
Microscopic Colitis and Celiac Disease: Sharing More than a Diagnostic Overlap

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
3. The Epidemiological Association between MC and CD
4. Common Ground and Sticking Points in Clinical Aspects between CD and MC
4.1. Risk Factors
4.2. Clinical Symptoms
4.3. Common Autoimmunity Background
4.4. Common Functional Bowel Disorders Overlapping
4.5. The Different Gluten Effect in Patients with MC
5. Common Ground in Pathogenetic Mechanisms between CD and MC
5.1. Genetic Factors
5.2. Impairment of Intestinal Epithelial Barrier Function in CD and MC
5.3. Mucosal Immunity Response
6. Diagnostic Work-Up and Practical Recommendation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miehlke, S.; Guagnozzi, D.; Zabana, Y.; Tontini, G.E.; Kanstrup Fiehn, A.M.; Wildt, S.; Bohr, J.; Bonderup, O.; Bouma, G.; D’Amato, M.; et al. European guidelines on microscopic colitis: United European Gastroenterology (UEG) and European Microscopic Colitis Group (EMCG) statements and recommendations. United Eur. Gastroenterol. J. 2021, 9, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Al-Toma, A.; Volta, U.; Auricchio, R.; Castillejo, G.; Sanders, D.S.; Cellier, C.; Mulder, C.; Lundin, K.E.A. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur. Gastroenterol. J. 2019, 7, 583–613. [Google Scholar] [CrossRef] [PubMed]
- Makharia, G.K.; Chauhan, A.; Singh, P.; Ahuja, V. Review article: Epidemiology of coeliac disease. Aliment. Pharmacol. Ther. 2022, 56, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, B.; Rubio-Tapia, A. Epidemiology, presentation and diagnosis of celiac disease. Gastroenterology 2021, 160, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Zabana, Y.; Tontini, G.; Hultgren-Hörnquist, E.; Zydecka-Skonieczna, K.; Latella, G.; Ostvik, A.E.; Marlicz, W.; D’Amato, M.; Arias, A.; Mielhke, S.; et al. Pathogenesis of Microscopic Colitis: A systematic review. J. Crohn’s Colitis 2022, 28, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Nimri, F.M.; Muhanna, A.; Almomani, Z.; Khazaaleh, S.; Alomari, M.; Almomani, L.; Likhitsup, A. The association between microscopic colitis and celiac disease; a systematic review and meta-analysis. Ann. Gastroenterol. 2022, 35, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.; Haghbin, H.; Khan, R.S.; Khan, Z.; Weissman, S.; Kamal, F.; Lee-Smith, W.; Chandan, S.; Feuerstein, J.D.; Adler, D.G. Celiac disease is associated with microscopic colitis in refractory cases in adults: A systematic review and meta-analysis of observational studies. Dig. Dis. Sci. 2022, 67, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Wildt, S.; Munk, L.K.; Winther-Jensen, M.; Jess, T.; Nyboe Andersen, N. Autoimmune diseases in microscopic colitis: A Danish nationwide case-control study. Aliment. Pharmacol. Ther. 2021, 54, 1454–1462. [Google Scholar] [CrossRef]
- Ebik, B.; Ekin, N.; Bacaksiz, F.; Uzel, A.; Akkuzu, M.Z.; Ucmak, F.; Kaya, M.; Goral, V. What is the incidence of celiac disease in patients with microscopic colitis? Why are these two diseases related? Gastroenterol. Rev. 2024, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Leeds, J.S.; Höroldt, B.S.; Sidhu, R.; Hopper, A.D.; Robinson, K.; Toulson, B.; Dixon, L.; Lobo, A.J.; McAlindon, M.E.; Huristone, D.P.; et al. Is There an association between coeliac disease and inflammatory bowel diseases? A study of relative prevalence in comparison with population controls. Scand. J. Gastroenterol. 2007, 42, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Bergman, D.; Khalili, H.; Lebwohl, B.; Roelstraete, B.; Green, P.H.; Ludvigsson, J.F. Celiac disease and risk of microscopic colitis: A nationwide population-based matched cohort study. United Eur. Gastroenterol. J. 2023, 11, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Ghoneim, S.; Paranji, N.; Waghray, N. Quantifying risk factors for microscopic colitis: A nationwide, retrospective cohort study. Indian J. Gastroenterol. 