
High Fat Diet-Induced Dysregulation of Tyrosine Kinases Is a Novel Player in Gut–Brain Axis in Alzheimer’s Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Brain–Gut Axis
3. Gut Microbiota–Brain Axis
3.1. Gut Microbiota
3.2. Mechanistic Insights into Gut Microbiota and Brain Communication
4. Compromised Gut–Brain Crosstalk in Alzheimer’s Disease
4.1. Gut Dysbiosis
4.2. High-Fat-Diet-Induced Gut Dysbiosis and Leaky Gut Are Involved in AD Pathogenesis
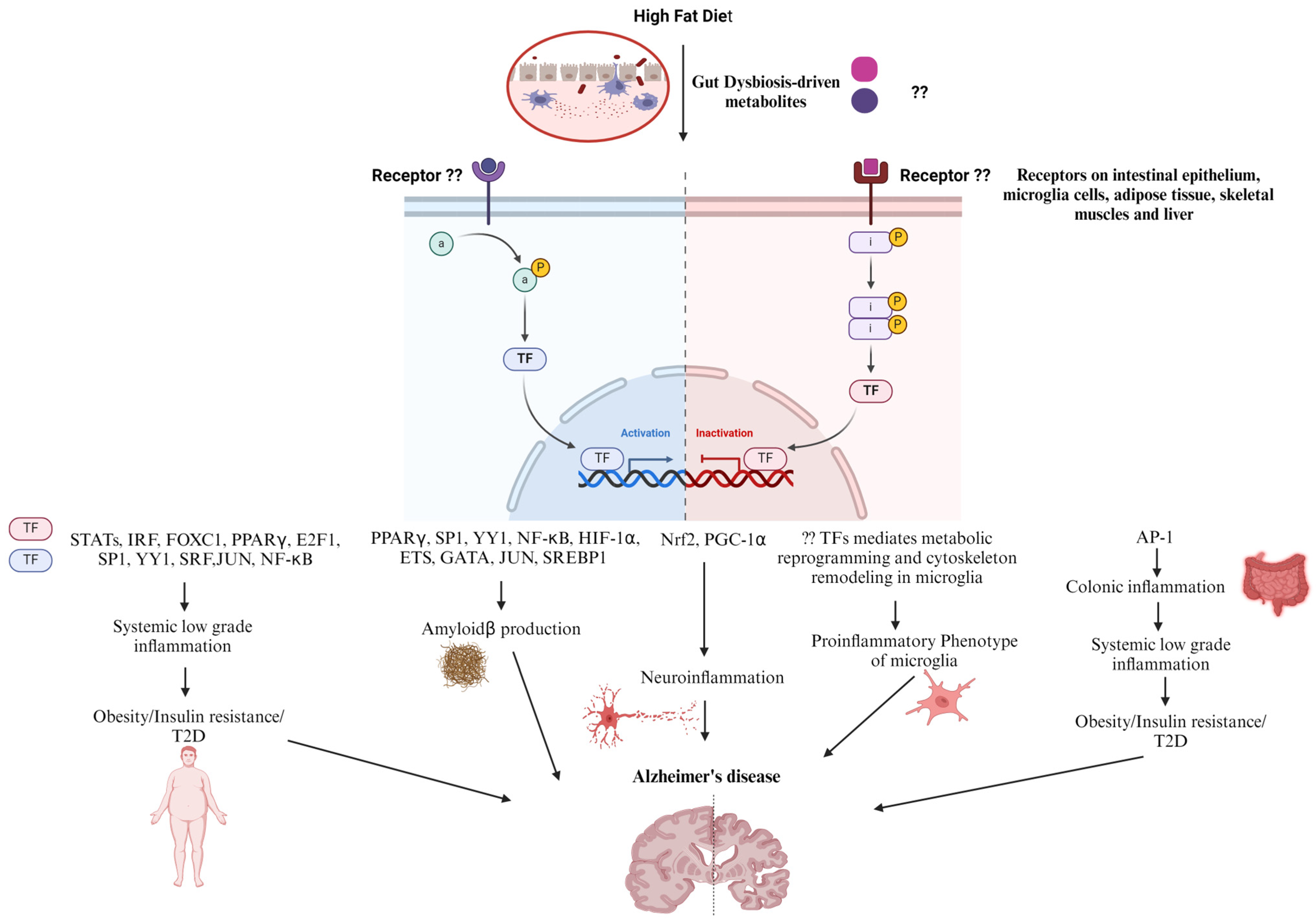
5. Diet and Transcription Factors in AD Progression
6. Role of Kinases in AD
7. Concluding Remarks and Future Work
Author Contributions
Funding
Conflicts of Interest
References
- Juan, S.M.A.; Adlard, P.A. Ageing and Cognition. In Biochemistry and Cell Biology of Ageing: Part II Clinical Science; Springer: Singapore, 2019; pp. 107–122. [Google Scholar] [CrossRef]
- Lunenfeld, B. An aging world–demographics and challenges. Gynecol. Endocrinol. 2008, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Kinsella, K.; Velkoff, V.A. The demographics of aging. Aging Clin. Exp. Res. 2002, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Põlluste, K.; Routasalo, P.; Fagerström, L.; Wagner, L. Advanced nursing practice for older people. Nurs. Res. Pract. 2013, 2013, 860167. [Google Scholar] [CrossRef]
- De Nardi, M.; French, E.; Jones, J.B.; McCauley, J. Medical spending of the US elderly. Fisc. Stud. 2016, 37, 717–747. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, M.; Zhou, R.; Ou, W.; Yao, P. How heavy is the medical expense burden among the older adults and what are the contributing factors? A literature review and problem-based analysis. Front. Public Health 2023, 11, 1165381. [Google Scholar] [CrossRef] [PubMed]
- Centers for Medicare & Medicaid Services. National Health Expenditure Fact Sheet. Available online: https://www.cms.gov/data-research/statistics-trends-and-reports/national-health-expenditure-data/nhe-fact-sheet#:~:text=Per%20person%20personal%20health%20care,-age%20person%20(%249%2C154) (accessed on 13 January 2024).
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Dementia [Fact Sheet]. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia#:~:text=Key%20facts,injuries%20that%20affect%20the%20brain (accessed on 13 January 2024).
- Wilbur, J. Dementia: Dementia Types. FP Essent. 2023, 534, 7–11. [Google Scholar]
- Haque, R.U.; Levey, A.I. Alzheimer′s disease: A clinical perspective and future nonhuman primate research opportunities. Proc. Natl. Acad. Sci. USA 2019, 116, 26224–26229. [Google Scholar] [CrossRef]
- Duong, S.; Patel, T.; Chang, F. Dementia: What pharmacists need to know. Can. Pharm. J. 2017, 150, 118–129. [Google Scholar] [CrossRef]
- Stefanacci, R.G. The costs of Alzheimer’s disease and the value of effective therapies. Am. J. Manag. Care 2011, 17, 356–362. [Google Scholar]
- Alzheimer’s Association Alzheimer’s-Dementia Facts and Figures. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 13 January 2024).
