as a Proinflammatory Regulator in Clinical and Experimental Arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 11193–11198. [Google Scholar] [CrossRef]
- Olsson, A.M.; Povoleri, G.A.M.; Somma, D.; Ridley, M.L.; Rizou, T.; Lalnunhlimi, S.; Macdonald, L.; Rajasekhar, M.; Martinez-Nunez, R.T.; Kurowska-Stolarska, M.; et al. miR-155-Overexpressing Monocytes Resemble HLAhighISG15+ Synovial Tissue Macrophages from Patients with Rheumatoid Arthritis and Induce Polyfunctional CD4+ T-Cell Activation. Clin. Exp. Immunol. 2022, 207, 188–198. [Google Scholar] [CrossRef]
- Radstake, T.R.D.J.; Roelofs, M.F.; Jenniskens, Y.M.; Oppers-Walgreen, B.; van Riel, P.L.C.M.; Barrera, P.; Joosten, L.A.B.; van den Berg, W.B. Expression of Toll-like Receptors 2 and 4 in Rheumatoid Synovial Tissue and Regulation by Proinflammatory Cytokines Interleukin-12 and Interleukin-18 via Interferon-γ. Arthritis Rheum. 2004, 50, 3856–3865. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, J.-Y.; Liu, J.-Q.; Yang, J.; Liu, Y.; Wang, C.; Ma, X.-N.; Liu, B.-L.; Xin, G.-Z.; Liu, L.-F. Succinate/NLRP3 Inflammasome Induces Synovial Fibroblast Activation: Therapeutical Effects of Clematichinenoside AR on Arthritis. Front. Immunol. 2016, 7, 532. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, W.; Wang, Y.; Wan, C.; Bai, Y.; Sun, X.; Ma, J.; Zheng, F. TNF-α/Calreticulin Dual Signaling Induced NLRP3 Inflammasome Activation Associated with HuR Nucleocytoplasmic Shuttling in Rheumatoid Arthritis. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. Al 2019, 68, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Feng, W.; Li, C.; Kou, Y.; Li, D.; Liu, S.; Hasegawa, T.; Li, M. LPS Induces Fibroblast-like Synoviocytes RSC-364 Cells to Pyroptosis through NF-κB Mediated Dual Signalling Pathway. J. Mol. Histol. 2021, 52, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Li, X.-F.; Yang, Y.-L.; Song, B.; Wu, S.; Niu, X.-N.; Wu, Y.-Y.; Shi, W.; Huang, C.; Li, J. PLCL1 Regulates Fibroblast-like Synoviocytes Inflammation via NLRP3 Inflammasomes in Rheumatoid Arthritis. Adv. Rheumatol. Lond. Engl. 2022, 62, 25. [Google Scholar] [CrossRef]
- Akhavani, M.A.; Madden, L.; Buysschaert, I.; Sivakumar, B.; Kang, N.; Paleolog, E.M. Hypoxia Upregulates Angiogenesis and Synovial Cell Migration in Rheumatoid Arthritis. Arthritis Res. Ther. 2009, 11, R64. [Google Scholar] [CrossRef]
- Nonomura, Y.; Mizoguchi, F.; Suzuki, A.; Nanki, T.; Kato, H.; Miyasaka, N.; Kohsaka, H. Hypoxia-Induced Abrogation of Contact-Dependent Inhibition of Rheumatoid Arthritis Synovial Fibroblast Proliferation. J. Rheumatol. 2009, 36, 698–705. [Google Scholar] [CrossRef]
- Hong, Z.; Zhang, X.; Zhang, T.; Hu, L.; Liu, R.; Wang, P.; Wang, H.; Yu, Q.; Mei, D.; Xue, Z.; et al. The ROS/GRK2/HIF-1α/NLRP3 Pathway Mediates Pyroptosis of Fibroblast-Like Synoviocytes and the Regulation of Monomer Derivatives of Paeoniflorin. Oxid. Med. Cell Longev. 2022, 2022, 4566851. [Google Scholar] [CrossRef]
- Tian, J.; Zhou, D.; Xiang, L.; Liu, X.; Zhang, H.; Wang, B.; Xie, B. MiR-223-3p Inhibits Inflammation and Pyroptosis in Monosodium Urate-Induced Rats and Fibroblast-like Synoviocytes by Targeting NLRP3. Clin. Exp. Immunol. 2021, 204, 396–410. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, X.-P.; Zhang, Q.; Zou, Y.-Y.; Ma, J.-D.; Chen, L.-F.; Zou, Y.-W.; Xue, J.-M.; Ma, R.-F.; Chen, Z.; et al. Gasdermin-E Mediated Pyroptosis-A Novel Mechanism Regulating Migration, Invasion and Release of Inflammatory Cytokines in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Front. Cell Dev. Biol. 2021, 9, 810635. [Google Scholar] [CrossRef]
- Amin, M.A.; Mansfield, P.J.; Pakozdi, A.; Campbell, P.L.; Ahmed, S.; Martinez, R.J.; Koch, A.E. Interleukin-18 Induces Angiogenic Factors in Rheumatoid Arthritis Synovial Tissue Fibroblasts via Distinct Signaling Pathways. Arthritis Rheum. 2007, 56, 1787–1797. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining Inflammatory Cell States in Rheumatoid Arthritis Joint Synovial Tissues by Integrating Single-Cell Transcriptomics and Mass Cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Mokuda, S.; Tokunaga, T.; Kohno, H.; Ishitoku, M.; Araki, K.; Sugimoto, T.; Yoshida, Y.; Yamamoto, T.; Matsumoto, M.; et al. Expression of Factor XIII Originating from Synovial Fibroblasts and Macrophages Induced by Interleukin-6 Signaling. Inflamm. Regen. 2023, 43, 2. [Google Scholar] [CrossRef] [PubMed]
- Demarco, B.; Danielli, S.; Fischer, F.A.; Bezbradica, J.S. How Pyroptosis Contributes to Inflammation and Fibroblast-Macrophage Cross-Talk in Rheumatoid Arthritis. Cells 2022, 11, 1307. [Google Scholar] [CrossRef] [PubMed]
- Zu, S.-Q.; Feng, Y.-B.; Zhu, C.-J.; Wu, X.-S.; Zhou, R.-P.; Li, G.; Dai, B.-B.; Wang, Z.-S.; Xie, Y.-Y.; Li, Y.; et al. Acid-Sensing Ion Channel 1a Mediates Acid-Induced Pyroptosis through Calpain-2/Calcineurin Pathway in Rat Articular Chondrocytes. Cell Biol. Int. 2020, 44, 2140–2152. [Google Scholar] [CrossRef]