2022, 41, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Green, P.H.; Yang, J.; Cheng, J.; Lee, A.R.; Harper, J.; Bhagat, G. An association between microscopic colitis and celiac disease. Clin. Gastroenterol. Hepatol. 2009, 7, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Vigren, L.; Tysk, C.; Ström, M.; Kilander, A.F.; Hjortswang, H.; Bohr, J.; Benoni, C.; Larson, L.; Sjöberg, K. Celiac disease and other autoimmune diseases in patients with collagenous colitis. Scandi J. Gastroenterol. 2013, 48, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Altawili, A.; Albalawi, M.A.; Albalawi, S.A.; Alyami, D.M.; Alatawi, A.A.; Albalawi, K.S.; Alghassab, M.; Alotaibi, T.F.O.; Althobaiti, A.A.H.; Abu-Zaid, A. Exploring the association between microscopic colitis and celiac disease: A comprehensive analysis using the national in -patient data (2016–2019). Saudi J. Gastroenterol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bañares, F.; Esteve, M.; Farré, C.; Salas, A.; Alsina, M.; Casalots, J.; Espinós, J.; Forné, M.; Viver, J.M. Predisposing HLA-DQ2 and HLA-DQ8 haplotypes of coeliac disease and associated enteropathy in microscopic colitis. Eur. J. Gastroenterol. Hepatol. 2005, 17, 1333–1338. [Google Scholar] [CrossRef]
- Bonagura, G.A.; Ribaldone, D.G.; Fagoonee, S.; Sapone, N.; Caviglia, G.P.; Saracco, G.M.; Astegiano, M.; Pellicano, R. Microscopic colitis in patients with mild duodenal damage: A new clinical and pathological entity (“lymphocytic enterocolitis”)? World J. Gastroenterol. Pathophysiol. 2016, 7, 307–313. [Google Scholar] [CrossRef]
- Fasano, A.; Berti, I.; Geraduzzi, T.; Not, T.; Colletti, R.B.; Drago, S.; Elitsur, Y.; Green, P.H.R.; GUandalini, S.; Hill, I.D.; et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: A large multicenter study. Arch. Intern. Med. 2003, 163, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Lou, S.; Panjawatanan, P.; Cheungpasitporn, W.; Pungpapong, S.; Lukens, F.J.; Ungprasert, P. Cigarette smoking and risk of celiac disease: A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2018, 6, 1285–1293. [Google Scholar] [CrossRef]
- Marild, K.; Tapia, G.; Midttun, O.; Ueland, P.M.; Magnus, M.C.; Rewers, M.; Stene, L.C.; Stordal, K. Smoking in pregnancy, cord blood cotinine and risk of celiac disease diagnosis in offspring. Eur. J. Epidemiol. 2019, 34, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Arora, S.; Lal, S.; Strand, T.A.; Makharia, G.K. Risk of celiac disease in the first- and second-degree relatives of patients with celiac disease: A systematic review and meta-analysis. Am. J. Gastroenterol. 2015, 110, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Wilckbom, A.; Nyhlin, N.; Montgomery, S.M.; Bohr, J.; Tysk, C. Family history, comorbidity, smoking and other risk factors in microscopic colitis: A case-control study. Eur. Gastroenterol. Hepatol. 2017, 29, 587–594. [Google Scholar] [CrossRef]
- Khalili, H.; Axelrad, J.E.; Roelstraete, B.; Olen, O.; D’Amato, M.; Ludvigsson, J.F. Gastrointestinal infection and risk of microscopic colitis: A nationwide case-control study in Sweden. Gastroenterology 2021, 160, 1599–1607. [Google Scholar] [CrossRef]
- Stene, L.C.; Honeyman, M.C.; Hoffenberg, E.J.; Haas, J.E.; Sokol, R.J.; Emery, L.; Taki, I.; Norris, J.M.; Erlich, H.A.; Eisenbarth, G.S.; et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: A longitudinal study. Am. J. Gastroenterol. 2006, 101, 2333–2340. [Google Scholar] [CrossRef]
