
Childhood Obesity, Hypothalamic Inflammation, and the Onset of Puberty: A Narrative Review
 , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Obesity and Early or Precocious Puberty
3. Obesity, High-Fat Diet, and Hypothalamic Inflammation
3.1. Phoenixin
3.2. Hypothalamic Microglial Cells

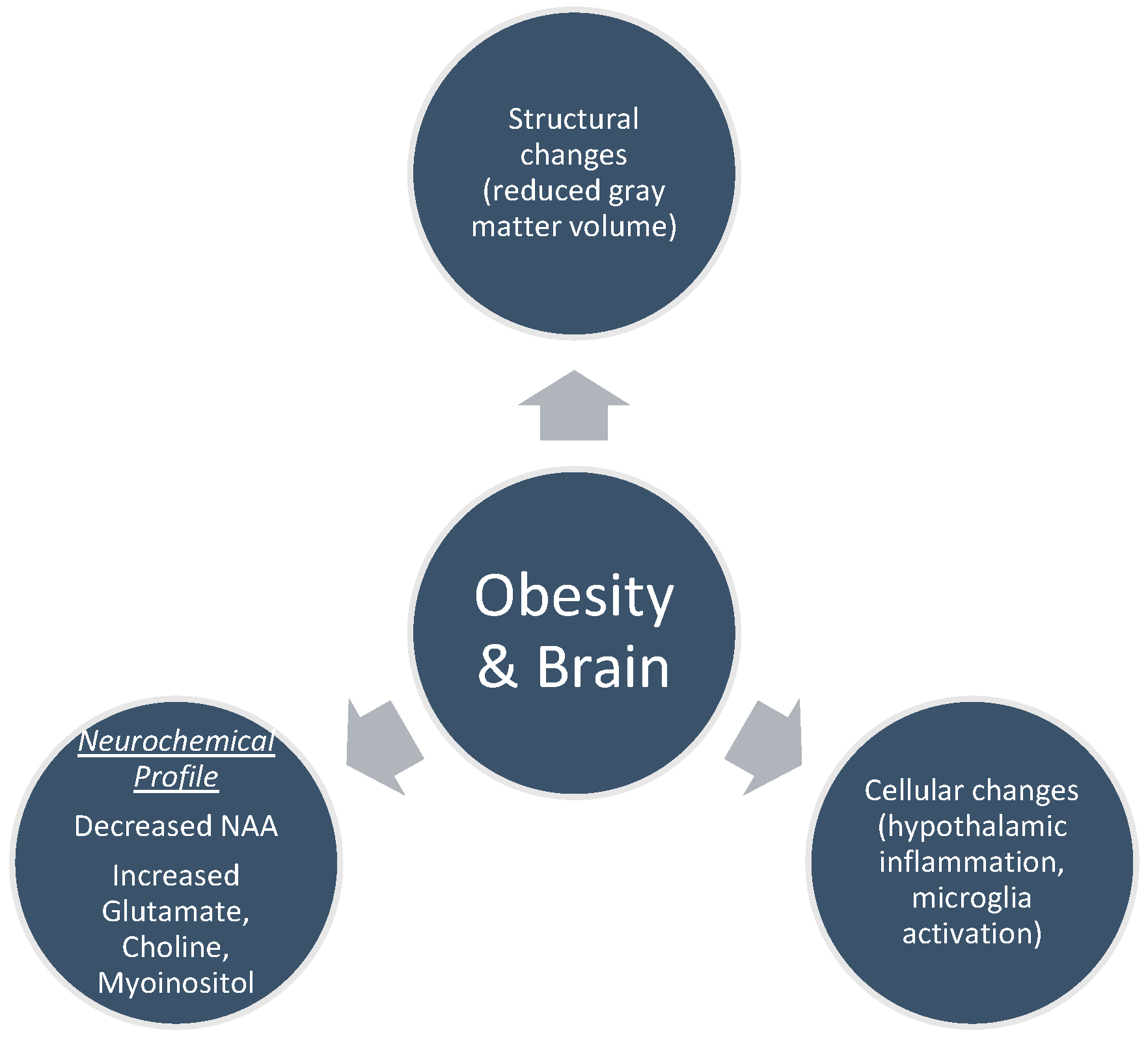
4. Brain MRI Findings in Obesity
4.1. Structural Changes
4.2. Cellular Changes
5. Magnetic Resonance Spectroscopy and Brain’s Neurochemical Profile
5.1. N-acetylaspartate (NAA)
5.2. Myoinositol (MI)
5.3. Choline
5.4. Glutamate (Glu)
5.5. Creatine
6. Discussion
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Latronico, A.C.; Brito, V.N.; Carel, J.C. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 2016, 4, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Manotas, M.C.; González, D.M.; Céspedes, C.; Forero, C.; Rojas Moreno, A.P. Genetic and Epigenetic Control of Puberty. Sex. Dev. 2022, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kalantaridou, S.N.; Chrousos, G.P. Clinical review 148: Monogenic disorders of puberty. J. Clin. Endocrinol. Metab. 2002, 87, 2481–2494. [Google Scholar] [CrossRef] [PubMed]
- Henriques-Neto, D.; Peralta, M.; Marques, A. Editorial: Puberty: Neurologic and physiologic development. Front. Endocrinol. 2023, 14, 1258656. [Google Scholar] [CrossRef] [PubMed]
- Kota, A.S.; Ejaz, S. Precocious Puberty. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chen, M.; Eugster, E.A. Central Precocious Puberty: Update on Diagnosis and Treatment. Paediatr. Drugs 2015, 17, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Maione, L.; Bouvattier, C.; Kaiser, U.B. Central precocious puberty: Recent advances in understanding the aetiology and in the clinical approach. Clin. Endocrinol. 2021, 95, 542–555. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Di Maio, S.; Soliman, N.; Elsedfy, H. Long-term effects and significant Adverse Drug Reactions (ADRs) associated with the use of Gonadotropin-Releasing Hormone analogs (GnRHa) for central precocious puberty: A brief review of literature. Acta Biomed. 2019, 90, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Bräuner, E.V.; Busch, A.S.; Eckert-Lind, C.; Koch, T.; Hickey, M.; Juul, A. Trends in the Incidence of Central Precocious Puberty and Normal Variant Puberty Among Children in Denmark, 1998 to 2017. JAMA Netw. Open 2020, 3, e2015665. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, T.; Li, X.; Pan, D.; Lai, X.; Chen, Y.; Wang, X.; Yu, X.; Fu, S.; Huang, S.; et al. Prevalence of precocious puberty among Chinese children: A school population-based study. Endocrine 2021, 72, 573–581. [Google Scholar] [CrossRef]
- Jebeile, H.; Kelly, A.S.; O’Malley, G.; Baur, L.A. Obesity in children and adolescents: Epidemiology, causes, assessment, and management. Lancet Diabetes Endocrinol. 2022, 10, 351–365. [Google Scholar] [CrossRef]
