
Nature of the Association between Rheumatoid Arthritis and Cervical Cancer and Its Potential Therapeutic Implications



Abstract
:1. Introduction
2. HPV-Induced CC Pathogenesis
3. Epidemiological Association between RA and CC
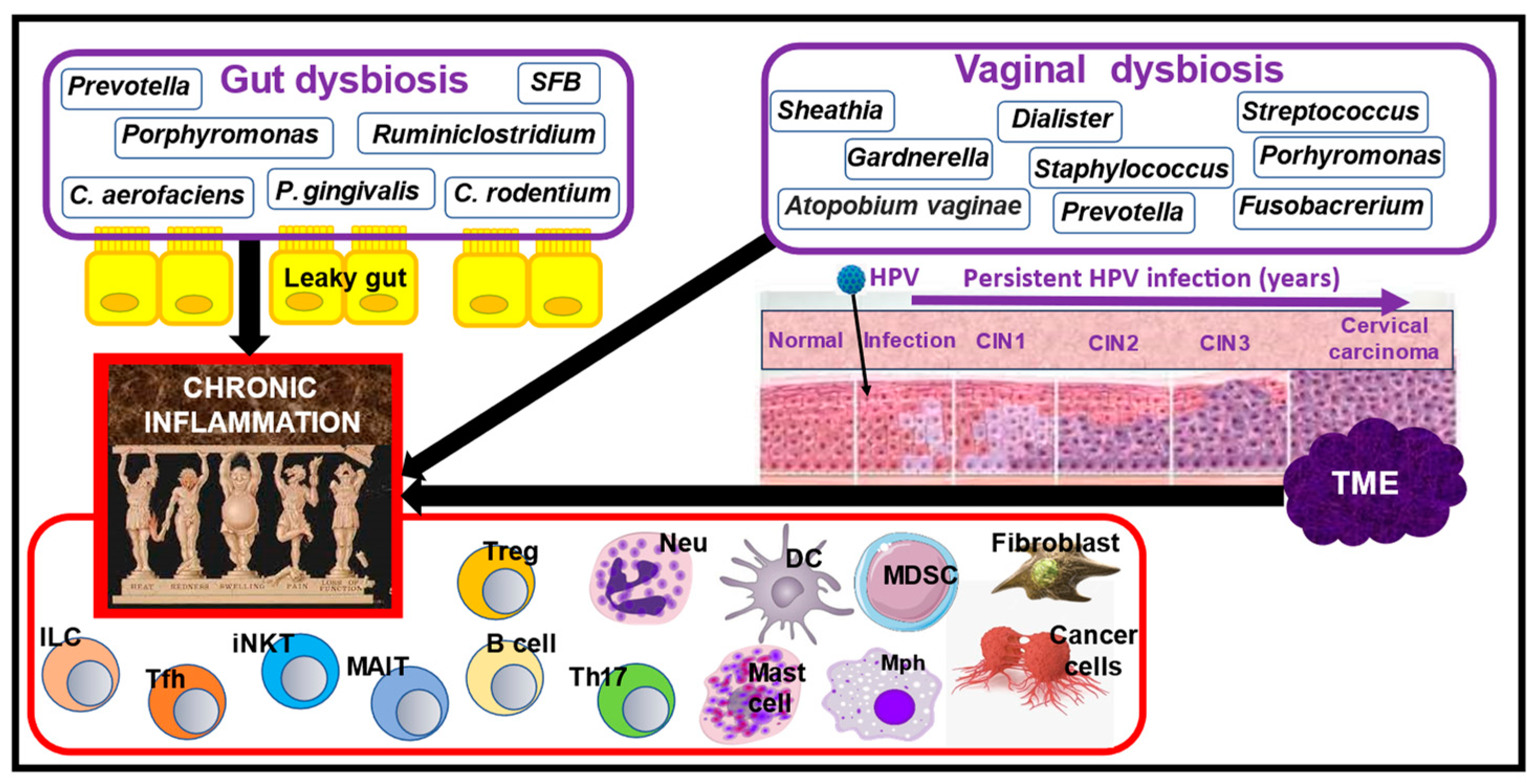
4. Nutrition and the Role of Gut Dysbiosis in RA Manifestations
5. Nutrition and the Role of Vaginal and Gut Dysbiosis in CC Pathogenesis
6. The Role of Chronic Inflammation in RA and CC Pathogenesis
7. Nutrition and the Role of Inflammasome Activation in RA and CC Pathogenesis
8. Nutrition and the Role of the cGAS–STING Signaling Pathway in RA and CC Pathogenesis
9. Failed Resolution of Chronic Inflammation Is a Common Mechanism in RA and CC Pathogenesis
10. Application of Inflammation-Resolving Agents for Attenuating RA and CC Manifestations
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| Acronym | Explanation |
| ATP | Adenosine triphosphate |
| CC | Cervical cancer |
| CDN | Cyclic dinucleotide |
| cGAMP | Cyclic guanosine monophosphate–adenosine monophosphate |
| DCs | Dendritic cells |
| CIN | Cervical intraepithelial neoplasia |
| DMARDs | Disease-modifying antirheumatic drugs |
| dsDNA | Double-stranded DNA |
| FLSs | Fibroblast-like synoviocytes |
| GALT | Gut-associated lymphoid tissue |
| GPCRs | G protein-coupled receptors |
| GTP | Guanosine triphosphate |
| HPV | Human papillomavirus |
| HR | Hazard ratio |
| IFN-I | Interferon type I |
| IRGs | IFN-I response genes |
| JAK-STAT | Janus kinase/signal transducers and activators of transcription |
| LRR | Leucine-rich repeat |
| LxCxE | Leucine-X- cysteine-X-Glutamic acid X is any amino acid |
| MAPK | Mitogen-activated protein kinase |
| MaR | Maresins |
| MDSCs | Myeloid-derived suppressor cells |
| MMPs | Metalloproteinases |
| mtDNA | Mitochondrial DNA |
| NAFLD | Nonalcoholic fatty liver disease |
| NF-kB | Nuclear factor kappa B |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| PUFAs | Polyunsaturated fatty acids |
| RA | Rheumatoid arthritis |
| RvE | E-series resolvins |
| RvD | D-series resolvins |
| SCFAs | Short-chain fatty acids |
| SFs | Synovial fibroblasts |
| SIR | Standardized incidence ratio |
| SNPs | Single nucleotide polymorphisms |
| SPMs | Specialized pro-resolving mediators |
| TAK1 | TGF-β-activated kinase 1 |
| TNFi | Tumor necrosis factor inhibitor |
| TLR | Toll-like receptor |
| TME | Tumor microenvironment |
| URR | Upstream regulatory HPV region |
References
- Beydon, M.; Pinto, S.; De Rycke, Y.; Fautrel, B.; Mariette, X.; Seror, R.; Tubach, F. Risk of Cancer for Patients with Rheumatoid Arthritis versus General Population: A National Claims Database Cohort Study. Lancet Reg. Health-Eur. 2023, 35, 100768. [Google Scholar] [CrossRef]