- Zai, Z.; Xu, Y.; Qian, X.; Li, Z.; Ou, Z.; Zhang, T.; Wang, L.; Ling, Y.; Peng, X.; Zhang, Y.; et al. Estrogen Antagonizes ASIC1a-Induced Chondrocyte Mitochondrial Stress in Rheumatoid Arthritis. J. Transl. Med. 2022, 20, 561. [Google Scholar] [CrossRef]
- Jiang, J.-M.; Mo, M.-L.; Long, X.-P.; Xie, L.-H. MiR-144-3p Induced by SP1 Promotes IL-1β-Induced Pyroptosis in Chondrocytes via PTEN/PINK1/Parkin Axis. Autoimmunity 2022, 55, 21–31. [Google Scholar] [CrossRef]
- Kiriakidou, M.; Ching, C.L. Systemic Lupus Erythematosus. Ann. Intern. Med. 2020, 172, ITC81–ITC96. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Bi, L. Role of the NLRP3 Inflammasome in Autoimmune Diseases (2020). Biomed. Pharmacother. 2020, 130, 110542. [Google Scholar] [CrossRef]
- Yang, C.-A.; Huang, S.-T.; Chiang, B.-L. Sex-Dependent Differential Activation of NLRP3 and AIM2 Inflammasomes in SLE Macrophages. Rheumatol. Oxf. Engl. 2015, 54, 324–331. [Google Scholar] [CrossRef]
- da Cruz, H.L.A.; Cavalcanti, C.A.J.; de Azêvedo Silva, J.; de Lima, C.A.D.; Fragoso, T.S.; Barbosa, A.D.; Dantas, A.T.; de Ataíde Mariz, H.; Duarte, A.L.B.P.; Pontillo, A.; et al. Differential Expression of the Inflammasome Complex Genes in Systemic Lupus Erythematosus. Immunogenetics 2020, 72, 217–224. [Google Scholar] [CrossRef]
- Huang, T.; Yin, H.; Ning, W.; Wang, X.; Chen, C.; Lin, W.; Li, J.; Zhou, Y.; Peng, Y.; Wang, M.; et al. Expression of Inflammasomes NLRP1, NLRP3 and AIM2 in Different Pathologic Classification of Lupus Nephritis. Clin. Exp. Rheumatol. 2020, 38, 680–690. [Google Scholar] [PubMed]
- Fu, R.; Guo, C.; Wang, S.; Huang, Y.; Jin, O.; Hu, H.; Chen, J.; Xu, B.; Zhou, M.; Zhao, J.; et al. Podocyte Activation of NLRP3 Inflammasomes Contributes to the Development of Proteinuria in Lupus Nephritis: Podocyte nlrp3 activation in lupus nephritis. Arthritis Rheumatol. 2017, 69, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Ishizawa, M.; Kubota, T. Monoclonal Anti-dsDNA Antibody 2C10 Escorts DNA to Intracellular DNA Sensors in Normal Mononuclear Cells and Stimulates Secretion of Multiple Cytokines Implicated in Lupus Pathogenesis. Clin. Exp. Immunol. 2020, 199, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil Extracellular Trap-Associated Protein Activation of the NLRP3 Inflammasome Is Enhanced in Lupus Macrophages. J. Immunol. Baltim. Md 1950 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Mende, R.; Vincent, F.B.; Kandane-Rathnayake, R.; Koelmeyer, R.; Lin, E.; Chang, J.; Hoi, A.Y.; Morand, E.F.; Harris, J.; Lang, T. Analysis of Serum Interleukin (IL)-1β and IL-18 in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1250. [Google Scholar] [CrossRef]
- Melamud, M.M.; Ermakov, E.A.; Boiko, A.S.; Kamaeva, D.A.; Sizikov, A.E.; Ivanova, S.A.; Baulina, N.M.; Favorova, O.O.; Nevinsky, G.A.; Buneva, V.N. Multiplex Analysis of Serum Cytokine Profiles in Systemic Lupus Erythematosus and Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 13829. [Google Scholar] [CrossRef]
- Chen, F.-F.; Liu, X.-T.; Tao, J.; Mao, Z.-M.; Wang, H.; Tan, Y.; Qu, Z.; Yu, F. Renal NLRP3 Inflammasome Activation Is Associated with Disease Activity in Lupus Nephritis. Clin. Immunol. 2023, 247, 109221. [Google Scholar] [CrossRef]
- Mähönen, K.; Hau, A.; Bondet, V.; Duffy, D.; Eklund, K.K.; Panelius, J.; Ranki, A. Activation of NLRP3 Inflammasome in the Skin of Patients with Systemic and Cutaneous Lupus Erythematosus. Acta Derm. Venereol. 2022, 102, adv00708. [Google Scholar] [CrossRef]
- Zhang, C.; Boini, K.M.; Xia, M.; Abais, J.M.; Li, X.; Liu, Q.; Li, P.-L. Activation of Nod-like Receptor Protein 3 Inflammasomes Turns on Podocyte Injury and Glomerular Sclerosis in Hyperhomocysteinemia. Hypertens. Dallas Tex 1979 2012, 60, 154–162. [Google Scholar] [CrossRef]
- Gupta, S.; Kaplan, M.J. Bite of the Wolf: Innate Immune Responses Propagate Autoimmunity in Lupus. J. Clin. Investg. 2021, 131, e144918. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, A.; Osada, A.; Kawaguchi, H.; Kurata, I.; Nishiyama, T.; Iwai, T.; Ishigami, A.; Kondo, Y.; Tsuboi, H.; Sumida, T.; et al. Specific Increase in Joint Neutrophil Extracellular Traps and Its Relation to Interleukin 6 in Autoimmune Arthritis. Int. J. Mol. Sci. 2021, 22, 7633. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, J.; Li, N.; Zhang, B.; Nguyen, V.D.; Yao, P.; Feng, J.; Liu, Q.; Chen, Y.; Li, G.; et al. NETosis Promotes Chronic Inflammation and Fibrosis in Systemic Lupus Erythematosus and COVID-19. Clin. Immunol. 2023, 254, 109687. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, X.; Song, R.; Li, S.; Zou, S.; Tan, Q.; Liu, T.; Luo, S.; Wu, Z.; Jie, H.; et al. Necrostatin-1 Alleviates Diffuse Pulmonary Haemorrhage by Preventing the Release of NETs via Inhibiting NE/GSDMD Activation in Murine Lupus. J. Immunol. Res. 2023, 2023, 4743975. [Google Scholar] [CrossRef]
- Kirchler, C.; Husar-Memmer, E.; Rappersberger, K.; Thaler, K.; Fritsch-Stork, R. Type I Interferon as Cardiovascular Risk Factor in Systemic and Cutaneous Lupus Erythematosus: A Systematic Review. Autoimmun. Rev. 2021, 20, 102794. [Google Scholar] [CrossRef]