- Lobstein, T. Obesity prevention and the Global Syndemic: Challenges and opportunities for the World Obesity Federation. Obes. Rev. 2019, 20, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Sanyaolu, A.; Okorie, C.; Qi, X.; Locke, J.; Rehman, S. Childhood and Adolescent Obesity in the United States: A Public Health Concern. Glob. Pediatr. Health 2019, 6, 2333794x19891305. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.A.; Samara, A.; Ray, M.K.; Rutlin, J.; Raji, C.A.; Shimony, J.S.; Sun, P.; Song, S.K.; Hershey, T.; Eisenstein, S.A. Childhood obesity is linked to putative neuroinflammation in brain white matter, hypothalamus, and striatum. Cereb. Cortex Commun. 2023, 4, tgad007. [Google Scholar] [CrossRef] [PubMed]
- Mendes, N.F.; Kim, Y.B.; Velloso, L.A.; Araújo, E.P. Hypothalamic Microglial Activation in Obesity: A Mini-Review. Front. Neurosci. 2018, 12, 846. [Google Scholar] [CrossRef] [PubMed]
- Dionysopoulou, S.; Charmandari, E.; Bargiota, A.; Vlahos, N.; Mastorakos, G.; Valsamakis, G. The Role of Hypothalamic Inflammation in Diet-Induced Obesity and Its Association with Cognitive and Mood Disorders. Nutrients 2021, 13, 498. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Guillén, L.; Argente, J. Central precocious puberty, functional and tumor-related. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101262. [Google Scholar] [CrossRef] [PubMed]
- Moise-Silverman, J.; Silverman, L.A. A review of the genetics and epigenetics of central precocious puberty. Front. Endocrinol. 2022, 13, 1029137. [Google Scholar] [CrossRef] [PubMed]
- Chirico, V.; Lacquaniti, A.; Salpietro, V.; Buemi, M.; Salpietro, C.; Arrigo, T. Central precocious puberty: From physiopathological mechanisms to treatment. J. Biol. Regul. Homeost. Agents 2014, 28, 367–375. [Google Scholar] [PubMed]
- Shearrer, G.E.; Sadler, J.R.; Papantoni, A.; Burger, K.S. Earlier onset of menstruation is related to increased body mass index in adulthood and altered functional correlations between visual, task control and somatosensory brain networks. J. Neuroendocrinol. 2020, 32, e12891. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Guo, J.; Zhang, X.; Lu, Y.; Miao, J.; Xue, H. Obesity is a risk factor for central precocious puberty: A case-control study. BMC Pediatr. 2021, 21, 509. [Google Scholar] [CrossRef]
- Frisch, R.E.; Revelle, R. Height and weight at menarche and a hypothesis of menarche. Arch. Dis. Child. 1971, 46, 695–701. [Google Scholar] [CrossRef]
- Calcaterra, V.; Magenes, V.C.; Hruby, C.; Siccardo, F.; Mari, A.; Cordaro, E.; Fabiano, V.; Zuccotti, G. Links between Childhood Obesity, High-Fat Diet, and Central Precocious Puberty. Children 2023, 10, 241. [Google Scholar] [CrossRef]
- Tenedero, C.B.; Oei, K.; Palmert, M.R. An Approach to the Evaluation and Management of the Obese Child with Early Puberty. J. Endocr. Soc. 2022, 6, bvab173. [Google Scholar] [CrossRef] [PubMed]
- Bo, T.; Liu, M.; Tang, L.; Lv, J.; Wen, J.; Wang, D. Effects of High-Fat Diet During Childhood on Precocious Puberty and Gut Microbiota in Mice. Front. Microbiol. 2022, 13, 930747. [Google Scholar] [CrossRef]
- Wang, L.; Xu, H.; Tan, B.; Yi, Q.; Liu, H.; Deng, H.; Chen, Y.; Wang, R.; Tian, J.; Zhu, J. Gut microbiota and its derived SCFAs regulate the HPGA to reverse obesity-induced precocious puberty in female rats. Front. Endocrinol. 2022, 13, 1051797. [Google Scholar] [CrossRef]