- De Cock, D.; Hyrich, K. Malignancy and Rheumatoid Arthritis: Epidemiology, Risk Factors and Management. Best Pract. Res. Clin. Rheumatol. 2018, 32, 869–886. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Glynn, R.J.; Giovannucci, E.; Hernández-Díaz, S.; Liu, J.; Feldman, S.; Karlson, E.W.; Schneeweiss, S.; Solomon, D.H. Risk of High-Grade Cervical Dysplasia and Cervical Cancer in Women with Systemic Inflammatory Diseases: A Population-Based Cohort Study. Ann. Rheum. Dis. 2015, 74, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Wadström, H.; Frisell, T.; Sparén, P.; Askling, J. Do RA or TNF Inhibitors Increase the Risk of Cervical Neoplasia or of Recurrence of Previous Neoplasia? A Nationwide Study from Sweden. Ann. Rheum. Dis. 2016, 75, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Lee, H. The Risk of Malignancy in Korean Patients with Rheumatoid Arthritis. Yonsei Med. J. 2019, 60, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Boccardo, E.; Lepique, A.P.; Villa, L.L. The Role of Inflammation in HPV Carcinogenesis. Carcinogenesis 2010, 31, 1905–1912. [Google Scholar] [CrossRef]
- Zhu, M.; Ding, Q.; Lin, Z.; Fu, R.; Zhang, F.; Li, Z.; Zhang, M.; Zhu, Y. New Targets and Strategies for Rheumatoid Arthritis: From Signal Transduction to Epigenetic Aspect. Biomolecules 2023, 13, 766. [Google Scholar] [CrossRef]
- Valle-Mendiola, A.; Gutiérrez-Hoya, A.; Soto-Cruz, I. JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus. Genes 2023, 14, 1141. [Google Scholar] [CrossRef]
- Li, K.; Qiu, J.; Pan, J.; Pan, J.P. Pyroptosis and Its Role in Cervical Cancer. Cancers 2022, 14, 5764. [Google Scholar] [CrossRef]
- Chen, P.-K.; Tang, K.-T.; Chen, D.-Y. The NLRP3 Inflammasome as a Pathogenic Player Showing Therapeutic Potential in Rheumatoid Arthritis and Its Comorbidities: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 626. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, J.; Neuhoff, M.-T.; Hoyler, T.; Noir, E.; Tessier, C.; Sarret, S.; Thorsen, T.N.; Littlewood-Evans, A.; Zhang, J.; Hasan, M.; et al. TNF Leads to MtDNA Release and CGAS/STING-Dependent Interferon Responses That Support Inflammatory Arthritis. Cell Rep. 2021, 37, 109977. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.; Huang, D.; Zhou, Z.; Shi, X.; Wu, M.; Rui, Y.; Su, J.; Zheng, W.; Yu, X. DNA Virus Oncoprotein HPV18 E7 Selectively Antagonizes CGAS-STING-triggered Innate Immune Activation. J. Med. Virol. 2023, 95, e28310. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.M.; Joyce Wu, H.-J.; Mauro, D.; Schett, G.; Ciccia, F. The Gut–Joint Axis in Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2021, 17, 224–237. [Google Scholar] [CrossRef]
- Chang, L.; Qiu, L.; Lei, N.; Zhou, J.; Guo, R.; Gao, F.; Dong, S.; Chen, M.; Wu, F.; Qin, B. Characterization of Fecal Microbiota in Cervical Cancer Patients Associated with Tumor Stage and Prognosis. Front. Cell. Infect. Microbiol. 2023, 13, 1145950. [Google Scholar] [CrossRef]