- Kennedy, W.P.; Maciuca, R.; Wolslegel, K.; Tew, W.; Abbas, A.R.; Chaivorapol, C.; Morimoto, A.; McBride, J.M.; Brunetta, P.; Richardson, B.C.; et al. Association of the Interferon Signature Metric with Serological Disease Manifestations but Not Global Activity Scores in Multiple Cohorts of Patients with SLE. Lupus Sci. Med. 2015, 2, e000080. [Google Scholar] [CrossRef]
- Lee, P.Y.; Li, Y.; Richards, H.B.; Chan, F.S.; Zhuang, H.; Narain, S.; Butfiloski, E.J.; Sobel, E.S.; Reeves, W.H.; Segal, M.S. Type I Interferon as a Novel Risk Factor for Endothelial Progenitor Cell Depletion and Endothelial Dysfunction in Systemic Lupus Erythematosus. Arthritis Rheum. 2007, 56, 3759–3769. [Google Scholar] [CrossRef]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.-Y.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN Triggers RIG-I/TLR3/NLRP3-Dependent Inflammasome Activation in Influenza A Virus Infected Cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef]
- Liu, J.; Berthier, C.C.; Kahlenberg, J.M. Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol. 2017, 69, 1840–1849. [Google Scholar] [CrossRef]
- Panchanathan, R.; Liu, H.; Leung, Y.-K.; Ho, S.; Choubey, D. Bisphenol A (BPA) Stimulates the Interferon Signaling and Activates the Inflammasome Activity in Myeloid Cells. Mol. Cell Endocrinol. 2015, 415, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-W.; Nedumaran, B.; Xie, Y.; Kim, D.-K.; Kim, Y.D.; Choi, H.-S. Bisphenol A Bis(2,3-Dihydroxypropyl) Ether (BADGE.2H2O) Induces Orphan Nuclear Receptor Nur77 Gene Expression and Increases Steroidogenesis in Mouse Testicular Leydig Cells. Mol. Cells 2008, 26, 74–80. [Google Scholar] [PubMed]
- Bordon, Y. Nur77 Senses LPS and dsDNA for Non-Canonical Inflammasome Activation. Nat. Rev. Immunol. 2023, 23, 271. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wu, D.; Lu, J.; Zhang, Y.; Yu, S.-M.; Xie, Y.; Li, H.; Yang, J.; Lai, D.-H.; Zeng, K.; et al. Inflammasome Activation Dampens Type I IFN Signaling to Strengthen Anti-Toxoplasma Immunity. mBio 2022, 13, e0236122. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, R.; Guo, C.; Huang, Y.; Wang, H.; Wang, S.; Zhao, J.; Yang, N. Anti-dsDNA Antibodies Bind to TLR4 and Activate NLRP3 Inflammasome in Lupus Monocytes/Macrophages. J. Transl. Med. 2016, 14, 156. [Google Scholar] [CrossRef]
- Leal, V.N.C.; Reis, E.C.; Fernandes, F.P.; Soares, J.L.d.S.; Oliveira, I.G.C.; de Lima, D.S.; Lara, A.N.; Lopes, M.H.; Pontillo, A. Common Pathogen-Associated Molecular Patterns Induce the Hyper-Activation of NLRP3 Inflammasome in Circulating B Lymphocytes of HIV-Infected Individuals. AIDS Lond. Engl. 2021, 35, 899–910. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, B.; Wang, S.; Zhou, M.; Huang, Y.; Guo, C.; Li, M.; Zhao, J.; Sung, S.-S.J.; Gaskin, F.; et al. Tfh Cells with NLRP3 Inflammasome Activation Are Essential for High-Affinity Antibody Generation, Germinal Centre Formation and Autoimmunity. Ann. Rheum. Dis. 2022, 81, 1006–1012. [Google Scholar] [CrossRef]
- Furini, F.; Giuliani, A.L.; Parlati, M.E.; Govoni, M.; Di Virgilio, F.; Bortoluzzi, A. P2X7 Receptor Expression in Patients With Serositis Related to Systemic Lupus Erythematosus. Front. Pharmacol. 2019, 10, 435. [Google Scholar] [CrossRef]
- Yang, Q.; Yu, C.; Yang, Z.; Wei, Q.; Mu, K.; Zhang, Y.; Zhao, W.; Wang, X.; Huai, W.; Han, L. Deregulated NLRP3 and NLRP1 Inflammasomes and Their Correlations with Disease Activity in Systemic Lupus Erythematosus. J. Rheumatol. 2014, 41, 444–452. [Google Scholar] [CrossRef]
- Ma, Z.-Z.; Sun, H.-S.; Lv, J.-C.; Guo, L.; Yang, Q.-R. Expression and Clinical Significance of the NEK7-NLRP3 Inflammasome Signaling Pathway in Patients with Systemic Lupus Erythematosus. J. Inflamm. Lond. Engl. 2018, 15, 16. [Google Scholar] [CrossRef]
- Loftus, S.N.; Liu, J.; Berthier, C.C.; Gudjonsson, J.E.; Gharaee-Kermani, M.; Tsoi, L.C.; Kahlenberg, J.M. Loss of Interleukin-1 Beta Is Not Protective in the Lupus-Prone NZM2328 Mouse Model. Front. Immunol. 2023, 14, 1162799. [Google Scholar] [PubMed]
- Andersen, K.; Eltrich, N.; Lichtnekert, J.; Anders, H.-J.; Vielhauer, V. The NLRP3/ASC Inflammasome Promotes T-Cell-Dependent Immune Complex Glomerulonephritis by Canonical and Noncanonical Mechanisms. Kidney Int. 2014, 86, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Lorenz, G.; Kulkarni, O.P.; Grosser, M.O.O.; Stigrot, N.; Darisipudi, M.N.; Günthner, R.; Wintergerst, M.W.M.; Anz, D.; Susanti, H.E.; et al. NLRP3 and ASC Suppress Lupus-like Autoimmunity by Driving the Immunosuppressive Effects of TGF-β Receptor Signalling. Ann. Rheum. Dis. 2015, 74, 2224–2235. [Google Scholar] [CrossRef]
- Cutolo, M.; Soldano, S.; Smith, V. Pathophysiology of Systemic Sclerosis: Current Understanding and New Insights. Expert Rev. Clin. Immunol. 2019, 15, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Jiang, Z.; Cao, L.; Zou, H.; Zhu, X. Role of NLRP3 Inflammasome in Systemic Sclerosis. Arthritis Res. Ther. 2022, 24, 196. [Google Scholar] [CrossRef]