- Ahmed, M.L.; Ong, K.K.; Dunger, D.B. Childhood obesity and the timing of puberty. Trends Endocrinol. Metab. 2009, 20, 237–242. [Google Scholar] [CrossRef]
- Marques, P.; Skorupskaite, K.; Rozario, K.S.; Anderson, R.A.; George, J.T. Physiology of GnRH and Gonadotropin Secretion. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.Com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Xie, Q.; Kang, Y.; Zhang, C.; Xie, Y.; Wang, C.; Liu, J.; Yu, C.; Zhao, H.; Huang, D. The Role of Kisspeptin in the Control of the Hypothalamic-Pituitary-Gonadal Axis and Reproduction. Front. Endocrinol. 2022, 13, 925206. [Google Scholar] [CrossRef]
- Plant, T.M. The neurobiological mechanism underlying hypothalamic GnRH pulse generation: The role of kisspeptin neurons in the arcuate nucleus. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Liang, H.; Zhao, Q.; Lv, S.; Ji, X. Regulation and physiological functions of phoenixin. Front. Mol. Biosci. 2022, 9, 956500. [Google Scholar] [CrossRef]
- Treen, A.K.; Luo, V.; Belsham, D.D. Phoenixin Activates Immortalized GnRH and Kisspeptin Neurons Through the Novel Receptor GPR173. Mol. Endocrinol. 2016, 30, 872–888. [Google Scholar] [CrossRef]
- McIlwraith, E.K.; Loganathan, N.; Belsham, D.D. Phoenixin Expression Is Regulated by the Fatty Acids Palmitate, Docosahexaenoic Acid and Oleate, and the Endocrine Disrupting Chemical Bisphenol A in Immortalized Hypothalamic Neurons. Front. Neurosci. 2018, 12, 838. [Google Scholar] [CrossRef] [PubMed]
- Valsamakis, G.; Arapaki, A.; Balafoutas, D.; Charmandari, E.; Vlahos, N.F. Diet-Induced Hypothalamic Inflammation, Phoenixin, and Subsequent Precocious Puberty. Nutrients 2021, 13, 3460. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, E.K.; Zhang, N.; Belsham, D.D. The Regulation of Phoenixin: A Fascinating Multidimensional Peptide. J. Endocr. Soc. 2022, 6, bvab192. [Google Scholar] [CrossRef]
- Fujioka, H.; Kakehashi, C.; Funabashi, T.; Akema, T. Immunohistochemical evidence for the relationship between microglia and GnRH neurons in the preoptic area of ovariectomized rats with and without steroid replacement. Endocr. J. 2013, 60, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef]
- Ding, H.; Chen, J.; Su, M.; Lin, Z.; Zhan, H.; Yang, F.; Li, W.; Xie, J.; Huang, Y.; Liu, X.; et al. BDNF promotes activation of astrocytes and microglia contributing to neuroinflammation and mechanical allodynia in cyclophosphamide-induced cystitis. J. Neuroinflamm. 2020, 17, 19. [Google Scholar] [CrossRef]
- Przybył, B.J.; Szlis, M.; Wójcik-Gładysz, A. Brain-derived neurotrophic factor (BDNF) affects the activity of the gonadotrophic axis in sheep. Horm. Behav. 2021, 131, 104980. [Google Scholar] [CrossRef]
- Patriarca, L.; Magerowski, G.; Alonso-Alonso, M. Functional neuroimaging in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 260–265. [Google Scholar] [CrossRef]
- Kroll, D.S.; Feldman, D.E.; Biesecker, C.L.; McPherson, K.L.; Manza, P.; Joseph, P.V.; Volkow, N.D.; Wang, G.J. Neuroimaging of Sex/Gender Differences in Obesity: A Review of Structure, Function, and Neurotransmission. Nutrients 2020, 12, 1942. [Google Scholar] [CrossRef]