- Gou, Y.; Zhang, J.; Li, C.; Liu, Y.; Hui, J.; Zhou, R.; Kang, M.; Liu, C.; Wang, B.; Shi, P.; et al. Causal Relationship between Gut Microbiota and Rheumatoid Arthritis: A Two-Sample Mendelian Randomisation Study. Clin. Exp. Rheumatol. 2023, 42, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhao, S.; Tan, Z.; Xu, J.; Lu, Q. Causal Associations between Gut Microbiota and Synovitis–Tenosynovitis: A Two-Sample Mendelian Randomization Study. Front. Microbiol. 2024, 15, 1355725. [Google Scholar] [CrossRef]
- Yang, H. The Causal Correlation between Gut Microbiota Abundance and Pathogenesis of Cervical Cancer: A Bidirectional Mendelian Randomization Study. Front. Microbiol. 2024, 15, 1336101. [Google Scholar] [CrossRef]
- Kong, Y.; Liu, S.; Wang, X.; Qie, R. Associations between Gut Microbiota and Gynecological Cancers: A Bi-Directional Two-Sample Mendelian Randomization Study. Medicine 2024, 103, e37628. [Google Scholar] [CrossRef]
- Philippou, E.; Petersson, S.D.; Rodomar, C.; Nikiphorou, E. Rheumatoid Arthritis and Dietary Interventions: Systematic Review of Clinical Trials. Nutr. Rev. 2021, 79, 410–428. [Google Scholar] [CrossRef]
- Chalif, J.; Wang, H.; Spakowicz, D.; Quick, A.; Arthur, E.K.; O’Malley, D.; Chambers, L.M. The Microbiome and Gynecologic Cancer: Cellular Mechanisms and Clinical Applications. Int. J. Gynecol. Cancer 2024, 34, 317–327. [Google Scholar] [CrossRef]
- Koshiyama, M. The Effects of the Dietary and Nutrient Intake on Gynecologic Cancers. Healthcare 2019, 7, 88. [Google Scholar] [CrossRef]
- Skoczyńska, M.; Świerkot, J. The Role of Diet in Rheumatoid Arthritis. Rheumatology 2018, 56, 259–267. [Google Scholar] [CrossRef]
- Liotti, F.; Marotta, M.; Melillo, R.M.; Prevete, N. The Impact of Resolution of Inflammation on Tumor Microenvironment: Exploring New Ways to Control Cancer Progression. Cancers 2022, 14, 3333. [Google Scholar] [CrossRef]
- Singh, D.; Vignat, J.; Lorenzoni, V.; Eslahi, M.; Ginsburg, O.; Lauby-Secretan, B.; Arbyn, M.; Basu, P.; Bray, F.; Vaccarella, S. Global Estimates of Incidence and Mortality of Cervical Cancer in 2020: A Baseline Analysis of the WHO Global Cervical Cancer Elimination Initiative. Lancet Glob. Health 2023, 11, e197–e206. [Google Scholar] [CrossRef]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human Papillomavirus and Cervical Cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Wilting, S.M.; Steenbergen, R.D.M. Molecular Events Leading to HPV-Induced High Grade Neoplasia. Papillomavirus Res. 2016, 2, 85–88. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef]
- Rosendo-Chalma, P.; Antonio-Véjar, V.; Ortiz Tejedor, J.G.; Ortiz Segarra, J.; Vega Crespo, B.; Bigoni-Ordóñez, G.D. The Hallmarks of Cervical Cancer: Molecular Mechanisms Induced by Human Papillomavirus. Biology 2024, 13, 77. [Google Scholar] [CrossRef]