- Myléus, A.; Hernell, O.; Gothefors, L.; Hammarström, M.L.; Persson, L.A.; Stenlud, H.; Ivarsson, A. Early infections are associated with increased risk celiac disease: An incident case-referent study. BMC Pediatr. 2012, 12, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Kahrs, C.R.; Chuda, K.; Tapia, G.; Stene, L.C.; Marild, K.; Rasmussen, T.; Ronningen, K.S.; Lundin, K.E.A.; Kramna, L.; Cinek, O.; et al. Enterovirus as trigger of coeliac disease: Nested case-control study within prospective birth cohort. BMJ 2019, 364, I231. [Google Scholar] [CrossRef] [PubMed]
- Iversen, R.; Sollid, L.M. The immunobiology and pathogenesis of celiac disease. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bañares, F.; Casanova, M.J.; Arguedas, Y.; Beltrán, B.; Busquets, D.; Farnández, J.M.; Fernández-Salazar, L.; García-Planella, E.; Guagnozzi, D.; Lucendo, A.J.; et al. Current concept son microscopic colitis: Evidence-based statements and recommendations of the Spanish Microscopic Colitis Group. Aliment. Pharmacol. Ther. 2016, 43, 400–426. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.; Gustafsson, R.J.; Ohlsson, B. Auto-antibodies and their association with clinical findings in women diagnosed with microscopic colitis. PLoS ONE 2013, 8, e66088. [Google Scholar]
- Savarino, E.; Zingone, F.; Barberio, B.; Marasco, G.; Akyuz, F.; Akpinar, H.; Barboi, O.; Bodini, G.; Bor, S.; Chiarioni, G.; et al. Functional bowel disorders with diarrea: Clinical guidelines of the United European Gastroenterology and European Society for Neurogastroenterology and Motility. United Eur. Gastroenterol. J. 2022, 10, 556–584. [Google Scholar] [CrossRef]
- Guagnozzi, D.; Arias, A.; Lucendo, A.J. Systematic review with meta-analysis: Diagnostic overlap of microscopic colitis and functional bowel disorders. Aliment. Pharmacol. Ther. 2016, 43, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, W.O., 3rd; Rubin, C.E. Studies of the Rectal Mucosa in Celiac Sprue. Gastroenterology 1964, 47, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Pardi, D.S.; Kelly, C.P. Microscopic colitis. Gastroenterology 2011, 140, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Green, P.H.R.; Paski, S.; Ko, C.; Rubio-Tapia, A. AGA Clinical Practice Update on Management of Refractory Celiac Disease: Expert Review. Gastroenterology 2022, 163, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-H.; Lebwohl, B.; Burke, K.E.; Ivey, K.L.; Ananthakrishnan, A.N.; Lochhead, P.; Olen, O.; Ludvigsson, J.F.; Richter, J.M.; Chan, A.T.; et al. Dietary gluten intake and risk of microscopic colitis among US women without celiac disease: A prospective cohort study. Am. J. Gastroenterol. 2019, 114, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Freeman, H.J. Failure of added dietary gluten to induce small intestinal histopathological changes in patients with watery diarrhea and lymphocytis colitis. Can. J. Gastroenterol. 1996, 10, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Aboulaghras, S.; Piancatelli, D.; Taghzouti, K.; Balahbib, A.; Alshahrani, M.M.; Al Awadh, A.A.; Goh, K.W.; Ming, L.C.; Bouyahya, A.; Oumhani, K. Meta-analysis and systematic review of HLA DQ2/DQ8 in adults with celiac disease. Int. J. Mol. Sci. 2023, 24, 1188. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Rewers, M.; Eisenbarth, G.S. Genetic testing: Who should do the testing and what is the role of genetic testing in the setting of celiac disease? Gastroenterology 2005, 128, S33–S37. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.C.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Adány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Trynka, G.; Hunt, K.A.; Bockett, N.A.; Romanos, J.; Mistry, V.; Szperl, A.; Bakker, S.F.; Bardella, M.T.; Bhaw-Rosun, L.; Castillejo, G.; et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011, 43, 1193–1201. [Google Scholar] [CrossRef]