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular calcium dysregulation by the Alzheimer’s disease-linked protein presenilin 2. Int. J. Mol. Sci. 2020, 21, 770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, C.; Mak, M.S.; Lu, J.; Wu, Z.; Chen, Q.; Han, Y.; Li, Y.; Pi, R. Advance of sporadic Alzheimer’s disease animal models. Med. Res. Rev. 2020, 40, 431–458. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The amyloid-β pathway in Alzheimer’s disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics, functions, and clinical impact of presenilin-1 (PSEN1) gene. Int. J. Mol. Sci. 2022, 23, 10970. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.D.; Diaz-Cintra, S.; et al. Alzheimer’s disease: An updated overview of its genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Chan, C.H.; Ma, Q.H.; Xu, X.H.; Xiao, Z.C.; Tan, E.K. The roles of amyloid precursor protein (APP) in neurogenesis: Implications to pathogenesis and therapy of Alzheimer disease. Cell Adhes. Migr. 2011, 5, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, F.; Zhang, S.; Xin, R.; Sun, Y. Genetic and environmental factors in Alzheimer’s and Parkinson’s diseases and promising therapeutic intervention via fecal microbiota transplantation. NPJ Park. Dis. 2021, 7, 70. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Janeiro, M.H.; Ramírez, M.J.; Solas, M. Dysbiosis and Alzheimer’s disease: Cause or treatment opportunity? Cell. Mol. Neurobiol. 2022, 42, 377–387. [Google Scholar] [CrossRef]
- Więckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef]
- Yoo, B.B.; Mazmanian, S.K. The Enteric network: Interactions between the immune and nervous Systems of the gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.t.; Mazmanian, S.K. The gut microbiota-brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef]
- Fleming, M.A., 2nd; Ehsan, L.; Moore, S.R.; Levin, D.E. The enteric nervous system and its emerging role as a therapeutic target. Gastroenterol. Res. Pract. 2020, 2020, 8024171. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B. Integrated neural and endocrine control of gastrointestinal function. Adv. Exp. Med. Biol. 2016, 891, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Schemann, M. Control of gastrointestinal motility by the “gut brain”—The enteric nervous system. J. Pediatr. Gastroenterol. Nutr. 2005, 41, S4–S6. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.D.; Xu, Q.J.; Chang, R.B. Vagal sensory neurons and gut-brain signaling. Curr. Opin. Neurobiol. 2020, 62, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.; Mayer, D.E.; Chen, S.; Mayer, E.A. Role of diet and its effects on the gut microbiome in the pathophysiology of mental disorders. Transl. Psychiatry 2022, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Appleton, J. The Gut-brain axis: Influence of microbiota on mood and mental health. Integr. Med. 2018, 17, 28–32. [Google Scholar]
- Farzi, a.; Fröhlich, E.E.; Holzer, P. Gut microbiota and the neuroendocrine system. Neurotherapeutics 2018, 15, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef]
- Yan, M.; Man, S.; Sun, B.; Ma, L.; Guo, L.; Huang, L.; Gao, W. Gut liver brain axis in diseases: The implications for therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 443. [Google Scholar] [CrossRef]
- Laterza, L.; Rizzatti, G.; Gaetani, E.; Chiusolo, P.; Gasbarrini, A. The gut microbiota and immune system relationship in human graft-versus-host disease. Mediterr. J. Hematol. Infect. Dis. 2016, 8, e2016025. [Google Scholar] [CrossRef] [PubMed]
- Auchtung, T.A.; Fofanova, T.Y.; Stewart, C.J.; Nash, A.K.; Wong, M.C.; Gesell, J.R.; Auchtung, J.M.; Ajami, N.J.; Petrosino, J.F. Investigating colonization of the healthy adult gastrointestinal tract by fungi. MSphere 2018, 3, e00092-00018. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef]
- Xu, M.; Cen, M.; Shen, Y.; Zhu, Y.; Cheng, F.; Tang, L.; Hu, W.; Dai, N. Deoxycholic acid-induced gut dysbiosis disrupts bile acid enterohepatic circulation and promotes intestinal inflammation. Dig. Dis. Sci. 2021, 66, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Chatzopoulos, D.; Zavos, C. Reactive oxygen metabolites and upper gastrointestinal diseases. Hepatogastroenterology 2001, 48, 743–751. [Google Scholar] [PubMed]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan metabolism and gut-brain homeostasis. Int. J. Mol. Sci. 2021, 22, 2972. [Google Scholar] [CrossRef]
- Mayer, E.A.; Knight, R.; Mazmanian, S.K.; Cryan, J.F.; Tillisch, K. Gut microbes and the brain: Paradigm shift in neuroscience. J. Neurosci. 2014, 34, 15490–15496. [Google Scholar] [CrossRef]
- Ratsika, A.; Cruz Pereira, J.S.; Lynch, C.M.K.; Clarke, G.; Cryan, J.F. Microbiota-immune-brain interactions: A lifespan perspective. Curr. Opin. Neurobiol. 2023, 78, 102652. [Google Scholar] [CrossRef]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Nishino, R.; Mikami, K.; Takahashi, H.; Tomonaga, S.; Furuse, M.; Hiramoto, T.; Aiba, Y.; Koga, Y.; Sudo, N. Commensal microbiota modulate murine behaviors in a strictly contamination-free environment confirmed by culture-based methods. Neurogastroenterol. Motil. 2013, 25, 521–528. [Google Scholar] [CrossRef]
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; McCoy, K.D.; et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 2011, 141, 599–609. [Google Scholar] [CrossRef]
- Al-Qudah, M.; Anderson, C.D.; Mahavadi, S.; Bradley, Z.L.; Akbarali, H.I.; Murthy, K.S.; Grider, J.R. Brain-derived neurotrophic factor enhances cholinergic contraction of longitudinal muscle of rabbit intestine via activation of phospholipase C. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G328–G337. [Google Scholar] [CrossRef]