- Zhang, X.; Han, L.; Lu, C.; McIntyre, R.S.; Teopiz, K.M.; Wang, Y.; Chen, H.; Cao, B. Brain structural and functional alterations in individuals with combined overweight/obesity and mood disorders: A systematic review of neuroimaging studies. J. Affect. Disord. 2023, 334, 166–179. [Google Scholar] [CrossRef]
- Woo, A.; Botta, A.; Shi, S.S.W.; Paus, T.; Pausova, Z. Obesity-Related Neuroinflammation: Magnetic Resonance and Microscopy Imaging of the Brain. Int. J. Mol. Sci. 2022, 23, 8790. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hu, Y.; Zhang, W.; Wang, J.; Ji, W.; Manza, P.; Volkow, N.D.; Zhang, Y.; Wang, G.J. Brain functional and structural magnetic resonance imaging of obesity and weight loss interventions. Mol. Psychiatry 2023, 28, 1466–1479. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, I.A.; Jansen, P.R.; Lamb, H.J. Obesity, Brain Volume, and White Matter Microstructure at MRI: A Cross-sectional UK Biobank Study. Radiology 2019, 291, 763–771. [Google Scholar] [CrossRef]
- Carnell, S.; Gibson, C.; Benson, L.; Ochner, C.N.; Geliebter, A. Neuroimaging and obesity: Current knowledge and future directions. Obes. Rev. 2012, 13, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.R.; Thapaliya, G.; Ranganath, K.; Gabay, A.; Chen, L.; Smith, K.R.; Osorio, R.S.; Convit, A.; Carnell, S. Paediatric obesity and metabolic syndrome associations with cognition and the brain in youth: Current evidence and future directions. Pediatr. Obes. 2023, 18, e13042. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.S.; Lepping, R.J.; Bruce, J.M.; Cherry, J.B.; Martin, L.E.; Davis, A.M.; Brooks, W.M.; Savage, C.R. Brain responses to food logos in obese and healthy weight children. J. Pediatr. 2013, 162, 759–764.e752. [Google Scholar] [CrossRef] [PubMed]
- Davids, S.; Lauffer, H.; Thoms, K.; Jagdhuhn, M.; Hirschfeld, H.; Domin, M.; Hamm, A.; Lotze, M. Increased dorsolateral prefrontal cortex activation in obese children during observation of food stimuli. Int. J. Obes. 2010, 34, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Reinert, K.R.; Po’e, E.K.; Barkin, S.L. The relationship between executive function and obesity in children and adolescents: A systematic literature review. J. Obes. 2013, 2013, 820956. [Google Scholar] [CrossRef]
- Alkan, I.; Altunkaynak, B.Z.; Gültekin, G.; Bayçu, C. Hippocampal neural cell loss in high-fat diet-induced obese rats-exploring the protein networks, ultrastructure, biochemical and bioinformatical markers. J. Chem. Neuroanat. 2021, 114, 101947. [Google Scholar] [CrossRef]
- Reichelt, A.C.; Lemieux, C.A.; Princz-Lebel, O.; Singh, A.; Bussey, T.J.; Saksida, L.M. Age-dependent and region-specific alteration of parvalbumin neurons, perineuronal nets and microglia in the mouse prefrontal cortex and hippocampus following obesogenic diet consumption. Sci. Rep. 2021, 11, 5593. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, V.I. The Impact of Obesity on Microglial Function: Immune, Metabolic and Endocrine Perspectives. Cells 2021, 10, 1584. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty food, fatty acids, and microglial priming in the adult and aged hippocampus and amygdala. Brain Behav. Immun. 