- Withoff, S.; Li, Y.; Jonkers, I.; Wijmenga, C. Understanding celiac disease by genomics. Trends Genet. 2016, 32, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Koskela, R.M.; Karttunen, T.J.; Niemelä, S.E.; Lehtola, J.K.; Bloigu, R.S.; Karttunen, R.A. Cytokine gene polymorphism in microscopic colitis association with the IL-6-174 GG genotype. Eur. J. Gastroenterol. Hepatol. 2011, 23, 607–613. [Google Scholar] [CrossRef]
- Madisch, A.; Hellmig, S.; Schreiber, S.; Bethke, B.; Stolte, M.; Miehlke, S. NOD2/CARD15 gene polymorphisms are not associated with collagenous colitis. Int. J. Color. Dis. 2007, 22, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Madisch, A.; Hellmig, S.; Schreiber, S.; Bethke, B.; Stolte, M.; Miehlke, S. Allelic variation of the matrix metalloproteinase-9 gene is associated with collagenous colitis. Inflamm. Bowel Dis. 2011, 17, 2295–2298. [Google Scholar] [CrossRef] [PubMed]
- Koskela, R.M.; Karttunen, T.J.; Niemela, S.E.; Lehtola, J.K.; Ilonen, J.; Karttunen, R.A. Human leucocyte antigen and TNFalpha polymorphism association in microscopic colitis. Eur. J. Gastroenterol. Hepatol. 2008, 20, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Fine, K.D.; Do, K.; Schulte, K.; Ogunji, F.; Guerra, R.; Osowski, L.; McCormack, J. High prevalence of celiac sprue-like HLA-DQ genes and enteropathy in patients with the microscopic colitis syndrome. Am. J. Gastroenterol. 2000, 95, 1974–1982. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Lazenby, A.J.; Yardley, J.H.; Bias, W.B.; Johnson, J.; Alianiello, R.G.; Bedine, M.S.; Bayless, T.M. Increased HLA A1 and diminished HLA A3 in lymphocytic colitis compared with controls and patients with collagenous colitis. Dig. Dis. Sci. 1992, 37, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Sikander, A.; Sinha, S.K.; Prasad, K.K.; Rana, S.V. Association of Serotonin Transporter Promoter Polymorphism (5-HTTLPR) with Microscopic Colitis and Ulcerative Colitis. Dig. Dis. Sci. 2015, 60, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Münch, A.; Söderholm, J.D.; Ost, A.; Ström, M. Increased transmucosal uptake of E. coli K12 in collagenous colitis persists after budesonide treatment. Am. J. Gastroenterol. 2009, 104, 679–685. [Google Scholar] [PubMed]
- Garner, C.; Ahn, R.; Ding, Y.C.; Steele, L.; Stoven, S.; Green, P.H.; Fasano, A.; Murray, J.A.; Neuhausen, S.L. Genome-wide association study of celiac disease in North America confirms FRMD4B as new celiac locus. PLoS ONE 2014, 9, e101428. [Google Scholar] [CrossRef] [PubMed]
- Green, H.; Beamunt, R.; Thomas, A.; Hamilton, B.; Wood, A.R.; Sharp, S.; Jones, S.E.; Tyrell, J.; Walker, G.; Goodhand, J.; et al. Genome-wide association study of microscopic colitis in the UK Biobank confirms immune-related pathogenesis. J. Crohns Colitis 2019, 13, 1578–1582. [Google Scholar] [CrossRef]
- Stahl, E.; Roda, G.; Dobbyn, A.; Hu, J.; Zhang, Z.; Westerlind, H.; Bonfiglio, F.; Ray, T.; Torres, J.; Chen, A.; et al. Collagenous colitis is associated with HLA signature and shares genetic risks with other immune-mediated diseases. Gastroenterology 2020, 159, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Westerlind, H.; Bonfiglio, F.; Mellander, M.R.; Hübenthal, M.; Brynedal, B.; Björk, J.; Törkvist, L.; Padyukov, L.; Ohlsson, B.; Löfberg, R.; et al. HLA associations distinguish collagenous from lymphocytic colitis. Am. J. Gastroenterol. 2016, 111, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Roda, G.; Zabana, Y.; Escudero-Hernández, C.; Liu, X.; Chen, Y.; Camargo Tavares, L.; Bonfiglio, F.; Mellander, M.R.; Janczewska, I.; et al. Human leukocyte antigen signature as pathophysiological discriminants of microscopic colitis subtypes. J. Crohn’s Colitis 2023, 18, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Van Elburg, R.M.; Uil, J.J.; Mulder, C.J.; Heymans, H.S. Intestinal permeability in patients with coeliac disease and relatives of patients with coeliac disease. Gut 1993, 34, 354–357. [Google Scholar] [CrossRef]