- Guo, C.; Qu, X.; Tang, X.; Song, Y.; Wang, J.; Hua, K.; Qiu, J. Spatiotemporally Deciphering the Mysterious Mechanism of Persistent HPV-induced Malignant Transition and Immune Remodelling from HPV-infected Normal Cervix, Precancer to Cervical Cancer: Integrating Single-cell RNA-sequencing and Spatial Transcriptome. Clin. Transl. Med. 2023, 13, e1219. [Google Scholar] [CrossRef]
- Graham, S.V. The Human Papillomavirus Replication Cycle, and Its Links to Cancer Progression: A Comprehensive Review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef]
- Starodubtseva, N.L.; Brzhozovskiy, A.G.; Bugrova, A.E.; Kononikhin, A.S.; Indeykina, M.I.; Gusakov, K.I.; Chagovets, V.V.; Nazarova, N.M.; Frankevich, V.E.; Sukhikh, G.T.; et al. Label-free Cervicovaginal Fluid Proteome Profiling Reflects the Cervix Neoplastic Transformation. J. Mass Spectrom. 2019, 54, 693–703. [Google Scholar] [CrossRef]
- Mukherjee, A.; Pednekar, C.B.; Kolke, S.S.; Kattimani, M.; Duraisamy, S.; Burli, A.R.; Gupta, S.; Srivastava, S. Insights on Proteomics-Driven Body Fluid-Based Biomarkers of Cervical Cancer. Proteomes 2022, 10, 13. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of Genomic Alterations in Cervical Carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The Human Papillomavirus Type 16 E7 Oncoprotein Induces a Transcriptional Repressor Complex on the Toll-like Receptor 9 Promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.H.; Wasson, C.W.; Watherston, O.; Doble, R.; Eric Blair, G.; Wittmann, M.; Macdonald, A. The Human Papillomavirus (HPV) E7 Protein Antagonises an Imiquimod-Induced Inflammatory Pathway in Primary Human Keratinocytes. Sci. Rep. 2015, 5, 12922. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses 2020, 12, 977. [Google Scholar] [CrossRef]
- Dugué, P.; Rebolj, M.; Hallas, J.; Garred, P.; Lynge, E. Risk of Cervical Cancer in Women with Autoimmune Diseases, in Relation with Their Use of Immunosuppressants and Screening: Population-based Cohort Study. Int. J. Cancer 2015, 136, E711–E719. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.; Malloy, M.J.; Jokubaitis, V.G.; Wrede, C.D.H.; Butzkueven, H.; Sasadeusz, J.; Van Doornum, S.; Macrae, F.; Unglik, G.; Brotherton, J.M.L.; et al. Increased Risk of Cervical Dysplasia in Females with Autoimmune Conditions—Results from an Australia Database Linkage Study. PLoS ONE 2020, 15, e0234813. [Google Scholar] [CrossRef]
- Simon, T.A.; Thompson, A.; Gandhi, K.K.; Hochberg, M.C.; Suissa, S. Incidence of Malignancy in Adult Patients with Rheumatoid Arthritis: A Meta-Analysis. Arthritis Res. Ther. 2015, 17, 212. [Google Scholar] [CrossRef]
- The American College of Obstetricians and Gynecologists (ACOG). Updated Cervical Cancer Screening Guidelines. Available online: https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2021/04/updated-cervical-cancer-screening-guidelines (accessed on 30 July 2024).