- Yang, H.; Shi, Y.; Liu, H.; Lin, F.; Qiu, B.; Feng, Q.; Wang, Y.; Yang, B. Pyroptosis Executor Gasdermin D Plays a Key Role in Scleroderma and Bleomycin-Induced Skin Fibrosis. Cell Death Discov. 2022, 8, 183. [Google Scholar] [CrossRef]
- Worrell, J.C.; O’Reilly, S. Bi-Directional Communication: Conversations between Fibroblasts and Immune Cells in Systemic Sclerosis. J. Autoimmun. 2020, 113, 102526. [Google Scholar] [CrossRef]
- Razonable, R.R.; Henault, M.; Paya, C.V. Stimulation of Toll-like Receptor 2 with Bleomycin Results in Cellular Activation and Secretion of pro-Inflammatory Cytokines and Chemokines. Toxicol. Appl. Pharmacol. 2006, 210, 181–189. [Google Scholar] [CrossRef]
- Hoshino, T.; Okamoto, M.; Sakazaki, Y.; Kato, S.; Young, H.A.; Aizawa, H. Role of Proinflammatory Cytokines IL-18 and IL-1beta in Bleomycin-Induced Lung Injury in Humans and Mice. Am. J. Respir. Cell Mol. Biol. 2009, 41, 661–670. [Google Scholar] [CrossRef]
- Martínez-Godínez, M.A.; Cruz-Domínguez, M.P.; Jara, L.J.; Domínguez-López, A.; Jarillo-Luna, R.A.; Vera-Lastra, O.; Montes-Cortes, D.H.; Campos-Rodríguez, R.; López-Sánchez, D.M.; Mejía-Barradas, C.M.; et al. Expression of NLRP3 Inflammasome, Cytokines and Vascular Mediators in the Skin of Systemic Sclerosis Patients. Isr. Med. Assoc. J. IMAJ 2015, 17, 5–10. [Google Scholar]
- Dziankowska-Bartkowiak, B.; Waszczykowska, E.; Zalewska, A.; Sysa-Jedrzejowska, A. Evaluation of Caspase 1 and sFas Serum Levels in Patients with Systemic Sclerosis: Correlation with Lung Dysfunction, Joint and Bone Involvement. Mediat. Inflamm. 2003, 12, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Vincent, F.B.; Sahhar, J.; Ngian, G.-S.; Kandane-Rathnayake, R.; Mende, R.; Morand, E.F.; Lang, T.; Harris, J. Analysis of Serum Interleukin(IL)-1α, IL-1β and IL-18 in Patients with Systemic Sclerosis. Clin. Transl. Immunol. 2019, 8, e1045. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Zhang, Q.; Yang, G.-J.; Tao, J.-H.; Wu, G.-C.; Huang, X.-L.; Duan, Y.; Li, X.-P.; Ye, D.-Q.; Wang, J. Elevated Serum Levels of Interleukin-1β and Interleukin-33 in Patients with Systemic Sclerosis in Chinese Population. Z. Rheumatol. 2018, 77, 151–159. [Google Scholar] [CrossRef]
- Mackiewicz, Z.; Hukkanen, M.; Povilenaite, D.; Sukura, A.; Fonseca, J.E.; Virtanen, I.; Konttinen, Y.T. Dual Effects of Caspase-1, Interleukin-1 Beta, Tumour Necrosis Factor-Alpha and Nerve Growth Factor Receptor in Inflammatory Myopathies. Clin. Exp. Rheumatol. 2003, 21, 41–48. [Google Scholar] [PubMed]
- Sambo, P.; Jannino, L.; Candela, M.; Salvi, A.; Donini, M.; Dusi, S.; Luchetti, M.M.; Gabrielli, A. Monocytes of Patients Wiht Systemic Sclerosis (Scleroderma Spontaneously Release in Vitro Increased Amounts of Superoxide Anion. J. Investig. Dermatol. 1999, 112, 78–84. [Google Scholar] [CrossRef]
- Umehara, H.; Kumagai, S.; Murakami, M.; Suginoshita, T.; Tanaka, K.; Hashida, S.; Ishikawa, E.; Imura, H. Enhanced Production of Interleukin-1 and Tumor Necrosis Factor Alpha by Cultured Peripheral Blood Monocytes from Patients with Scleroderma. Arthritis Rheum. 1990, 33, 893–897. [Google Scholar] [CrossRef]
- Zakrzewska, K.; Arvia, R.; Torcia, M.G.; Clemente, A.M.; Tanturli, M.; Castronovo, G.; Sighinolfi, G.; Giuggioli, D.; Ferri, C. Effects of Parvovirus B19 In Vitro Infection on Monocytes from Patients with Systemic Sclerosis: Enhanced Inflammatory Pathways by Caspase-1 Activation and Cytokine Production. J. Investg. Dermatol. 2019, 139, 2125–2133.e1. [Google Scholar] [CrossRef]
- Bhandari, R.; Ball, M.S.; Martyanov, V.; Popovich, D.; Schaafsma, E.; Han, S.; ElTanbouly, M.; Orzechowski, N.M.; Carns, M.; Arroyo, E.; et al. Profibrotic Activation of Human Macrophages in Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 1160–1169. [Google Scholar] [CrossRef]
- Mohamed, M.E.; Gamal, R.M.; El-Mokhtar, M.A.; Hassan, A.T.; Abozaid, H.S.M.; Ghandour, A.M.; Abdelmoez A Ismail, S.; A Yousef, H.; H El-Hakeim, E.; S Makarem, Y.; et al. Peripheral Cells from Patients with Systemic Sclerosis Disease Co-Expressing M1 and M2 Monocyte/Macrophage Surface Markers: Relation to the Degree of Skin Involvement. Hum. Immunol. 2021, 82, 634–639. [Google Scholar] [CrossRef]
- Ototake, Y.; Yamaguchi, Y.; Asami, M.; Komitsu, N.; Akita, A.; Watanabe, T.; Kanaoka, M.; Kurotaki, D.; Tamura, T.; Aihara, M. Downregulated IRF8 in Monocytes and Macrophages of Patients with Systemic Sclerosis May Aggravate the Fibrotic Phenotype. J. Investig. Dermatol. 2021, 141, 1954–1963. [Google Scholar] [CrossRef]
- Karki, R.; Lee, E.; Sharma, B.R.; Banoth, B.; Kanneganti, T.-D. IRF8 Regulates Gram-Negative Bacteria-Mediated NLRP3 Inflammasome Activation and Cell Death. J. Immunol. Baltim. Md 1950 2020, 204, 2514–2522. [Google Scholar] [CrossRef]
- Laurent, P.; Lapoirie, J.; Leleu, D.; Levionnois, E.; Grenier, C.; Jurado-Mestre, B.; Lazaro, E.; Duffau, P.; Richez, C.; Seneschal, J.; et al. Interleukin-1β–Activated Microvascular Endothelial Cells Promote DC-SIGN–Positive Alternatively Activated Macrophages as a Mechanism of Skin Fibrosis in Systemic Sclerosis. Arthritis Rheumatol. 2022, 74, 1013–1026. [Google Scholar] [CrossRef]