2020, 89, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.H.; Kim, M.S.; Lee, I.K.; Yu, R.; Suk, K. Interglial Crosstalk in Obesity-Induced Hypothalamic Inflammation. Front. Neurosci. 2018, 12, 939. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Daniele, G.; Campi, B.; Saba, A.; Codini, S.; Ciccarone, A.; Giusti, L.; Del Prato, S.; Esterline, R.L.; Ferrannini, E. Plasma N-Acetylaspartate Is Related to Age, Obesity, and Glucose Metabolism: Effects of Antidiabetic Treatment and Bariatric Surgery. Front. Endocrinol. 2020, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Barker, P.B. MR spectroscopy and spectroscopic imaging of the brain. Methods Mol. Biol. 2011, 711, 203–226. [Google Scholar] [CrossRef]
- Lizarbe, B.; Campillo, B.; Guadilla, I.; López-Larrubia, P.; Cerdán, S. Magnetic resonance assessment of the cerebral alterations associated with obesity development. J. Cereb. Blood Flow Metab. 2020, 40, 2135–2151. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wen, L.; Chen, J.; Chang, Y.; Huang, R.; Lin, Y.; Shen, G.; Feng, J. Discover boy specific-biomarkers and reveal gender-related metabolic differences in central precocious puberty. J. Steroid Biochem. Mol. Biol. 2023, 231, 106305. [Google Scholar] [CrossRef]
- Warepam, M.; Ahmad, K.; Rahman, S.; Rahaman, H.; Kumari, K.; Singh, L.R. N-Acetylaspartate Is an Important Brain Osmolyte. Biomolecules 2020, 10, 286. [Google Scholar] [CrossRef]
- Baranovicova, E.; Kalenska, D.; Kaplan, P.; Kovalska, M.; Tatarkova, Z.; Lehotsky, J. Blood and Brain Metabolites after Cerebral Ischemia. Int. J. Mol. Sci. 2023, 24, 17302. [Google Scholar] [CrossRef] [PubMed]
- Perdue, M.V.; DeMayo, M.M.; Bell, T.K.; Boudes, E.; Bagshawe, M.; Harris, A.D.; Lebel, C. Changes in brain metabolite levels across childhood. Neuroimage 2023, 274, 120087. [Google Scholar] [CrossRef] [PubMed]
- Coplan, J.D.; Fathy, H.M.; Abdallah, C.G.; Ragab, S.A.; Kral, J.G.; Mao, X.; Shungu, D.C.; Mathew, S.J. Reduced hippocampal N-acetyl-aspartate (NAA) as a biomarker for overweight. Neuroimage Clin. 2014, 4, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Haris, M.; Cai, K.; Singh, A.; Hariharan, H.; Reddy, R. In vivo mapping of brain myo-inositol. Neuroimage 2011, 54, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Cichocka, M.; Bereś, A. From fetus to older age: A review of brain metabolic changes across the lifespan. Ageing Res. Rev. 2018, 46, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Bolo, N.R.; Jacobson, A.M.; Musen, G.; Simonson, D.C. Hyperglycemia and hyperinsulinemia effects on anterior cingulate cortex myoinositol-relation to brain network functional connectivity in healthy adults. J. Neurophysiol. 2022, 127, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Haley, A.P.; Gonzales, M.M.; Tarumi, T.; Tanaka, H. Dyslipidemia links obesity to early cerebral neurochemical alterations. Obesity 2013, 21, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Derbyshire, E.; Obeid, R. Choline, Neurological Development and Brain Function: A Systematic Review Focusing on the First 1000 Days. Nutrients 2020, 12, 1731. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.C.; Blusztajn, J.K.; Caudill, M.A.; Klatt, K.C.; Natker, E.; Zeisel, S.H.; Zelman, K.M. Choline: The Underconsumed and Underappreciated Essential Nutrient. Nutr. Today 2018, 53, 240–253. [Google Scholar] [CrossRef]