- The American Cancer Society (ACS). The American Cancer Society Guidelines for the Prevention and Early Detection of Cervical Cancer. Available online: https://www.cancer.org/cancer/types/cervical-cancer/detection-diagnosis-staging/cervical-cancer-screening-guidelines.html (accessed on 30 July 2024).
- United States Preventive Services Task Force (USPSTF). Cervical Cancer: Screening. Available online: https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/cervical-cancer-screening (accessed on 30 July 2024).
- Kim, S.C.; Feldman, S.; Moscicki, A.-B. Risk of Human Papillomavirus Infection in Women with Rheumatic Disease: Cervical Cancer Screening and Prevention. Rheumatology 2018, 57, v26–v33. [Google Scholar] [CrossRef] [PubMed]
- Gavinski, K.; DiNardo, D. Cervical Cancer Screening. Med. Clin. N. Am. 2023, 107, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, L.; Kearsley-Fleet, L.; Brown, N.; Watson, K.D.; Lunt, M.; Symmons, D.P.M.; Hyrich, K.L. Cervical Screening Uptake and Rates of Cervical Dysplasia in the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Rheumatology 2020, 59, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Takeda, K. Host–Microbiota Interactions in Rheumatoid Arthritis. Exp. Mol. Med. 2019, 51, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wei, Y.; Zhu, Y.; Xie, Z.; Hai, Q.; Li, Z.; Qin, D. Gut Microbiota and Rheumatoid Arthritis: From Pathogenesis to Novel Therapeutic Opportunities. Front. Immunol. 2022, 13, 1007165. [Google Scholar] [CrossRef]
- Godha, Y.; Kumar, S.; Wanjari, A. Role of Gut Microbiota in the Development and Management of Rheumatoid Arthritis: A Narrative Review. Cureus 2023, 15, e49458. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, B.T.P.; Tadeo, R.Y.T.; Lamacchia, C.; Studer, O.; Courvoisier, D.; Raes, J.; Finckh, A. Gut Micro-biome and Intestinal Inflammation in Preclinical Stages of Rheumatoid Arthritis. RMD Open 2024, 10, e003589. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, B.; Liu, X.; Zhao, J.; Ross, R.P.; Stanton, C.; Zhang, H.; Chen, W. Microbiota-Assisted Therapy for Systemic Inflammatory Arthritis: Advances and Mechanistic Insights. Cell. Mol. Life Sci. 2022, 79, 470. [Google Scholar] [CrossRef]
- Tsetseri, M.-N.; Silman, A.J.; Keene, D.J.; Dakin, S.G. The Role of the Microbiome in Rheumatoid Arthritis: A Review. Rheumatol. Adv. Pract. 2023, 7, rkad034. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, K.; Xiong, Q.; Zhang, J.; Cai, B.; Huang, Z.; Yang, B.; Wei, B.; Chen, J.; Niu, Q. Gut Microbiota in Pre-Clinical Rheumatoid Arthritis: From Pathogenesis to Preventing Progression. J. Autoimmun. 2023, 141, 103001. [Google Scholar] [CrossRef]