- Mélanie, G.G.; Eytan, W.; David, M.R.; Joon Ho, C.; Mark, T.W.; Dana, J.P.; Glenda, M.; Philip, M.S. Bacterial infection causes stress-induced memory dysfunction in mice. Gut 2011, 60, 307. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, R.; Bouzari, B.; Hosseini-Fard, S.R.; Mazaheri, M.; Ahmadyousefi, Y.; Abdi, M.; Jalalifar, S.; Karimitabar, Z.; Teimoori, A.; Keyvani, H.; et al. Role of microbiota-derived short-chain fatty acids in nervous system disorders. Biomed. Pharmacother. 2021, 139, 111661. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Mahowald, M.A.; Rey, F.E.; Seedorf, H.; Turnbaugh, P.J.; Fulton, R.S.; Wollam, A.; Shah, N.; Wang, C.; Magrini, V.; Wilson, R.K.; et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci. USA 2009, 106, 5859–5864. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Vijay, N.; Morris, M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.; Colombo, J.P.; Berüter, J. Short chain fatty acids in plasma and brain: Quantitative determination by gas chromatography. Clin. Chim. Acta 1979, 92, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, Y.; Yoshioka, N.; Nozaki, K.; Ito, H.; Oda, K.; Harada, K.; Shirawachi, S.; Asano, S.; Aizawa, H.; Yamawaki, S.; et al. Sodium butyrate abolishes lipopolysaccharide-induced depression-like behaviors and hippocampal microglial activation in mice. Brain Res. 2018, 1680, 13–38. [Google Scholar] [CrossRef]
- Spichak, S.; Donoso, F.; Moloney, G.M.; Gunnigle, E.; Brown, J.M.; Codagnone, M.; Dinan, T.G.; Cryan, J.F. Microbially-derived short-chain fatty acids impact astrocyte gene expression in a sex-specific manner. Brain Behav. Immun. Health 2021, 16, 100318. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Caetano-Silva, M.E.; Rund, L.; Hutchinson, N.T.; Woods, J.A.; Steelman, A.J.; Johnson, R.W. Inhibition of inflammatory microglia by dietary fiber and short-chain fatty acids. Sci. Rep. 2023, 13, 2819. [Google Scholar] [CrossRef] [PubMed]
- Patnala, R.; Arumugam, T.V.; Gupta, N.; Dheen, S.T. HDAC Inhibitor Sodium Butyrate-Mediated Epigenetic Regulation Enhances Neuroprotective Function of Microglia During Ischemic Stroke. Mol. Neurobiol. 2017, 54, 6391–6411. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Agis-Balboa, R.C.; Walter, J.; Sananbenesi, F.; Fischer, A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimer’s Dis. 2011, 26, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, K.; Li, X.; Xu, L.; Yang, Z. Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem. Biol. Interact. 2021, 341, 109452. [Google Scholar] [CrossRef] [PubMed]
- Barichello, T.; Generoso, J.S.; Simões, L.R.; Faller, C.J.; Ceretta, R.A.; Petronilho, F.; Lopes-Borges, J.; Valvassori, S.S.; Quevedo, J. Sodium Butyrate Prevents Memory Impairment by Re-establishing BDNF and GDNF Expression in Experimental Pneumococcal Meningitis. Mol. Neurobiol. 2015, 52, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, M.; Nishimura, Y.; Kurebayashi, R.; Minamihata, T.; Kawabe, K.; Takano, K.; Nakamura, Y. Acetate suppresses lipopolysaccharide-stimulated nitric oxide production in primary rat microglia but not in BV-2 microglia cells. Curr. Mol. Pharmacol. 2021, 14, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, J.; Yang, B.; Chen, K.; Kong, Y.; Fang, N.; Gong, T.; Wang, F.; Ling, Z.; Liu, J. Effect of clostridium butyricum against microglia-mediated neuroinflammation in Alzheimer’s disease via regulating gut microbiota and metabolites butyrate. Mol. Nutr. Food Res. 2020, 64, e1900636. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Gong, T.; Chen, W.; Mao, S.; Kong, Y.; Yu, J.; Sun, J. Anti-neuroinflammatory effect of short-chain fatty acid acetate against Alzheimer’s disease via upregulating GPR41 and inhibiting ERK/JNK/NF-κB. J. Agric. Food Chem. 2020, 68, 7152–7161. [Google Scholar] [CrossRef]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, 53–65. [Google Scholar] [CrossRef]
- Bolognini, D.; Tobin, A.B.; Milligan, G.; Moss, C.E. The pharmacology and function of receptors for short-chain fatty acids. Mol. Pharmacol. 2016, 89, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Sittipo, P.; Choi, J.; Lee, S.; Lee, Y.K. The function of gut microbiota in immune-related neurological disorders: A review. J. Neuroinflamm. 2022, 19, 154. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan metabolism by gut microbiome and gut-brain-axis: An in silico analysis. Front. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.; Hess, M.; Weimer, B.C. Microbial-derived tryptophan metabolites and their role in neurological disease: Anthranilic acid and anthranilic acid derivatives. Microorganisms 2023, 11, 1825. [Google Scholar] [CrossRef] [PubMed]
- Juricek, L.; Coumoul, X. The aryl hydrocarbon receptor and the nervous system. Int. J. Mol. Sci. 2018, 19, 2504. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Osadchiy, V.; Martin, C.R.; Mayer, E.A. The gut-brain axis and the microbiome: Mechanisms and clinical implications. Clin. Gastroenterol. Hepatol. 2019, 17, 322–332. [Google Scholar] [CrossRef]
- Wei, G.Z.; Martin, K.A.; Xing, P.Y.; Agrawal, R.; Whiley, L.; Wood, T.K.; Hejndorf, S.; Ng, Y.Z.; Low, J.Z.Y.; Rossant, J.; et al. Tryptophan-metabolizing gut microbes regulate adult neurogenesis via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2021, 118, e2021091118. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.; Kong, Y.; Ye, T.; Yu, Q.; Kumaran Satyanarayanan, S.; Su, K.P.; Liu, J. Microbiota-derived metabolite Indoles induced aryl hydrocarbon receptor activation and inhibited neuroinflammation in APP/PS1 mice. Brain Behav. Immun. 2022, 106, 76–88. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.A.; Park, C.J.; Shaughnessy, M.P.; Cowles, R.A. Serotonin as a Mitogen in the Gastrointestinal Tract: Revisiting a Familiar Molecule in a New Role. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Aaldijk, E.; Vermeiren, Y. The role of serotonin within the microbiota-gut-brain axis in the development of Alzheimer’s disease: A narrative review. Ageing Res. Rev. 2022, 75, 101556. [Google Scholar] [CrossRef] [PubMed]