- Baños-Hernández, C.J.; Bucala, R.; Hernández-Bello, J.; Navarro-Zarza, J.E.; Villanueva-Pérez, M.A.; Godínez-Rubí, M.; Parra-Rojas, I.; Vázquez-Villamar, M.; Pereira-Suárez, A.L.; Muñoz Valle, J.F. Expression of Macrophage Migration Inhibitory Factor and Its Receptor CD74 in Systemic Sclerosis. Cent.-Eur. J. Immunol. 2021, 46, 375–383. [Google Scholar] [CrossRef]
- Lang, T.; Lee, J.P.W.; Elgass, K.; Pinar, A.A.; Tate, M.D.; Aitken, E.H.; Fan, H.; Creed, S.J.; Deen, N.S.; Traore, D.A.K.; et al. Macrophage Migration Inhibitory Factor Is Required for NLRP3 Inflammasome Activation. Nat. Commun. 2018, 9, 2223. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I.; Katsiari, C.G.; Daoussis, D.; Bogdanos, D.P. The Role of B Cells in the Pathogenesis of Systemic Sclerosis: An Update. Rheumatol. Oxf. Engl. 2023, 62, 1780–1786. [Google Scholar] [CrossRef]
- François, A.; Gombault, A.; Villeret, B.; Alsaleh, G.; Fanny, M.; Gasse, P.; Adam, S.M.; Crestani, B.; Sibilia, J.; Schneider, P.; et al. B Cell Activating Factor Is Central to Bleomycin- and IL-17-Mediated Experimental Pulmonary Fibrosis. J. Autoimmun. 2015, 56, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.-H.; Chen, L.-C.; Hsu, K.; Chang, C.-C.; Chang, C.-Y.; Kao, C.-W.; Chang, Y.-F.; Chang, M.-C.; Chen, C.G. BAFF-Driven NLRP3 Inflammasome Activation in B Cells. Cell Death Dis. 2020, 11, 820. [Google Scholar] [CrossRef]
- Alsaleh, G.; François, A.; Philippe, L.; Gong, Y.-Z.; Bahram, S.; Cetin, S.; Pfeffer, S.; Gottenberg, J.-E.; Wachsmann, D.; Georgel, P.; et al. MiR-30a-3p Negatively Regulates BAFF Synthesis in Systemic Sclerosis and Rheumatoid Arthritis Fibroblasts. PLoS ONE 2014, 9, e111266. [Google Scholar] [CrossRef]
- Ali, M.F.; Dasari, H.; Van Keulen, V.P.; Carmona, E.M. Canonical Stimulation of the NLRP3 Inflammasome by Fungal Antigens Links Innate and Adaptive B-Lymphocyte Responses by Modulating IL-1β and IgM Production. Front. Immunol. 2017, 8, 1504. [Google Scholar] [CrossRef]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J.; et al. The Role of Endothelial Cells in the Vasculopathy of Systemic Sclerosis: A Systematic Review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef]
- Dri, E.; Lampas, E.; Lazaros, G.; Lazarou, E.; Theofilis, P.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Endothelial Dysfunction. Life 2023, 13, 1420. [Google Scholar] [CrossRef] [PubMed]
- Alyaseer, A.A.A.; de Lima, M.H.S.; Braga, T.T. The Role of NLRP3 Inflammasome Activation in the Epithelial to Mesenchymal Transition Process During the Fibrosis. Front. Immunol. 2020, 11, 883. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Kietzmann, T. Reactive Oxygen Species and Fibrosis: Further Evidence of a Significant Liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Postlethwaite, A.E.; Raghow, R.; Stricklin, G.P.; Poppleton, H.; Seyer, J.M.; Kang, A.H. Modulation of Fibroblast Functions by Interleukin 1: Increased Steady-State Accumulation of Type I Procollagen Messenger RNAs and Stimulation of Other Functions but Not Chemotaxis by Human Recombinant Interleukin 1α and β. J. Cell Biol. 1988, 106, 311–318. [Google Scholar] [CrossRef]
- Ji, J.; Hou, J.; Xia, Y.; Xiang, Z.; Han, X. NLRP3 Inflammasome Activation in Alveolar Epithelial Cells Promotes Myofibroblast Differentiation of Lung-Resident Mesenchymal Stem Cells during Pulmonary Fibrogenesis. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2021, 1867, 166077. [Google Scholar] [CrossRef]
- Cao, S.; Weaver, C.M. Bioactives in the Food Supply: Effects on CVD Health. Curr. Atheroscler. Rep. 2022, 24, 655–661. [Google Scholar] [CrossRef]
- Daryabor, G.; Gholijani, N.; Kahmini, F.R. A Review of the Critical Role of Vitamin D Axis on the Immune System. Exp. Mol. Pathol. 2023, 132–133, 104866. [Google Scholar] [CrossRef]
- Rao, Z.; Chen, X.; Wu, J.; Xiao, M.; Zhang, J.; Wang, B.; Fang, L.; Zhang, H.; Wang, X.; Yang, S.; et al. Vitamin D Receptor Inhibits NLRP3 Activation by Impeding Its BRCC3-Mediated Deubiquitination. Front. Immunol. 2019, 10, 2783. [Google Scholar] [CrossRef]
- Dong, X.; He, Y.; Ye, F.; Zhao, Y.; Cheng, J.; Xiao, J.; Yu, W.; Zhao, J.; Sai, Y.; Dan, G.; et al. Vitamin D3 Ameliorates Nitrogen Mustard-Induced Cutaneous Inflammation by Inactivating the NLRP3 Inflammasome through the SIRT3-SOD2-mtROS Signaling Pathway. Clin. Transl. Med. 2021, 11, e312. [Google Scholar] [CrossRef]