- Kalinkovich, A.; Gabdulina, G.; Livshits, G. Autoimmunity, Inflammation, and Dysbiosis Mutually Gov-ern the Transition from the Preclinical to the Clinical Stage of Rheumatoid Arthritis. Immunol. Res. 2018, 66, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Shinebaum, R.; Neumann, V.C.; Cooke, E.M.; Wright, V. Comparison of Faecal Florae in Patients with Rheumatoid Arthritis and Controls. Rheumatology 1987, 26, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Bucci, L.; Schett, G.; Zaiss, M.M. Crossing the Barriers: Revisiting the Gut Feeling in Rheumatoid Arthritis. Eur. J. Immunol. 2021, 51, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Marietta, E.V.; Murray, J.A.; Luckey, D.H.; Jeraldo, P.R.; Lamba, A.; Patel, R.; Luthra, H.S.; Mangalam, A.; Taneja, V. Suppression of Inflammatory Arthritis by Human Gut-Derived Prevotella Histicola in Humanized Mice. Arthritis Rheumatol. 2016, 68, 2878–2888. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Takeda, K. Role of Gut Microbiota in Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Zhang, R.; Wang, X. The Gut-Joint Axis: Genetic Evidence for a Causal Association between Gut Microbiota and Seropositive Rheumatoid Arthritis and Seronegative Rheumatoid Arthritis. Medicine 2024, 103, e37049. [Google Scholar] [CrossRef] [PubMed]
- Mörbe, U.M.; Jørgensen, P.B.; Fenton, T.M.; von Burg, N.; Riis, L.B.; Spencer, J.; Agace, W.W. Human Gut-Associated Lymphoid Tissues (GALT); Diversity, Structure, and Function. Mucosal Immunol. 2021, 14, 793–802. [Google Scholar] [CrossRef]
- Seo, G.-Y.; Giles, D.A.; Kronenberg, M. The Role of Innate Lymphoid Cells in Response to Microbes at Mucosal Surfaces. Mucosal Immunol. 2020, 13, 399–412. [Google Scholar] [CrossRef]
- Van de Wiele, T.; Van Praet, J.T.; Marzorati, M.; Drennan, M.B.; Elewaut, D. How the Microbiota Shapes Rheumatic Diseases. Nat. Rev. Rheumatol. 2016, 12, 398–411. [Google Scholar] [CrossRef]
- Horta-Baas, G.; Romero-Figueroa, M.D.S.; Montiel-Jarquín, A.J.; Pizano-Zárate, M.L.; García-Mena, J.; Ramírez-Durán, N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J. Immunol. Res. 2017, 2017, 4835189. [Google Scholar] [CrossRef]
- Kalinkovich, A.; Livshits, G. A Cross Talk between Dysbiosis and Gut-Associated Immune System Gov-erns the Development of Inflammatory Arthropathies. Semin. Arthritis Rheum. 2019, 49, 474–484. [Google Scholar] [CrossRef]
- Nikiphorou, E.; Philippou, E. Nutrition and Its Role in Prevention and Management of Rheumatoid Arthritis. Autoimmun. Rev. 2023, 22, 103333. [Google Scholar] [CrossRef] [PubMed]
- Schönenberger, K.A.; Schüpfer, A.-C.; Gloy, V.L.; Hasler, P.; Stanga, Z.; Kaegi-Braun, N.; Reber, E. Effect of Anti-Inflammatory Diets on Pain in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Nutrients 2021, 13, 4221. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.N.; Halse, A.-K.; Skeie, E.; Lein, R.K.; Nilsen, R.M.; Tangvik, R.J. Effect of Dietary Interventions on Nutritional Status in Patients with Rheumatoid Arthritis and Spondyloarthritis—A Systematic Re-view and Meta-Analysis. Clin. Nutri. 