- Sorgdrager, F.J.H.; Naudé, P.J.W.; Kema, I.P.; Nollen, E.A.; Deyn, P.P. Tryptophan metabolism in Inflammaging: From biomarker to therapeutic target. Front. Immunol. 2019, 10, 2565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, H.; Yang, G.; Yang, Y.; Li, W.; Song, M.; Shao, M.; Su, X.; Lv, L. Associations between expression of indoleamine 2, 3-dioxygenase enzyme and inflammatory cytokines in patients with first-episode drug-naive schizophrenia. Transl. Psychiatry 2021, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.A.; Goertz, J.E.; Ren, T.; Rich, S.S.; Onengut-Gumuscu, S.; Farber, E.; Wu, M.; Overall, C.C.; Kipnis, J.; Gaultier, A. Microbiota alteration is associated with the development of stress-induced despair behavior. Sci. Rep. 2017, 7, 43859. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Solvang, S.H.; Nordrehaug, J.E.; Aarsland, D.; Lange, J.; Ueland, P.M.; McCann, A.; Midttun, Ø.; Tell, G.S.; Giil, L.M. Kynurenines, neuropsychiatric symptoms, and cognitive prognosis in patients with mild dementia. Int. J. Tryptophan Res. 2019, 12, 1178646919877883. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef]
- Choi, H.; Mook-Jung, I. Functional effects of gut microbiota-derived metabolites in Alzheimer’s disease. Curr. Opin. Neurobiol. 2023, 81, 102730. [Google Scholar] [CrossRef] [PubMed]
- Hsuchou, H.; Pan, W.; Kastin, A.J. Fibroblast growth factor 19 entry into brain. Fluids Barriers CNS 2013, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Nance, K.; Chen, S. The gut-brain axis. Annu. Rev. Med. 2022, 73, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Lee, S.; Ma, L.; Zhang, D.; Schlessinger, J.; Shulman, G.I. FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat. Commun. 2015, 6, 6980. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-gut-microbiome axis. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Keshava, H.B.; Mowla, A.; Heinberg, L.J.; Schauer, P.R.; Brethauer, S.A.; Aminian, A. Bariatric surgery may reduce the risk of Alzheimer’s diseases through GLP-1 mediated neuroprotective effects. Med. Hypotheses 2017, 104, 4–9. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Bernaez, I.; Del Rio, M.; Hernanz, A. Stimulation of murine peritoneal macrophage functions by neuropeptide Y and peptide YY. Involvement of protein kinase C. Immunology 1993, 80, 259–265. [Google Scholar] [PubMed]
- Wu, X.; Lv, Y.G.; Du, Y.F.; Chen, F.; Reed, M.N.; Hu, M.; Suppiramaniam, V.; Tang, S.S.; Hong, H. Neuroprotective effects of INT-777 against Aβ(1–42)-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice. Brain Behav. Immun. 2018, 73, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lv, Y.G.; Du, Y.F.; Hu, M.; Reed, M.N.; Long, Y.; Suppiramaniam, V.; Hong, H.; Tang, S.S. Inhibitory effect of INT-777 on lipopolysaccharide-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 88, 360–374. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Chenodeoxycholic acid ameliorates AlCl(3)-induced Alzheimer’s disease neurotoxicity and cognitive deterioration via enhanced insulin signaling in rats. Molecules 2019, 24, 1992. [Google Scholar] [CrossRef]
- Li, D.; Ke, Y.; Zhan, R.; Liu, C.; Zhao, M.; Zeng, A.; Shi, X.; Ji, L.; Cheng, S.; Pan, B.; et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell 2018, 17, e12768. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.W.; Gao, X.B.; et al. A neural circuit for gut-induced reward. Cell 2018, 175, 887–888. [Google Scholar] [CrossRef] [PubMed]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eeat5236. [Google Scholar] [CrossRef] [PubMed]
- Bellono, N.W.; Bayrer, J.R.; Leitch, D.B.; Castro, J.; Zhang, C.; O’Donnell, T.A.; Brierley, S.M.; Ingraham, H.A.; Julius, D. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 2017, 170, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron 2019, 101, 246–259. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Grasset, E.; Mannerås Holm, L.; Karsenty, G.; Macpherson, A.J.; Olofsson, L.E.; Bäckhed, F. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc. Natl. Acad. Sci. USA 2018, 115, 6458–6463. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.K.; Kasper, D.L.; Wang, B.; Forsythe, P.; Bienenstock, J.; Kunze, W.A. Bacteroides fragilis polysaccharide A is necessary and sufficient for acute activation of intestinal sensory neurons. Nat. Commun. 2013, 4, 1465. [Google Scholar] [CrossRef]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Pérez, A.; Peterson, V.; et al. Microbiota-related changes in bile acid & tryptophan metabolism are associated with gastrointestinal dysfunction in a mouse model of autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Schneeberger, M.; Matheis, F.; Wang, P.; Kerner, Z.; Ilanges, A.; Pellegrino, K.; Del Mármol, J.; Castro, T.B.R.; Furuichi, M.; et al. Microbiota modulate sympathetic neurons via a gut-brain circuit. Nature 2020, 583, 441–446. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of neurotransmitters by the gut microbiota and effects on cognition in neurological disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef]
- Miri, S.; Yeo, J.; Abubaker, S.; Hammami, R. Neuromicrobiology, an emerging neurometabolic facet of the gut microbiome? Front. Microbiol. 2023, 14, 1098412. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Varlamov, O. Western-style diet, sex steroids and metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1147–1155. [Google Scholar] [CrossRef]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-style diet, systemic inflammation, and gut microbiota: A narrative review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef]
- Herath, M.; Hosie, S.; Bornstein, J.C.; Franks, A.E.; Hill-Yardin, E.L. The role of the gastrointestinal mucus system in intestinal homeostasis: Implications for neurological disorders. Front. Cell Infect. Microbiol. 2020, 10, 248. [Google Scholar] [CrossRef]
- Wei, Z.; Cui, Y.; Tian, L.; Liu, Y.; Yu, Y.; Jin, X.; Li, H.; Wang, K.; Sun, Q. Probiotic Lactiplantibacillus plantarum N-1 could prevent ethylene glycol-induced kidney stones by regulating gut microbiota and enhancing intestinal barrier function. FASEB J. 2021, 35, e21937. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide regulation of intestinal tight junction permeability is mediated by TLR4 signal transduction pathway activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Delzenne, N.M. The gut microbiome as therapeutic target. Pharmacol. Ther. 2011, 130, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Tanabe, S.; Suzuki, T. High-fat diet-induced intestinal hyperpermeability is associated with increased bile acids in the large intestine of mice. J. Food Sci. 2016, 81, H216–H222. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Safratowich, B.D.; Cheng, W.H.; Larson, K.J.; Briske-Anderson, M. Deoxycholic acid modulates cell-junction gene expression and increases intestinal barrier dysfunction. Molecules 2022, 27, 723. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.; Detloff, S.J.; Ao, M.; Khan, N.; French, S.; Sirajuddin, H.; Nair, T.; Rao, M.C. The Yin and Yang of bile acid action on tight junctions in a model colonic epithelium. Physiol. Rep. 2017, 5, e13294. [Google Scholar] [CrossRef]