- Cardoso-Silva, D.; Delbue, D.; Itzlinger, A.; Moerkens, R.; Withoff, S.; Branchi, F.; Schumann, M. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients 2019, 11, 2325. [Google Scholar] [CrossRef]
- Schulzke, J.D.; Bentzel, C.J.; Schulzke, I.; Riecken, E.O.; Fromm, M. Epithelial tight junction structure in the jejunum of children with acute and treated celiac sprue. Pediatr. Res. 1998, 43, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.; Rozing, J.; Sapone, A.; Lammers, K.; Fasano, A. Tight junctions, intestinal permeability and autoimmunity: Celiac disease and type 1 diabetes paradigms. Ann. N. Y. Acad. Sci. 2009, 1165, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Demin, O.O.; Smirnov, S.; Sokolov, V.; Cucurull-Sanchez, L.; Pichardo-Almarza, C.; Flores, M.; Benson, N.; Demin, O.V. Modeling of celiac disease immune response and the therapeutic effect of potential drugs. BMC Syst. Biol. 2013, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.; De Virgiliis, S.; Kang, J.; Macatagney, R.; Musu, M.; Di Pierro, M.; Drago, S.; Congia, M.; Fasano, A. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 2003, 52, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Sander, G.; Cummins, A.; Henshall, T.; Powell, B. Rapid disruption of intestinal barrier function by gliadin involves altered expression of apical junctional proteins. FEBS Lett. 2005, 579, 4851–4855. [Google Scholar] [CrossRef] [PubMed]
- Jauregi-Miguel, A. The tight junction and the epithelial barrier in coeliac disease. Int. Rev. Cell Mol. Biol. 2021, 358, 105–132. [Google Scholar] [PubMed]
- Sturgeon, C.; Fasano, A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 2016, 4, e1251384. [Google Scholar] [CrossRef] [PubMed]
- Armandi, A.; Pellicano, R.; Caviglia, G.P. Tight junction regulation in celiac disease: Role of larazotide acetate. Minerva Gastroenterol. 2022, 68, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Barmeyer, C.; Erko, I.; Fromm, A.; Bojarski, C.; Allers, K.; Moos, V.; Zeit, M.; Fromm, M.; Schulzke, J.D. Ion transport and barrier function are disturbed in microscopic colitis. Ann. N. Y. Acad. Sci. 2012, 1258, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Wildt, S.; Madsen, J.L.; Rumessen, J.J. Small-bowel permeability in collagenous colitis. Scand. J. Gastroenterol. 2006, 41, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Tagkalidis, P.P.; Gibson, P.R.; Bhathal, P.S. Microscopic colitis demonstrates a T helper cell type 1 mucosal cytokine profile. J. Clin. Pathol. 2007, 60, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Burgel, N.; Bojarski, C.; Mankertz, J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Mechanisms of diarrhea in collagenous colitis. Gastroenterology 2002, 123, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Norén, E.; Mellander, M.R.; Almer, S.; Soderman, J. Genetic variation and gene expression levels of tight junction genes indicate relationships between PTEN as well as MAGI1 and microscopic colitis. Dig. Dis. Sci. 2018, 63, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Järnerot, G.; Tysk, C.; Bohr, J.; Eriksson, S. Collagenous colitis and fecal stream diversion. Gastroenterology 1995, 109, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, J.F.; Larvol, L.; Barge, J.; Dubois, S.; De La Bigne, G.; Hénin, D.; Cerf, M. Lymphocytic and collagenous colitis: An immunohistochemical study. Am. J. Gastroenterol. 1996, 91, 709–713. [Google Scholar] [PubMed]