- Aslam, M.M.; John, P.; Bhatti, A.; Jahangir, S.; Kamboh, M.I. Vitamin D as a Principal Factor in Mediating Rheumatoid Arthritis-Derived Immune Response. BioMed Res. Int. 2019, 2019, e3494937. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Zhao, Y.; Xu, N.; Lv, E.; Ci, C.; Li, X. 1,25-Dihydroxyvitamin D3 Ameliorates Lupus Nephritis through Inhibiting the NF-κB and MAPK Signalling Pathways in MRL/Lpr Mice. BMC Nephrol. 2022, 23, 243. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, T. Compound K—An Immunomodulator of Macrophages in Inflammation. Life Sci. 2023, 323, 121700. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Hua, K.-F.; Hsu, W.-H.; Suzuki, Y.; Chu, L.J.; Lee, Y.-C.; Takahata, A.; Lee, S.-L.; Wu, C.-C.; Nikolic-Paterson, D.J.; et al. IgA Nephropathy Benefits from Compound K Treatment by Inhibiting NF-κB/NLRP3 Inflammasome and Enhancing Autophagy and SIRT1. J. Immunol. Baltim. Md 1950 2020, 205, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-J.; Wu, C.-Y.; Tsai, P.-Y.; Hsu, W.-H.; Hua, K.-F.; Chu, C.-L.; Lee, Y.-C.; Chen, A.; Lee, S.-L.; Lin, Y.-J.; et al. Accelerated and Severe Lupus Nephritis Benefits From M1, an Active Metabolite of Ginsenoside, by Regulating NLRP3 Inflammasome and T Cell Functions in Mice. Front. Immunol. 2019, 10, 1951. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qian, P.; Guo, Y.; Gu, L.; Jurat, J.; Bai, Y.; Zhang, D. Myrtenal and β-Caryophyllene Oxide Screened from Liquidambaris Fructus Suppress NLRP3 Inflammasome Components in Rheumatoid Arthritis. BMC Complement. Med. Ther. 2021, 21, 242. [Google Scholar] [CrossRef]
- Ling, Y.; Xiao, M.; Huang, Z.-W.; Xu, H.; Huang, F.-Q.; Ren, N.-N.; Chen, C.-M.; Lu, D.M.; Yao, X.-M.; Xiao, L.-N.; et al. Jinwujiangu Capsule Treats Fibroblast-Like Synoviocytes of Rheumatoid Arthritis by Inhibiting Pyroptosis via the NLRP3/CAPSES/GSDMD Pathway. Evid.-Based Complement. Altern. Med. ECAM 2021, 2021, 4836992. [Google Scholar] [CrossRef]
- Li, W.; Mao, X.; Wang, X.; Liu, Y.; Wang, K.; Li, C.; Li, T.; Zhang, Y.; Lin, N. Disease-Modifying Anti-Rheumatic Drug Prescription Baihu-Guizhi Decoction Attenuates Rheumatoid Arthritis via Suppressing Toll-Like Receptor 4-Mediated NLRP3 Inflammasome Activation. Front. Pharmacol. 2021, 12, 743086. [Google Scholar] [CrossRef]
- Su, J.; Tao, Y.; Liu, J.; Sun, J.; Zeng, Y.; Meng, X.; Fan, G.; Zhang, Y. Tibetan Medicine Qi-Sai-Er-Sang-Dang-Song Decoction Inhibits TNF-α-Induced Rheumatoid Arthritis in Human Fibroblast-like Synoviocytes via Regulating NOTCH1/NF-κB/NLRP3 Pathway. J. Ethnopharmacol. 2023, 310, 116402. [Google Scholar] [CrossRef]
- Xu, L.; Wang, H.; Yu, Q.-Q.; Ge, J.-R.; Zhang, X.-Z.; Mei, D.; Liang, F.-Q.; Cai, X.-Y.; Zhu, Y.; Shu, J.-L.; et al. The Monomer Derivative of Paeoniflorin Inhibits Macrophage Pyroptosis via Regulating TLR4/NLRP3/GSDMD Signaling Pathway in Adjuvant Arthritis Rats. Int. Immunopharmacol. 2021, 101, 108169. [Google Scholar] [CrossRef]
- Cao, J.; Ni, Y.; Ning, X.; Zhang, H. Wedelolactone Ameliorates Synovial Inflammation and Cardiac Complications in a Murine Model of Collagen-Induced Arthritis by Inhibiting NF-κB/NLRP3 Inflammasome Activation. Folia Histochem. Cytobiol. 2022, 60, 301–310. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, X.; Su, X.; Liu, X.; Ren, K.; Ning, C.; Zhang, Q.; Zhang, S. Daphnes Cortex and Its Licorice-Processed Products Suppress Inflammation via the TLR4/NF-κB/NLRP3 Signaling Pathway and Regulation of the Metabolic Profile in the Treatment of Rheumatoid Arthritis. J. Ethnopharmacol. 2022, 283, 114657. [Google Scholar] [CrossRef]
- Li, W.; Wang, K.; Liu, Y.; Wu, H.; He, Y.; Li, C.; Wang, Q.; Su, X.; Yan, S.; Su, W.; et al. A Novel Drug Combination of Mangiferin and Cinnamic Acid Alleviates Rheumatoid Arthritis by Inhibiting TLR4/NFκB/NLRP3 Activation-Induced Pyroptosis. Front. Immunol. 2022, 13, 912933. [Google Scholar] [CrossRef]
- Ge, G.; Bai, J.; Wang, Q.; Liang, X.; Tao, H.; Chen, H.; Wei, M.; Niu, J.; Yang, H.; Xu, Y.; et al. Punicalagin Ameliorates Collagen-Induced Arthritis by Downregulating M1 Macrophage and Pyroptosis via NF-κB Signaling Pathway. Sci. China Life Sci. 2022, 65, 588–603. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Song, H.; Xiao, X.; Yang, Y.; Huang, Q.; Yu, J.; Yu, J.; Liu, Y.; Han, T.; Zhang, D.; et al. Tectoridin Ameliorates Proliferation and Inflammation in TNF-α-Induced HFLS-RA Cells via Suppressing the TLR4/NLRP3/NF-κB Signaling Pathway. Tissue Cell 2022, 77, 101826. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xin, D.; Liang, X.-D.; Tang, Y. Effect of a Combination of <em>Atractylodes Macrocephala</Em> Extract with Strychnine on the TLR4/NF-κB/NLRP3 Pathway in MH7A Cells. Exp. Ther. Med. 2023, 25, 1–15. [Google Scholar] [CrossRef]
- Gan, W.; Li, X.; Cui, Y.; Xiao, T.; Liu, R.; Wang, M.; Wei, Y.; Cui, M.; Ren, S.; Helian, K.; et al. Pinocembrin Relieves Lipopolysaccharide and Bleomycin Induced Lung Inflammation via Inhibiting TLR4-NF-κB-NLRP3 Inflammasome Signaling Pathway. Int. Immunopharmacol. 2021, 90, 107230. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Xu, M.; Jing, X.; Qiu, J.; Huang, S.; Yan, H.; Yin, L.; Lou, J.; Zhao, L.; Fan, Y.; et al. Honokiol Suppresses the Aberrant Interactions between Renal Resident Macrophages and Tubular Epithelial Cells in Lupus Nephritis through the NLRP3/IL-33/ST2 Axis. Cell Death Dis. 2023, 14, 174. [Google Scholar] [CrossRef]
- Tian, J.; Huang, T.; Chen, J.; Wang, J.; Chang, S.; Xu, H.; Zhou, X.; Yang, J.; Xue, Y.; Zhang, T.; et al. SIRT1 Slows the Progression of Lupus Nephritis by Regulating the NLRP3 Inflammasome through ROS/TRPM2/Ca2+ Channel. Clin. Exp. Med. 2023. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Y.; Wang, Z.; Peng, C.; Li, Q.; Wu, J.; Wang, L.; Liu, D.; Yue, Y.; Qing, Q.; et al. The Method of Yiqi Yangyin Tongluo Can Attenuate the Pyroptosis of Rheumatoid Arthritis Chondrocytes through the ASIC1a/NLRP3 Signaling Pathway. Ann. Transl. Med. 2022, 10, 145. [Google Scholar] [CrossRef]