- Edwards, C.G.; Walk, A.M.; Cannavale, C.N.; Flemming, I.R.; Thompson, S.V.; Reeser, G.R.; Holscher, H.D.; Khan, N.A. Dietary choline is related to neural efficiency during a selective attention task among middle-aged adults with overweight and obesity. Nutr. Neurosci. 2021, 24, 269–278. [Google Scholar] [CrossRef]
- Dobransky, T.; Rylett, R.J. Functional regulation of choline acetyltransferase by phosphorylation. Neurochem. Res. 2003, 28, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, I.; Tayebati, S.K.; Roy, P.; Micioni Di Bonaventura, M.V.; Moruzzi, M.; Cifani, C.; Amenta, F.; Tomassoni, D. Obesity-Related Brain Cholinergic System Impairment in High-Fat-Diet-Fed Rats. Nutrients 2022, 14, 1243. [Google Scholar] [CrossRef] [PubMed]
- Petroff, O.A. GABA and glutamate in the human brain. Neuroscientist 2002, 8, 562–573. [Google Scholar] [CrossRef]
- Maltais-Payette, I.; Boulet, M.M.; Prehn, C.; Adamski, J.; Tchernof, A. Circulating glutamate concentration as a biomarker of visceral obesity and associated metabolic alterations. Nutr. Metab. 2018, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Neves, T.M.G.; Simoes, E.; Otaduy, M.C.G.; Calfat, E.L.B.; Bertolazzi, P.; da Costa, N.A.; Duran, F.L.S.; Correia-Lima, J.; Martin, M.; Seelander, M.C.L.; et al. Inverse Association Between Hypothalamic N-Acetyl Aspartate/Creatine Ratio and Indices of Body Mass in Adolescents with Obesity. J. Nutr. 2022, 152, 663–670. [Google Scholar] [CrossRef]
- Du, D.; Zhang, Y.; Zhu, C.; Chen, H.; Sun, J. Metabolic Regulation of Hypoxia-Inducible Factors in Hypothalamus. Front. Endocrinol. 2021, 12, 650284. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef]
- Rhea, E.M.; Salameh, T.S.; Logsdon, A.F.; Hanson, A.J.; Erickson, M.A.; Banks, W.A. Blood-Brain Barriers in Obesity. AAPS J. 2017, 19, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Rauf, N.; Nabi, G.; Yi, S.; Yu-Dong, Z.; Fu, J. Mechanistic insight into high-fat diet-induced metabolic inflammation in the arcuate nucleus of the hypothalamus. Biomed. Pharmacother. 2021, 142, 112012. [Google Scholar] [CrossRef]
- Stathori, G.; Tzounakou, A.M.; Mastorakos, G.; Vlahos, N.F.; Charmandari, E.; Valsamakis, G. Alterations in Appetite-Regulating Hormones in Girls with Central Early or Precocious Puberty. Nutrients 2023, 15, 4306. [Google Scholar] [CrossRef]
- Purkayastha, S.; Zhang, G.; Cai, D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-β and NF-κB. Nat. Med. 2011, 17, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.; Pham, M.; Maloney, E.; Rizzo, N.O.; Morton, G.J.; Wisse, B.E.; Kirk, E.A.; Chait, A.; Schwartz, M.W. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Liu, M.; Bezy, O.; Almind, K.; Shapiro, H.; Kasif, S.; Kahn, C.R. A systems biology approach identifies inflammatory abnormalities between mouse strains prior to development of metabolic disease. Diabetes 2010, 59, 2960–2971. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Shefer, G.; Marcus, Y.; Stern, N. Is obesity a brain disease? Neurosci. Biobehav. Rev. 2013, 37, 2489–2503. [Google Scholar] [CrossRef] [PubMed]