- Halstensen, T.S.; Scott, H.; Brandtzaeg, P. Intraepithelial T cells of the TcRγδ+CD8− and Vδ1/Jδ1+ phenotypes are increased in coeliac disease. Scand. J. Immunol. 1989, 30, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, T.; Brousse, N.; Rambaud, C.; Le Deist, F.; Schmitz, J.; Cerf-Bensussan, N. Numbers of T cell receptor (TCR) αβ+ but not of TcR γδ+ intraepithelial lymphocytes correlate with the grade of villous atrophy in coeliac patients on a long term normal diet. Gut 1993, 34, 208–214. [Google Scholar] [CrossRef]
- Roy, G.; Fernandez-Bañares, F.; Corzo, M.; Gomez-Aguililla, S.; Garcia-Hoz, C.; Nuñez, C. Intestinal and blood lymphograms as new diagnostic tests for celiac disease. Front. Immunol. 2023, 13, 1081955. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Curran, S.A.; Ciszewski, C.; Orbelyan, G.; Setty, M.; Bhagat, G.; Lee, L.; Tretiakova, M.; Semrad, C.; Kistner, E.; et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. Exp. Med. 2006, 203, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Hüe, S.; Mention, J.J.; Monteiro, R.C.; Zhang, S.; Cellier, C.; Schmitz, J.; Verkarre, V.; Fodil, N.; Bahram, S.; Cerf-Bensussan, N.; et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 2004, 21, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, A.K.; Strid, H.; Elgbratt, K.; Tysk, C.; Bohr, J.; Hultgren Hörnquist, E. Microscopic colitis patients have increased proportions of Ki67+ proliferating and CD45RO+ active/memory CD8+ and CD4+8+ mucosal T cells. J. Crohns Colitis 2013, 7, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Halstensen, T.S.; Brandtzaeg, P. Activated T lymphocytes in the celiac lesion: Non-proliferative activation (CD25) of CD4+ α/β cells in the lamina propria but proliferation (Ki-67) of α/β and γ/δ cells in the epithelium. Eur. J. Immunol. 1993, 23, 505–510. [Google Scholar] [CrossRef]
- Göranzon, C.; Kumawat, A.K.; Hultgren-Hörnqvist, E.; Tysk, C.; Eriksson, S.; Bohr, J.; Nyhlin, N. Immunohistochemical characterization of lymphocytes in microscopic colitis. J. Crohns Colitis 2013, 7, e434–e442. [Google Scholar] [CrossRef]
- Lahat, N.; Shapiro, S.; Karban, A.; Gerstein, R.; Kinarty, A.; Lerner, A. Cytokine profile in coeliac disease. Scand. J. Immunol. 1999, 49, 441–446. [Google Scholar] [CrossRef]
- Hansson, T.; Ulfgren, A.K.; Lindroos, E.; DannAEus, A.; Dahlbom, I.; Klareskog, L. Transforming growth factor-β (TGF-β) and tissue transglutaminase expression in the small intestine in children with coeliac disease. Scand. J. Immunol. 2002, 56, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Salvati, V.M.; Mazzarella, G.; Gianfrani, C.; Levings, M.K.; Stefanile, R.; De Giulio, B.; Iaquinto, G.; Giardullo, N.; Auricchio, S.; Roncarolo, M.G. Recombinant human interleukin 10 suppresses gliadin dependent T cell activation in ex vivo cultured coeliac intestinal mucosa. Gut 2005, 54, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, G.; Hernell, O.; Hammarström, S.; Hammarström, M.L. Concomitant increase of IL-10 and pro-inflammatory cytokines in intraepithelial lymphocyte subsets in celiac disease. Int. Immunol. 2007, 19, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Gianfrani, C.; Levings, M.K.; Sartirana, C.; Mazzarella, G.; Barba, G.; Zanzi, D.; Camarca, A.; Iaquinto, G.; Giardullo, N.; Auricchio, S. Gliadin-specific type 1 regulatory T cells from the intestinal mucosa of treated celiac patients inhibit pathogenic T cells. J. Immunol. 2006, 177, 4178–4186. [Google Scholar] [CrossRef]