- Liu, L.; Xu, J.; Xu, X.; Mao, T.; Niu, W.; Wu, X.; Lu, L.; Zhou, H. Intestinal stem cells damaged by deoxycholic acid via AHR pathway contributes to mucosal barrier dysfunction in high-fat feeding mice. Int. J. Mol. Sci. 2022, 23, 15578. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, Q.; Yu, K.; Fan, X.; Xiao, W.; Cai, Y.; Xu, P.; Yu, M.; Yang, H. 6-Formylindolo(3,2-b)carbazole induced aryl hydrocarbon receptor activation prevents intestinal barrier dysfunction through regulation of claudin-2 expression. Chem. Biol. Interact. 2018, 288, 83–90. [Google Scholar] [CrossRef]
- Ji, T.; Xu, C.; Sun, L.; Yu, M.; Peng, K.; Qiu, Y.; Xiao, W.; Yang, H. Aryl hydrocarbon receptor activation down-regulates IL-7 and reduces inflammation in a mouse model of DSS-induced colitis. Dig. Dis. Sci. 2015, 60, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, J.; Wang, S.; Xiong, J.; Chen, Y.; Liu, Y.; Xiao, T.; Li, Y.; He, T.; Li, Y.; et al. Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics 2020, 10, 7384–7400. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Ruocco, M.; Rapa, S.F.; Piaz, F.D.; Raffaele Di Iorio, B.; Popolo, A.; Autore, G.; Nishijima, F.; Pinto, A.; Marzocco, S. Effect of indoxyl sulfate on the repair and intactness of intestinal epithelial cells: Role of reactive oxygen species’ release. Int. J. Mol. Sci. 2019, 20, 2280. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative effects of a high-fat diet on intestinal permeability: A review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Rescigno, M. The gut immune barrier and the blood-brain barrier: Are they so different? Immunity 2009, 31, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat. Rev. Immunol. 2021, 21, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Anand, N.; Gorantla, V.R.; Chidambaram, S.B. The Role of Gut Dysbiosis in the Pathophysiology of Neuropsychiatric Disorders. Cells 2022, 12, 54. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Secretory products of the human GI tract microbiome and their potential impact on Alzheimer’s disease (AD): Detection of lipopolysaccharide (LPS) in AD hippocampus. Front. Cell Infect. Microbiol. 2017, 7, 318. [Google Scholar] [CrossRef]
- Zhao, Y.; Cong, L.; Jaber, V.; Lukiw, W.J. Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front. Immunol. 2017, 8, 1064. [Google Scholar] [CrossRef]
- Megur, A.; Baltriukienė, D.; Bukelskienė, V.; Burokas, A. The microbiota-gut-brain axis and Alzheimer’s disease: Neuroinflammation Is to blame? Nutrients 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Yanguas-Casás, N.; Torres-Fuentes, C.; Crespo-Castrillo, A.; Diaz-Pacheco, S.; Healy, K.; Stanton, C.; Chowen, J.A.; Garcia-Segura, L.M.; Arevalo, M.A.; Cryan, J.F.; et al. High-fat diet alters stress behavior, inflammatory parameters and gut microbiota in Tg APP mice in a sex-specific manner. Neurobiol. Dis. 2021, 159, 105495. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Han, Y.; Zheng, Z.; Peng, G.; Liu, P.; Yue, S.; Zhu, S.; Chen, J.; Lv, H.; Shao, L.; et al. Altered gut microbial metabolites in Amnestic mild cognitive impairment and Alzheimer’s disease: Signals in host-microbe interplay. Nutrients 2021, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Jiang, Y.; Zhao, Q.; Zhu, Z.; Liang, X.; Zhang, K.; Wu, W.; Dong, Q.; An, Y.; Tang, H.; et al. Metabolomics and incident dementia in older Chinese adults: The Shanghai aging study. Alzheimer’s Dement. 2020, 16, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.X.; Tong, Y.; Wang, J.; Yin, G.; Huang, H.S.; Zeng, L.; Wang, P.; Li, J.P.; Bi, K.S.; Wang, T.J. Determination and comparison of short-chain fatty acids in serum and colon content samples: Alzheimer’s disease rat as a case study. Molecules 2020, 25, 5739. [Google Scholar] [CrossRef] [PubMed]
- Cuervo-Zanatta, D.; Garcia-Mena, J.; Perez-Cruz, C. Gut microbiota alterations and cognitive impairment are sexually dissociated in a transgenic mice model of Alzheimer’s disease. J. Alzheimer’s Dis. 2021, 82, S195–S214. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Golovko, S.; Golovko, M.Y.; Singh, S.; Darland, D.C.; Combs, C.K. Effects of probiotic supplementation on short chain fatty acids in the AppNL-G-F mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2020, 76, 1083–1102. [Google Scholar] [CrossRef]
- Chen, C.; Liao, J.; Xia, Y.; Liu, X.; Jones, R.; Haran, J.; McCormick, B.; Sampson, T.R.; Alam, A.; Ye, K. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut 2022, 71, 2233–2252. [Google Scholar] [CrossRef]
- Vogt, N.M.; Romano, K.A.; Darst, B.F.; Engelman, C.D.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B.; et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 124. [Google Scholar] [CrossRef]
- Huguenard, C.J.C.; Cseresznye, A.; Evans, J.E.; Darcey, T.; Nkiliza, A.; Keegan, A.P.; Luis, C.; Bennett, D.A.; Arvanitakis, Z.; Yassine, H.N.; et al. APOE ε4 and Alzheimer’s disease diagnosis associated differences in L-carnitine, GBB, TMAO, and acylcarnitines in blood and brain. Curr. Res. Transl. Med. 2023, 71, 103362. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Li, N.; Li, D.; Song, B.; Li, L. The presence of elevated circulating trimethylamine N-oxide exaggerates postoperative cognitive dysfunction in aged rats. Behav. Brain Res. 2019, 368, 111902. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Wang, Y.; Wang, X.; Fu, S.; Zhang, X.; Wang, R.T.; Zhang, X. Decreased levels of circulating trimethylamine N-oxide alleviate cognitive and pathological deterioration in transgenic mice: A potential therapeutic approach for Alzheimer’s disease. Aging 2019, 11, 8642–8663. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Lin, C.H.; Tsai, T.H.; Huang, C.H.; Li, J.L.; Chen, L.K.; Li, C.H.; Tsai, T.F.; Wang, P.N. Discovery of a metabolic signature predisposing high risk patients with mild cognitive impairment to converting to Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 10903. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A. Activation of aryl hydrocarbon receptor (AhR) in Alzheimer’s disease: Role of tryptophan metabolites generated by gut host-microbiota. J. Mol. Med. 2023, 101, 201–222. [Google Scholar] [CrossRef] [PubMed]