- Puig, J.; Blasco, G.; Daunis, I.E.J.; Molina, X.; Xifra, G.; Ricart, W.; Pedraza, S.; Fernández-Aranda, F.; Fernández-Real, J.M. Hypothalamic damage is associated with inflammatory markers and worse cognitive performance in obese subjects. J. Clin. Endocrinol. Metab. 2015, 100, E276–E281. [Google Scholar] [CrossRef]
- Sewaybricker, L.E.; Huang, A.; Chandrasekaran, S.; Melhorn, S.J.; Schur, E.A. The Significance of Hypothalamic Inflammation and Gliosis for the Pathogenesis of Obesity in Humans. Endocr. Rev. 2023, 44, 281–296. [Google Scholar] [CrossRef]
- Sewaybricker, L.E.; Kee, S.; Melhorn, S.J.; Schur, E.A. Greater radiologic evidence of hypothalamic gliosis predicts adiposity gain in children at risk for obesity. Obesity 2021, 29, 1770–1779. [Google Scholar] [CrossRef]
- Merz, E.C.; Monk, C.; Bansal, R.; Sawardekar, S.; Lee, S.; Feng, T.; Spann, M.; Foss, S.; McDonough, L.; Werner, E.; et al. Neonatal brain metabolite concentrations: Associations with age, sex, and developmental outcomes. PLoS ONE 2020, 15, e0243255. [Google Scholar] [CrossRef]
- DelParigi, A.; Chen, K.; Salbe, A.D.; Hill, J.O.; Wing, R.R.; Reiman, E.M.; Tataranni, P.A. Persistence of abnormal neural responses to a meal in postobese individuals. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 370–377. [Google Scholar] [CrossRef]
- Plachta-Danielzik, S.; Kehden, B.; Landsberg, B.; Schaffrath Rosario, A.; Kurth, B.M.; Arnold, C.; Graf, C.; Hense, S.; Ahrens, W.; Müller, M.J. Attributable risks for childhood overweight: Evidence for limited effectiveness of prevention. Pediatrics 2012, 130, e865–e871. [Google Scholar] [CrossRef] [PubMed]
- Cintra, D.E.; Ropelle, E.R.; Moraes, J.C.; Pauli, J.R.; Morari, J.; Souza, C.T.; Grimaldi, R.; Stahl, M.; Carvalheira, J.B.; Saad, M.J.; et al. Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS ONE 2012, 7, e30571. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzounakou, A.-M.; Stathori, G.; Paltoglou, G.; Valsamakis, G.; Mastorakos, G.; Vlahos, N.F.; Charmandari, E. Childhood Obesity, Hypothalamic Inflammation, and the Onset of Puberty: A Narrative Review. Nutrients 2024, 16, 1720. https://doi.org/10.3390/nu16111720
Tzounakou A-M, Stathori G, Paltoglou G, Valsamakis G, Mastorakos G, Vlahos NF, Charmandari E. Childhood Obesity, Hypothalamic Inflammation, and the Onset of Puberty: A Narrative Review. Nutrients. 2024; 16(11):1720. https://doi.org/10.3390/nu16111720
Chicago/Turabian StyleTzounakou, Anastasia-Maria, Galateia Stathori, George Paltoglou, Georgios Valsamakis, George Mastorakos, Nikolaos F. Vlahos, and Evangelia Charmandari. 2024. "Childhood Obesity, Hypothalamic Inflammation, and the Onset of Puberty: A Narrative Review" Nutrients 16, no. 11: 1720. https://doi.org/10.3390/nu16111720
APA StyleTzounakou, A.-M., Stathori, G., Paltoglou, G., Valsamakis, G., Mastorakos, G., Vlahos, N. F., & Charmandari, E. (2024). Childhood Obesity, Hypothalamic Inflammation, and the Onset of Puberty: A Narrative Review. Nutrients, 16(11), 1720. https://doi.org/10.3390/nu16111720