- Wu, D.; Xu, J.; Jiao, W.; Liu, L.; Yu, J.; Zhang, M.; Chen, G. Suppression of Macrophage Activation by Sodium Danshensu via HIF-1 Alpha/STAT3/NLRP3 Pathway Ameliorated Collagen-Induced Arthritis in Mice. Molecules 2023, 28, 1551. [Google Scholar] [CrossRef]
- Dai, X.; Yang, D.; Bao, J.; Zhang, Q.; Ding, J.; Liu, M.; Ding, M.; Liu, M.; Liang, J.; Jia, X. Er Miao San, a Traditional Chinese Herbal Formula, Attenuates Complete Freund’s Adjuvant-Induced Arthritis in Rats by Regulating Th17/Treg Cells. Pharm. Biol. 2020, 58, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Meng, X.; Xuan, Z.; Chen, S.; Wang, J.; Chen, Z.; Wang, J.; Jia, X. Effect of Er Miao San on Peritoneal Macrophage Polarisation through the miRNA-33/NLRP3 Signalling Pathway in a Rat Model of Adjuvant Arthritis. Pharm. Biol. 2022, 60, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.; Chen, S.; Meng, X.; Cheng, Z.; Wang, J.; Tan, Y.; Su, W.; Lu, Z.; Zhang, M.; et al. Exploring the Effect of Er Miao San-Containing Serum on Macrophage Polarization through miR-33/NLRP3 Pathway. J. Ethnopharmacol. 2023, 307, 116178. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; An, Q.; Ju, B.-M.; Zhang, J.; Fan, P.; He, L.; Wang, L. Role of Vitamin D/VDR Nuclear Translocation in down-Regulation of NF-κB/NLRP3/Caspase-1 Axis in Lupus Nephritis. Int. Immunopharmacol. 2021, 100, 108131. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Yan, T.; Shen, J.; Shi, X.; Luo, F.; Ren, Y. Sulforaphene Targets NLRP3 Inflammasome to Suppress M1 Polarization of Macrophages and Inflammatory Response in Rheumatoid Arthritis. J. Biochem. Mol. Toxicol. 2023, 37, e23362. [Google Scholar] [CrossRef]
- Jing, M.; Yang, J.; Zhang, L.; Liu, J.; Xu, S.; Wang, M.; Zhang, L.; Sun, Y.; Yan, W.; Hou, G.; et al. Celastrol Inhibits Rheumatoid Arthritis through the ROS-NF-κB-NLRP3 Inflammasome Axis. Int. Immunopharmacol. 2021, 98, 107879. [Google Scholar] [CrossRef]
- Zhou, M.; Tan, W.; Hasimu, H.; Liu, J.; Gu, Z.; Zhao, J. Euphorbium Total Triterpenes Improve Freund’s Complete Adjuvant-Induced Arthritis through PI3K/AKT/Bax and NF-κB/NLRP3 Signaling Pathways. J. Ethnopharmacol. 2023, 306, 116146. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, D.; Xu, J.; Liu, L.; Jiao, W.; Yu, J.; Chen, G. Suppression of NLRP3 Inflammasome by Dihydroarteannuin via the HIF-1α and JAK3/STAT3 Signaling Pathway Contributes to Attenuation of Collagen-Induced Arthritis in Mice. Front. Pharmacol. 2022, 13, 884881. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTM | Effect on NLRP3 Activation | Enzyme | Site on NLRP3 | Reference |
|---|---|---|---|---|
| Dephosphorylation | Promote | PP2A | Ser5 in PYD domain | [54] |
| PTEN | Tyr32 in PYD domain | [55] | ||
| Phosphorylation | Inhibit | CSNK1A1 | Ser803 in LRR domain (mouse) | [56] |
| EphA2 | Tyr136 (Tyr132 in mouse) | [57] | ||
| Serine/threonine protein kinase AKT | Ser5 in PYD domain | [58] | ||
| Promote | Stress-activated protein kinase JNK1 | Ser198 (Ser194 in mouse) | [59] | |
| BTK | Tyr136, Tyr140, Tyr143, and Tyr168 | [60] | ||
| Pak1 | Thr659 | [61] | ||
| PKD | Ser295 in NACHT domain | [62] |
| PTM | Effect on NLRP3 | Enzyme | Site on NLRP3 | Reference |
|---|---|---|---|---|
| Ubiquitination | Promote | BRCC3 | Unknown | [63] |
| Deubiquitination | Promote | USP7/USP47 | Unknown | [64] |
| HUWE1 | Lys27 | [65] | ||
| Ubc13 | Lys567, Lys 689 (Lys 565, Lys 687 in mouse) | [66] | ||
| Inhibit | ARIH2 | Unknown | [67] | |
| gp78 | Unknown | [68] | ||
| Cbl-b | Lys 496 (Lys 492 in mouse) | [69] | ||
| β-TrCP1 | Lys384 (Lys380 in mouse) | [70] | ||
| Cullin1 | Lys 689 (Lys687 in mouse) | [71] |
| Mechanism | Bioactive Components | Note | Disease | Model | Effects |
|---|---|---|---|---|---|
| Inhibiting the activation of the NLRP3 inflammasome | Vitamin D | - | RA | HEK293T cells [188] and RA patients | Reducing the levels of inflammatory cytokines and ROS in RA patients [190] |
| VDR agonist/VD3 [214] | - | SLE | MRL/Lpr mice | Decreasing urine protein and serum anti-dsDNA antibody levels | |
| Compound K [193,194] | Major absorbable intestinal bacterial metabolite of ginsenosides | SLE | ASLN mice | Improving renal function, albuminuria, and renal lesions, and reducing serum levels of anti-dsDNA | |
| Myrtenal and β-caryophyllene oxide [195] | Screened from Liquidambaris Fructus | RA | AIA mice | Reducing IL-1β and TNF-α in serum; attenuating the upregulation of NLRP3 and IL-1β expression in the synovial tissue | |
| Sulforaphene [215] | RA | CIA mice; mouse synovial macrophages | Specifically binding to NLRP3 and inhibiting its activation in tissues; alleviating arthritis and suppressing M1 macrophage | ||
| Regulating the TLR4/NLRP3/NF-KB/GSDMD signaling pathway | Jinwujiangu capsule [196] | A traditional Chinese medicine | RA | RA-FLS | Decreasing the expression of caspase-1, GSDMD, NLRP3, and ASC, suppressing the expression of IL-1β and IL-18 |