- Zanzi, D.; Stefanile, R.; Santagata, S.; Iaffaldano, L.; Iaquinto, G.; Giardullo, N.; Lania, G.; Vigliano, I.; Vera, A.R.; Ferrara, K. IL-15 interferes with suppressive activity of intestinal regulatory T cells expanded in Celiac Disease. Am. J. Gastroenterol. 2011, 106, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Dahal-Koirala, S.; Risnes, L.F.; Sollid, L.M. Pathogenesis of coeliac disease—A disorder driven by gluten-specific CD4+ T cells. In Coeliac Disease and Gluten-Related Disorders; Schiepatti, A., Sanders, D., Eds.; Academic Press: London, UK, 2022; pp. 41–68. [Google Scholar]
- Carrasco, A.; Esteve, M.; Salas, A.; Pedrosa, E.; Rosinach, M.; Aceituno, M.; Zabana, Y.; Fernández-Bañares, F. Immunological Differences between Lymphocytic and Collagenous Colitis. J. Crohns Colitis 2016, 10, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rubio, A.; Santin, I.; Irastorza, I.; Castaño, L.; Carlos Vitoria, J.; Ramon Bilbao, J. TH17 (and TH1) signatures of intestinal biopsies of CD patients in response to gliadin. Autoimmunity 2009, 42, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Abadie, V.; Jabri, B. IL-15: A central regulator of celiac disease immunopathology. Immunol. Rev. 2014, 260, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, A.K.; Strid, H.; Tysk, C.; Bohr, J.; Hörnquist, E.H. Microscopic colitis patients demonstrate a mixed Th17/Tc17 and Th1/Tc1 mucosal cytokine profile. Mol. Immunol. 2013, 55, 355–364. [Google Scholar] [CrossRef] [PubMed]
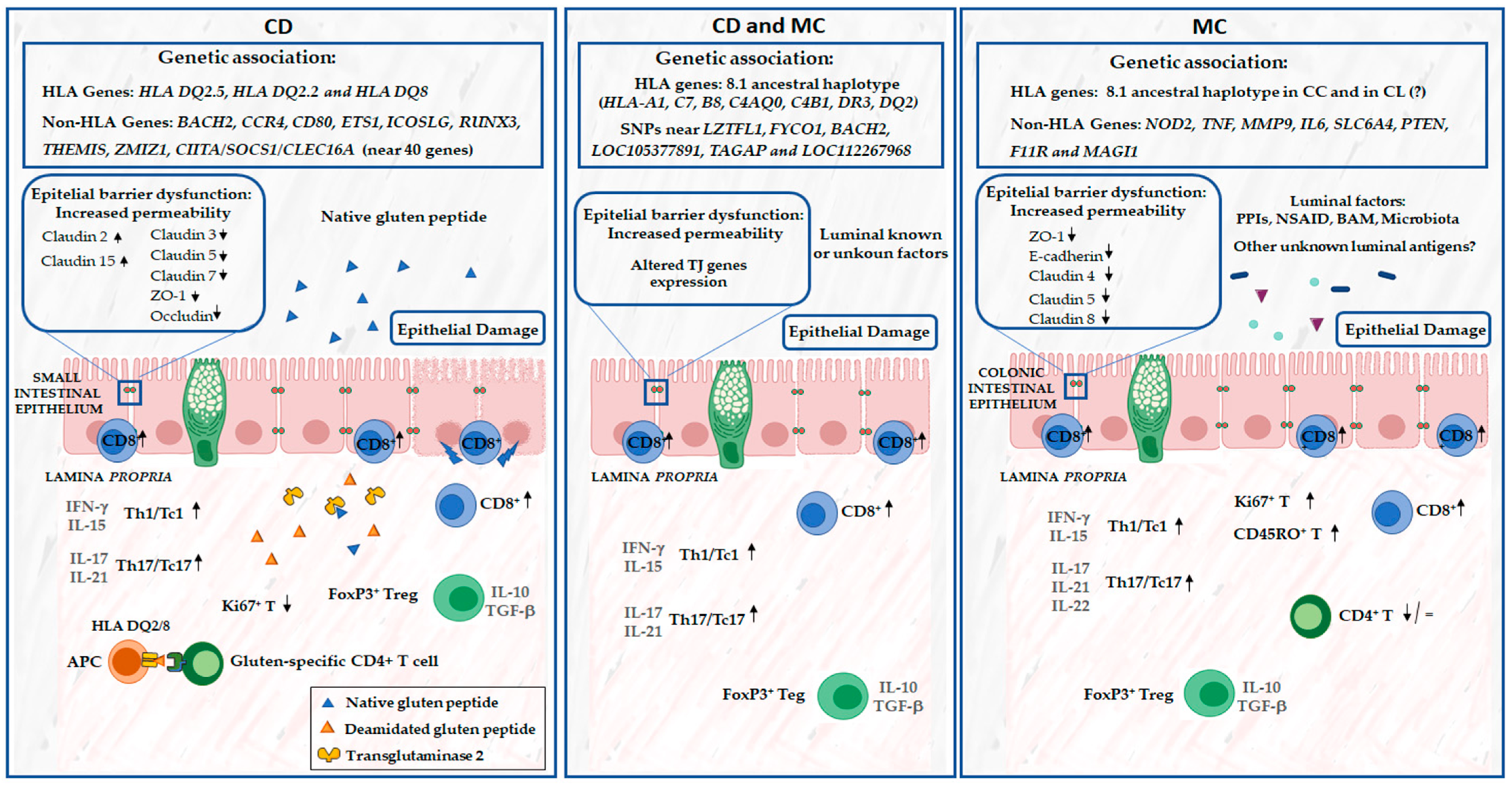

{kind=link}
| Gene (Chromosome) | Genetic Variants | Study Design | Study Cohort | Gene Function | Reference |
|---|---|---|---|---|---|
| HLA (6p) | (LC) HLA-A1 frequency: 0.666 (66.6%) part of 8.1 ancestral haplotype HLA-A3 frequency: 0.00 (0%) (CC) HLA-A1 (part of 8.1 ancestral haplotype) and HLA-A3 frequencies with no significant difference | Genetic Association Study | LC n = 24, CC n = 47, controls n = 3.942 | Antigen presentation | [42] |
| HLA-DQ2 frequency: 0.64 (64%) part of 8.1 ancestral haplotype HLA-DQ1,3 frequency: DQ1,7 0.47 (47%); DQ1,8 0.3 (30%); DQ1,9 0.2 (20%) | Genetic Association study | MC n = 53, controls n = 429 | [43] | ||
| (CC) HLA-DQ2 frequency: 0.480 (48%) part of 8.1 ancestral haplotype | Genetic Association Study | LC n = 25, CC n = 34, controls n = 70 | [16] | ||
| (CC) HLA DR3-DQ2 frequency: 0.438 (43.8%) part of 8.1 ancestral haplotype HLA DR4-DQ8 frequency: 0.138 (13.8%) | Genetic Association Study | MC n = 80 (CC n = 29, LC = 51), controls n = 3.627 | [44] | ||
| (CC) 8.1 ancestral haplotype | Genetic Association Study | LC n = 116, controls n = 1.995 | [45] | ||
| (CC) 8.1 ancestral haplotype related HLA class I (A*01:01, B*08:01, C*07:01) and class II (DRB1*03:01, DQA1*05:01, DQB1*02:01) | Genetic Association Study | CC n = 1.051, controls n = 27.101 | [46] | ||
| (CC) 8.1 ancestral haplotype DRB1*03:01: strongest association (LC) no GWAS-significant signal detected for LC | GWAS meta-analyses | Europe and USA LC n = 373, CC n = 1.498, controls n = 13.487; UK Biobank and FinnGen MC n = 2.599, controls n = 552.343 | [47] |
| Gene (Chromosome) | Genetic Variants | Study Design | Study Cohort | Gene Function | Reference |
|---|---|---|---|---|---|
| NOD2 (16q12.1) | (CC) NOD2 allele (carriage frequency): SNP 8 (9.5%), SNP 12 (1.3%), SNP 13 (8.1%) | Genetic Association Study | CC n = 75, controls n = 534 | Immune response to intracellular bacterial lipopolysaccharides (LPS) | [43] |
| TNF (6p21.33) | TNFα, genotype (carriage frequency): 1.1 (53.8%); 1.2 (39.7%); 2.2 (6.4%) TNF-2 allele carriage frequency: 46.2% | Genetic Association Study | MC n = 78, controls n = 178 | Multifunctional pro-inflammatory cytokine | [44] |
| MMP9 (20q13.12) | (CC) TNF-2 allele carriage frequency: 24% | Genetic Association Study | CC n = 75, controls n = 334 | Breakdown of extracellular matrix | [44] |
| IL6 (7p15.3) | Polymorphism IL-6-174: Susceptibility allele GG | Genetic Association Study | MC n = 81, controls n = 178 | Inflammation and maturation of B cells | [42] |