- van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Zetterberg, H.; Goozee, K.; Lim, C.K.; Jacobs, K.R.; Ashton, N.J.; Hye, A.; Pedrini, S.; Sohrabi, H.R.; Shah, T.; et al. Plasma neurofilament light chain and amyloid-β are associated with the kynurenine pathway metabolites in preclinical Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 186. [Google Scholar] [CrossRef]
- Jena, P.K.; Sheng, L.; Di Lucente, J.; Jin, L.W.; Maezawa, I.; Wan, Y.Y. Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J. 2018, 32, 2866–2877. [Google Scholar] [CrossRef]
- Chen, Q.; Ma, H.; Guo, X.; Liu, J.; Gui, T.; Gai, Z. Farnesoid X receptor (FXR) aggravates amyloid-β-triggered apoptosis by modulating the cAMP-response element-binding protein (CREB)/brain-derived neurotrophic factor (BDNF) pathway in vitro. Med. Sci. Monit. 2019, 25, 9335–9345. [Google Scholar] [CrossRef]
- MahmoudianDehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimer’s Dement. 2019, 15, 76–92. [Google Scholar] [CrossRef]
- Xia, Y.; Xiao, Y.; Wang, Z.-H.; Liu, X.; Alam, A.M.; Haran, J.P.; McCormick, B.A.; Shu, X.; Wang, X.; Ye, K. Bacteroides fragilis in the gut microbiomes of Alzheimer’s disease activates microglia and triggers pathogenesis in neuronal C/EBPβ transgenic mice. Nat. Commun. 2023, 14, 5471. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Yilmaz, Ö.H. Molecular pathways: Dietary regulation of stemness and tumor Initiation by the PPAR-δ pathway. Clin. Cancer Res. 2016, 22, 5636–5641. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Gerding, A.; van Dijk, T.H.; Ciapaite, J.; Bleeker, A.; van Eunen, K.; Havinga, R.; Groen, A.K.; Reijngoud, D.J.; Bakker, B.M. Protection against the metabolic syndrome by guar gum-derived short-chain fatty acids depends on peroxisome proliferator-activated receptor γ and glucagon-like peptide-1. PLoS ONE 2015, 10, e0136364. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Fei, N.; Wu, G.; Zhang, C.; Zhao, L.; Zhang, M. Regulated inflammation and lipid metabolism in colon MRNA expressions of obese germfree mice responding to Enterobacter cloacae B29 combined with the high fat diet. Front. Microbiol. 2016, 7, 1786. [Google Scholar] [CrossRef] [PubMed]
- Granados, J.C.; Falah, K.; Koo, I.; Morgan, E.W.; Perdew, G.H.; Patterson, A.D.; Jamshidi, N.; Nigam, S.K. AHR is a master regulator of diverse pathways in endogenous metabolism. Sci. Rep. 2022, 12, 16625. [Google Scholar] [CrossRef] [PubMed]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Hazlett, H.F.; Nemani, K.V.; Trask, H.W.; West, R.J.; Lupien, L.E.; Collins, A.J.; Ringelberg, C.S.; et al. Inhibition of the aryl hydrocarbon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicol. Appl. Pharmacal. 2016, 300, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Rojas, I.Y.; Moyer, B.J.; Ringelberg, C.S.; Tomlinson, C.R. Reversal of obesity and liver steatosis in mice via inhibition of aryl hydrocarbon receptor and altered gene expression of CYP1B1, PPARα, SCD1, and osteopontin. Int. J. Obes. 2020, 44, 948–963. [Google Scholar] [CrossRef]
- Mogensen, T.H. IRF and stat transcription factors-from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front. Immunol. 2019, 9, 426889. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Investig. 2016, 126, 2839–2854. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Kochumon, S.; Thomas, R.; Bennakhi, A.; Al-Mulla, F.; Ahmad, R. Enhanced Adipose Expression of Interferon Regulatory Factor (IRF)-5 Associates with the Signatures of Metabolic Inflammation in Diabetic Obese Patients. Cells 2020, 9, 730. [Google Scholar] [CrossRef] [PubMed]
- Preeti, K.; Sood, A.; Fernandes, V.; Khan, I.; Khatri, D.K.; Singh, S.B. Experimental Type 2 diabetes and lipotoxicity-associated neuroinflammation involve mitochondrial DNA-mediated cGAS/STING axis: Implication of Type-1 interferon response in cognitive impairment. Mol. Neurobiol. 2024, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.A.; Zhang, R.; Zhang, S.; Deng, S.; Jiang, D.; Zhong, J.; Yang, L.; Wang, T.; Hong, S.; Guo, S.; et al. Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E485–E495. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and Type 2 diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lomeli, J.; Deol, P.; Deans, J.R.; Jiang, T.; Ruegger, P.; Borneman, J.; Sladek, F.M. Impact of various high fat diets on gene expression and the microbiome across the mouse intestines. Sci. Rep. 2023, 13, 22758. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.H.; Peng, S.; Hu, X.; Chen, C.; Rahman, M.R.; Uddin, S.; Quinn, J.M.W.; Moni, M.A. A network-based bioinformatics approach to identify molecular biomarkers for Type 2 diabetes that Are linked to the progression of neurological diseases. Int. J. Environ. Res. Public Health 2020, 17, 1053. [Google Scholar] [CrossRef]
- Fu, X.; Qin, T.; Yu, J.; Jiao, J.; Ma, Z.; Fu, Q.; Deng, X.; Ma, S. Formononetin ameliorates cognitive disorder via PGC-1α pathway in neuroinflammation conditions in high-fat diet-induced mice. CNS Neurol. Disord. Drug Targets 2019, 18, 566–577. [Google Scholar] [CrossRef]
- Zhang, X.L.; Hollander, C.M.; Khan, M.Y.; D’Silva, M.; Ma, H.; Yang, X.; Bai, R.; Keeter, C.K.; Galkina, E.V.; Nadler, J.L.; et al. Myeloid cell deficiency of the inflammatory transcription factor Stat4 protects long-term synaptic plasticity from the effects of a high-fat, high-cholesterol diet. Commun. Biol. 2023, 6, 967. [Google Scholar] [CrossRef]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood-brain barrier disruption, neuroinflammation, amyloidogenic gene expression, and cognitive decline in mice, mimicking the aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, Y.; Yao, S.; Bin Zhao, B. Increases in β-amyloid protein in the hippocampus caused by diabetic metabolic disorder are blocked by minocycline through inhibition of NF-κB pathway activation. Pharmacol. Rep. 2011, 63, 381–391. [Google Scholar] [CrossRef] [PubMed]