| Baihu-Guizhi decoction [197] | A traditional Chinese medicine-originated disease-modifying anti-rheumatic drug prescription | RA | AA rats | Reducing levels of TLR4, NLRP3, IL-1β, and IL-18; attenuating the redness and swelling of joints, arthritis incidence, and diameter of the limb | |
| Qi-Sai-Er-Sang-Dang-Song Decoction [198] | A Tibetan classical herbal formula | RA | RA-FLS | Down-regulating the levels of NLRP3 and relative cytokines | |
| Monomer derivative of paeoniflorin [199] | Paeoniflorin could inhibit the development and progression of arthritis in experimental animal models of arthritis | RA | AA rats | Inhibiting macrophage polarization and pyroptosis; attenuating bone erosion, soft tissue swelling, and joint space narrowing | |
| Wedelolactone [200] | Derived from Eclipta alba | RA | CIA rats | Ameliorating ankle joint swelling and cartilage degradation; decreasing the release of pro-inflammatory cytokines | |
| Licorice-processed DGN products [201] | Daphnes Cortex is a popular traditional Chinese herbal medicine for traumatic injuries and RA | RA | CIA rats; LPS-induced RAW264.7 cells | Ameliorating RA symptoms; regulating inflammatory cytokines, matrix metalloproteinases, and vascular endothelial growth factor | |
| Combination of mangiferin and cinnamic acid [202] | May be active components of the Baihu-Guizhi decoction | RA | AA rats RAW264.7cells; MH7A cells | Ameliorating arthritis severity, suppressing NLRP3 inflammasome-induced pyroptosis | |
| Punicalagin [203] | Active substance extracted from pomegranate peel | RA | CIA rats | Alleviating the high expression of inflammatory cytokines in synovial tissue; shifting macrophages to the M2 phenotype | |
| Tectoridin [204] | Isolated from the dry rhizome of iris | RA | RA-FLS | Hindering cell proliferation; markedly promoting apoptosis rates | |
| Strychnine combined with Atractylodes Macrocephala [205] | - | RA | MH7A cells | Promoting the apoptosis of synovial cell; reducing the level of TLR4and NLRP3 | |
| Celastrol [216] | Extracted from Tripterygium wilfordii | RA | AA rats; THP-1 cells | Decreasing the arthritis index score; ameliorating joint swelling and synovial hyperplasia | |
| Euphorbium total triterpenes [217] | Mainly characteristic constituents of euphorbium | RA | AA rats | Relieving swelling; decreasing inflammatory cytokines | |
| Pinocembrin [206] | A flavonoid with anti-inflammatory effects | SSc | RAW264.7 and J774A.1 cells; BLM induced mice | Relieving pulmonary inflammatory response | |
| Regulating the SIRT1/autophagy/NLRP3 axis and inhibiting the NLRP3/IL-33/ST2 axis | Honokiol | A major anti-inflammatory bioactive compound in Magnolia officinalis | SLE | NZB/WF1mice; MRL/Lpr mice | Improving renal function, albuminuria, and renal pathology; regulating T cell functions and reducing anti-dsDNA autoantibodies in serum |
| Regulating the ASIC1a/NLRP3 signaling pathway | Yiqi Yangyin Tongluo | Raw astragalus 30 g, dendrobium 15 g, polygala 15 g, achyranthes bidentata 25 g, honeysuckle 15 g | RA | AA rats | Significantly reducing the infiltration of inflammatory cells in the soft tissues; relieving the swelling of the ankle joints |
| Regulating the HIF-1α/NLRP3 pathway | Monomeric derivatives of paeoniflorin | - | RA | RA-FLS | Decreasing levels of HIF-1α and GSDMD-N; inhibiting FLS pyroptosis |
| Sodium Danshensu | A structurally representative water-soluble derivative of Danshen | RA | CIA mice | Reducing the serum levels of IL-1β and IL-6; ameliorating paw oedema and bone destruction | |
| Dihydroarteannuin [218] | Extracted from the traditional Chinese herb Artemisia annua L. | RA | CIA mice; THP-1 cells | Reducing the serum levels of IL-1β and IL-6; alleviating paw oedema and bone destruction | |
| Regulating the GBP5/P2X7/NLRP3 pathway | Sinomenine | Isolated from Sinomenii Caulis | RA | CIA mice; RAW264.7 cells | Alleviating arthritis symptoms; reducing the levels of inflammatory cytokines |
| Regulating the miRNA-33/NLRP3 pathway | Er Miao San | A traditional Chinese medicine composed of Atractylodis Rhizoma and Phellodendri Cortex in a 1:1 weight ratio | RA | AA rats, RAW264.7 cells, MH7A cells | Decreasing the paw volume and polyarthritis index; alleviating ankle joint histopathology, regulating Th17/Treg and M1polarization |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.; Wang, Y.; Chen, G. New Potentiality of Bioactive Substances: Regulating the NLRP3 Inflammasome in Autoimmune Diseases. Nutrients 2023, 15, 4584. https://doi.org/10.3390/nu15214584
Chen B, Wang Y, Chen G. New Potentiality of Bioactive Substances: Regulating the NLRP3 Inflammasome in Autoimmune Diseases. Nutrients. 2023; 15(21):4584. https://doi.org/10.3390/nu15214584
Chicago/Turabian StyleChen, Baixi, Yuhua Wang, and Guangjie Chen. 2023. "New Potentiality of Bioactive Substances: Regulating the NLRP3 Inflammasome in Autoimmune Diseases" Nutrients 15, no. 21: 4584. https://doi.org/10.3390/nu15214584