| SLC6A4 (17q11.2) | Polymorphism 5-HTTLPR (SS) frequency: 12% [lower than control (30%)] | Genetic Association Study | MC n = 41, controls n = 100 | Serotonin transporter | [48] |
| PTEN (10q23.31) | rs1234224: susceptibility allele G | Genetic Association Study | MC n = 25 (CC n = 10, LC n = 14), controls n = 58 | Tight junction | [49] |
| F11R (1q23.3) | (CC) rs790055: susceptibility allele G | Tight junction | |||
| MAGI1 (3p14.1) | rs17417230: susceptibility allele C | Tight junctions, scaffolding protein at cell–cell junctions | |||
| CLEC16A (16p13.13) | rs35099084: susceptibility allele C | GWAS meta-analyses | Europe and USA LC n = 373, CC n = 1.498, controls n = 13.487; UK Biobank and FinnGen MC n = 2.599, controls n = 552.343 | Membrane-associated endosomal protein | [47] |
| RMI2 (16p13.13) | DNA repair and genome stability |
| Gene (Chromosome) | Genetic Variants | Study Design | Study Cohort | Gene Function | Reference |
|---|---|---|---|---|---|
| HLA (6p) | HLA-DQ1,3: DQ1,7; DQ1,8; DQ1,9 HLA-DQ2 Part of 8.1 ancestral haplotype | Genetic Association Study | MC n = 53, CD n = 25, controls n = 429 | Antigen presentation | [43] |
| HLA-DR3-DQ2 frequency: 13 (86.7%). Part of 8.1 ancestral haplotype HLA DR4-DQ8 frequency: 1 (6.7%) HLA-DR3-DQ2 and/or HLA-DR4-DQ8 frequency: 14 (93.3%) | Genetic Association Study | MC with CD n = 15, controls n = 3627 | [44] | ||
| HLA-DRB1*04:01: CC protective alleles and strong risk for CD | Genetic Association Study | CC patients n = 1051 and controls n = 27,101 | [46] | ||
| LZTFL1 (3p21.31) | rs4683148 frequency: 0.39 (39%) | GWAS | MC cases n = 69 (with concomitant celiac disease), CD cases n = 1550 of North American and controls n = 3084 | Protein trafficking | [50] |
| FYCO1 (3p21.31) | rs1072755 frequency: 0.39 (39%) rs4535265 frequency: 0.39(39%) rs2234358 frequency: 0.49(49%) rs3796375 frequency: 0.42 (42%) rs737452 frequency: 0.42 (42%) | Microtubule transport of autophagosomes | |||
| None reported (3p21.31) | rs2373154 frequency: 0.43 (43%) | Not described | |||
| BACH2 (6q15) | rs207270 frequency: 0.46 (46%) rs4142967 frequency: 0.46 (46%) rs12212193 frequency: 0.46 (46%) | Adaptive immune response of T and B cell | |||
| LOC105377891 (6q15) | rs285640 frequency: 0.33 (33%) rs1847473 frequency: 0.27 (27%) | RNA gene, ncRNA class | |||
| TAGAP (6q25.3) | rs1738074 frequency: 0.43 (43%) | T cell activation | |||
| LOC112267968 6q25.3 | rs1738074 frequency: 0.43 (43%) | Not described | |||
| None reported (1q24.3) | rs2227203 frequency: 0.46 (46%) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Castro, A.M.; Fernández-Bañares, F.; Zabana, Y.; Farago-Pérez, G.; Ortega-Barrionuevo, J.; Expósito, E.; Guagnozzi, D. Microscopic Colitis and Celiac Disease: Sharing More than a Diagnostic Overlap. Nutrients 2024, 16, 2233. https://doi.org/10.3390/nu16142233
González-Castro AM, Fernández-Bañares F, Zabana Y, Farago-Pérez G, Ortega-Barrionuevo J, Expósito E, Guagnozzi D. Microscopic Colitis and Celiac Disease: Sharing More than a Diagnostic Overlap. Nutrients. 2024; 16(14):2233. https://doi.org/10.3390/nu16142233
Chicago/Turabian StyleGonzález-Castro, Ana María, Fernando Fernández-Bañares, Yamile Zabana, Georgina Farago-Pérez, Jonathan Ortega-Barrionuevo, Elba Expósito, and Danila Guagnozzi. 2024. "Microscopic Colitis and Celiac Disease: Sharing More than a Diagnostic Overlap" Nutrients 16, no. 14: 2233. https://doi.org/10.3390/nu16142233
APA StyleGonzález-Castro, A. M., Fernández-Bañares, F., Zabana, Y., Farago-Pérez, G., Ortega-Barrionuevo, J., Expósito, E., & Guagnozzi, D. (2024). Microscopic Colitis and Celiac Disease: Sharing More than a Diagnostic Overlap. Nutrients, 16(14), 2233. https://doi.org/10.3390/nu16142233