- FangFang; Li, H.; Qin, T.; Li, M.; Ma, S. Thymol improves high-fat diet-induced cognitive deficits in mice via ameliorating brain insulin resistance and upregulating NRF2/HO-1 pathway. Metab. Brain Dis. 2017, 32, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Zhang, Z.; Ye, K. C/EBPβ/AEP Signaling drives Alzheimer’s disease pathogenesis. Neurosci. Bull. 2023, 39, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Z.H.; Kang, S.S.; Liu, X.; Xia, Y.; Chan, C.B.; Ye, K. High-fat diet-induced diabetes couples to Alzheimer’s disease through inflammation-activated C/EBPβ/AEP pathway. Mol. Psychiatry 2022, 27, 3396–3409. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Claycombe-Larson, K.; Lund, J.; Ghribi, O. Palmitate-Induced SREBP1 expression and activation underlies the increased BACE 1 activity and amyloid beta genesis. Mol. Neurobiol. 2019, 56, 5256–5269. [Google Scholar] [CrossRef] [PubMed]
- Roßner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression—Implications for Alzheimer’s disease. Progress Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Zhang, Y.W.; Xu, H.; Bu, G. Transcriptional regulation and its misregulation in Alzheimer’s disease. Mol. Brain 2013, 6, 44. [Google Scholar] [CrossRef]
- Pastorcic, M.; Das, H.K. Ets transcription factors ER81 and Elk1 regulate the transcription of the human presenilin 1 gene promoter. Brain Res. Mol. Brain Res. 2003, 113, 57–66. [Google Scholar] [CrossRef]
- Das, H.K. Transcriptional regulation of the presenilin-1 gene: Implication in Alzheimer’s disease. Front. Biosci. 2008, 13, 822–832. [Google Scholar] [CrossRef]
- Abyadeh, M.; Tofigh, N.; Hosseinian, S.; Hasan, M.; Amirkhani, A.; Fitzhenry, M.J.; Gupta, V.; Chitranshi, N.; Salekdeh, G.H.; Haynes, P.A.; et al. Key Genes and Biochemical Networks in Various Brain Regions Affected in Alzheimer’s Disease. Cells 2022, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.X.; Xu, J.; Cheng, F.; Ruppin, E.; Schäffer, A.A. Altered gene expression in excitatory neurons is associated with Alzheimer’s disease and its higher incidence in women. Alzheimer’s Dement. 2023, 9, e12373. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.R.; Islam, T.; Turanli, B.; Zaman, T.; Faruquee, H.M.; Rahman, M.M.; Mollah, M.N.H.; Nanda, R.K.; Arga, K.Y.; Gov, E.; et al. Network-based approach to identify molecular signatures and therapeutic agents in Alzheimer’s disease. Comput. Biol. Chem. 2019, 78, 431–439. [Google Scholar] [CrossRef]
- Haim, Y.; Blüher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef]
- Choi, Y.; Jang, S.; Choi, M.S.; Ryoo, Z.Y.; Park, T. Increased expression of FGF1-mediated signaling molecules in adipose tissue of obese mice. J. Physiol. Biochem. 2016, 72, 157–167. [Google Scholar] [CrossRef]
- Ali Beg, M.M.; Verma, A.K.; Saleem, M.; Saud Alreshidi, F.; Alenazi, F.; Ahmad, H.; Joshi, P.C. Role and significance of circulating biomarkers: miRNA and E2F1 mRNA expression and their association with Type-2 diabetic complications. Int. J. Endocrinol. 2020, 2020, 6279168. [Google Scholar] [CrossRef] [PubMed]
- Tuerxun, M.; Muhda, A.; Yin, L. The molecular mechanisms of signal pathway activating effect of E2F-1/NF-κB/GSK-3β on cognitive dysfunction of Alzheimer rats. Bioengineered 2021, 12, 10000–10008. [Google Scholar] [CrossRef]
- Verdaguer, E.; Susana Gde, A.; Clemens, A.; Pallàs, M.; Camins, A. Implication of the transcription factor E2F-1 in the modulation of neuronal apoptosis. Biomed Pharmacother. 2007, 61, 390–399. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia phenotypes in aging and neurodegenerative diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Biswas, K. Microglia mediated neuroinflammation in neurodegenerative diseases: A review on the cell signaling pathways involved in microglial activation. J. Neuroimmunol. 2023, 383, 578180. [Google Scholar] [CrossRef]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef]
- Lepiarz-Raba, I.; Gbadamosi, I.; Florea, R.; Paolicelli, R.C.; Jawaid, A. Metabolic regulation of microglial phagocytosis: Implications for Alzheimer’s disease therapeutics. Transl. Neurodegener. 2023, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Sangineto, M.; Ciarnelli, M.; Cassano, T.; Radesco, A.; Moola, A.; Bukke, V.N.; Romano, A.; Villani, R.; Kanwal, H.; Capitanio, N.; et al. Metabolic reprogramming in inflammatory microglia indicates a potential way of targeting inflammation in Alzheimer’s disease. Redox Biol. 2023, 66, 102846. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.P.; York, E.M.; MacVicar, B.A. Immunometabolism in the brain: How metabolism shapes microglial function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Herrera, O.M.; Hong, B.V.; Ruiz Mendiola, U.; Maezawa, I.; Jin, L.W.; Lebrilla, C.B.; Harvey, D.J.; Zivkovic, A.M. Cholesterol, amyloid beta, fructose, and LPS influence ROS and ATP concentrations and the phagocytic capacity of HMC3 human microglia cell line. Int. J. Mol. Sci. 2023, 24, 10396. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-κB signalling. Br. J. Nutr. 2012, 107, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Medrano-Jiménez, E.; Jiménez-Ferrer Carrillo, I.; Pedraza-Escalona, M.; Ramírez-Serrano, C.E.; Álvarez-Arellano, L.; Cortés-Mendoza, J.; Herrera-Ruiz, M.; Jiménez-Ferrer, E.; Zamilpa, A.; Tortoriello, J.; et al. Malva parviflora extract ameliorates the deleterious effects of a high fat diet on the cognitive deficit in a mouse model of Alzheimer’s disease by restoring microglial function via a PPAR-γ-dependent mechanism. J. Neuroinflamm. 2019, 16, 143. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Basu, R.; Chatterjee, D.; Templin, A.T.; Flak, J.N.; Johnson, T.S. Disease-associated astrocytes and microglia markers are upregulated in mice fed high fat diet. Sci. Rep. 2023, 13, 12919. [Google Scholar] [CrossRef] [PubMed]
- Desale, S.E.; Chinnathambi, S. Phosphoinositides signaling modulates microglial actin remodeling and phagocytosis in Alzheimer’s disease. Cell Commun. Signal 2021, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Franco-Bocanegra, D.K.; McAuley, C.; Nicoll, J.A.R.; Boche, D. Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease. Cells 2019, 8, 639. [Google Scholar] [CrossRef]
- Uhlemann, R.; Gertz, K.; Boehmerle, W.; Schwarz, T.; Nolte, C.; Freyer, D.; Kettenmann, H.; Endres, M.; Kronenberg, G. Actin dynamics shape microglia effector functions. Brain Struct. Funct. 2016, 221, 2717–2734. [Google Scholar] [CrossRef] [PubMed]
- Antequera, D.; Vargas, T.; Ugalde, C.; Spuch, C.; Molina, J.A.; Ferrer, I.; Bermejo-Pareja, F.; Carro, E. Cytoplasmic gelsolin increases mitochondrial activity and reduces Abeta burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 36, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Silacci, P.; Mazzolai, L.; Gauci, C.; Stergiopulos, N.; Yin, H.L.; Hayoz, D. Gelsolin superfamily proteins: Key regulators of cellular functions. Cell Mol. Life Sci. 2004, 61, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Knöll, B. Serum response factor mediated gene activity in physiological and pathological processes of neuronal motility. Front. Mol. Neurosci. 2011, 4, 49. [Google Scholar] [CrossRef] [PubMed]
- Vartiainen, M.K.; Guettler, S.; Larijani, B.; Treisman, R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 2007, 316, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.S.; Jin, Y.; Graham, D.K.; Witthuhn, B.A.; Ihle, J.N.; Liu, E.T. A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells. J. Biol. Chem. 1995, 270, 25028–25036. [Google Scholar] [CrossRef] [PubMed]
- Verbsky, J.W.; Bach, E.A.; Fang, Y.F.; Yang, L.; Randolph, D.A.; Fields, L.E. Expression of Janus kinase 3 in human endothelial and other non-lymphoid and non-myeloid cells. J. Biol. Chem. 1996, 271, 13976–13980. [Google Scholar] [CrossRef] [PubMed]
- Gurniak, C.B.; Berg, L.J. Murine JAK3 is preferentially expressed in hematopoietic tissues and lymphocyte precursor cells. Blood 1996, 87, 3151–3160. [Google Scholar] [CrossRef]
- Harpur, A.G.; Andres, A.C.; Ziemiecki, A.; Aston, R.R.; Wilks, A.F. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 1992, 7, 1347–1353. [Google Scholar]
- Kawamura, M.; McVicar, D.W.; Johnston, J.A.; Blake, T.B.; Chen, Y.Q.; Lal, B.K.; Lloyd, A.R.; Kelvin, D.J.; Staples, J.E.; Ortaldo, J.R.; et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 6374–6378. [Google Scholar] [CrossRef]
- Rane, S.G.; Reddy, E.P. JAK3: A novel JAK kinase associated with terminal differentiation of hematopoietic cells. Oncogene 1994, 9, 2415–2423. [Google Scholar] [PubMed]
- Agashe, R.P.; Lippman, S.M.; Kurzrock, R. JAK: Not just another kinase. Mol. Cancer Ther. 2022, 21, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Karanki, S.S.; Kumar, N. Identification of molecular switch regulating interactions of Janus kinase 3 with cytoskeletal proteins. J. Biol. Chem. 2012, 287, 41386–41391. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kuang, L.; Villa, R.; Kumar, P.; Mishra, J. Mucosal epithelial jak kinases in health and diseases. Mediat. Inflamm. 2021, 2021, 6618924. [Google Scholar] [CrossRef]
- Mishra, J.; Verma, R.K.; Alpini, G.; Meng, F.; Kumar, N. Role of janus kinase 3 in predisposition to obesity-associated metabolic syndrome. J. Biol. Chem. 2015, 290, 29301–29312. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Chung, J.I.; Kim, A.Y.; Lee, S.Y.; Lee, S.H.; Baik, E.J. Jak kinase 3 signaling in microgliogenesis from the spinal nestin+ progenitors in both development and response to injury. Neuroreport 2017, 28, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wang, Q.; Zhang, R.; Zhang, N. Ketogenic diet attenuates neuroinflammation and induces conversion of M1 microglia to M2 in an EAE model of multiple sclerosis by regulating the NF-κB/NLRP3 pathway and inhibiting HDAC3 and P2X7R activation. Food Funct. 2023, 14, 7247–7269. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Sun, D.; Feng, J.; Tan, W.; Fang, X.; Zhao, M.; Zhao, X.; Pu, Y.; Huang, A.; Xiang, Z.; et al. MSX3 Switches Microglia Polarization and Protects from Inflammation-Induced Demyelination. J. Neurosci. 2015, 35, 6350–6365. [Google Scholar] [CrossRef]
- Ferreira, R.; Lively, S.; Schlichter, L.C. IL-4 type 1 receptor signaling up-regulates KCNN4 expression, and increases the KCa3.1 current and its contribution to migration of alternative-activated microglia. Front. Cell Neurosci. 2014, 8, 183. [Google Scholar] [CrossRef]
- Kumar, P.; Mishra, J.; Kumar, N. Mechanistic role of jak3 in obesity-associated cognitive impairments. Nutrients 2022, 14, 3715. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Momen, Y.; Kumar, P.; Kumar, N. Gut-brain Communication in Pathogenesis of Alzheimer’s Disease. In Proceedings of the PATHOBIOLOGY Mechanisms of Disease, Annual Meeting of the American Society for Investigative Pathology, Baltimore, MD, USA, 20–23 April 2024; Abstract Number 039. ASIP: Baltimore, MD, USA, 2024; p. 42. [Google Scholar]
- Aringer, M.; Hofmann, S.R.; Frucht, D.M.; Chen, M.; Centola, M.; Morinobu, A. Characterization and analysis of the proximal Janus kinase 3 promoter. J. Immunol. 2003, 170, 6057–6064. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Momen, Y.S.; Mishra, J.; Kumar, N. High Fat Diet-Induced Dysregulation of Tyrosine Kinases Is a Novel Player in Gut–Brain Axis in Alzheimer’s Disease. Nutrients 2024, 16, 2558. https://doi.org/10.3390/nu16152558
Momen YS, Mishra J, Kumar N. High Fat Diet-Induced Dysregulation of Tyrosine Kinases Is a Novel Player in Gut–Brain Axis in Alzheimer’s Disease. Nutrients. 2024; 16(15):2558. https://doi.org/10.3390/nu16152558
Chicago/Turabian StyleMomen, Yomna S., Jayshree Mishra, and Narendra Kumar. 2024. "High Fat Diet-Induced Dysregulation of Tyrosine Kinases Is a Novel Player in Gut–Brain Axis in Alzheimer’s Disease" Nutrients 16, no. 15: 2558. https://doi.org/10.3390/nu16152558