2024, 43, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Bae, S.-C.; Song, G.-G. Omega-3 Polyunsaturated Fatty Acids and the Treatment of Rheumatoid Arthritis: A Meta-Analysis. Arch. Med. Res. 2012, 43, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Gioxari, A.; Kaliora, A.C.; Marantidou, F.; Panagiotakos, D.P. Intake of ω-3 Polyunsaturated Fatty Acids in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Nutrition 2018, 45, 114–124.e4. [Google Scholar] [CrossRef] [PubMed]
- Tański, W.; Świątoniowska-Lonc, N.; Tabin, M.; Jankowska-Polańska, B. The Relationship between Fatty Acids and the Development, Course and Treatment of Rheumatoid Arthritis. Nutrients 2022, 14, 1030. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert Consensus Document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) Consensus Statement on the Definition and Scope of Prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef]
- Wilkins, T.; Sequoia, J.; Jennings, W.; Dorn, B. Probiotics for Gastrointestinal Conditions: A Summary of the Evidence. Am. Fam. Physician 2017, 96, 170–178. [Google Scholar]
- De Luca, F.; Shoenfeld, Y. The Microbiome in Autoimmune Diseases. Clin. Exp. Immunol. 2018, 195, 74–85. [Google Scholar] [CrossRef]
- Marietta, E.; Mangalam, A.K.; Taneja, V.; Murray, J.A. Intestinal Dysbiosis in, and Enteral Bacterial Therapies for, Systemic Autoimmune Diseases. Front. Immunol. 2020, 11, 573079. [Google Scholar] [CrossRef] [PubMed]
- Horta-Baas, G.; Sandoval-Cabrera, A.; Romero-Figueroa, M.D.S. Modification of Gut Microbiota in Inflammatory Arthritis: Highlights and Future Challenge. Curr. Rheumatol. Rep. 2021, 23, 67. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hong, Q.; Zhang, X.; Liu, Z. Rheumatoid Arthritis and the Intestinal Microbiome: Probiotics as a Potential Therapy. Front. Immunol. 2024, 15, 1331486. [Google Scholar] [CrossRef] [PubMed]
- Aqaeinezhad Rudbane, S.M.; Rahmdel, S.; Abdollahzadeh, S.M.; Zare, M.; Bazrafshan, A.; Mazloomi, S.M. The Efficacy of Probiotic Supplementation in Rheumatoid Arthritis: A Meta-Analysis of Randomized, Controlled Trials. Inflammopharmacology 2018, 26, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Letarouilly, J.-G.; Nguyen, Y.; Sigaux, J.; Barnetche, T.; Czernichow, S.; Flipo, R.-M.; Sellam, J.; Daïen, C. Efficacy of Probiotics in Rheumatoid Arthritis and Spondyloarthritis: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2022, 14, 354. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Li, Y.; Wu, Y.; Liang, X.; Zhou, Y.; Liao, Z.; Wen, J.; Cheng, L.; Luo, Y.; Liu, Y. Effects of Microecological Regulators on Rheumatoid Arthritis: A Systematic Review and Meta-Analysis of Randomized, Controlled Trials. Nutrients 2023, 15, 1102. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Deng, Y.; He, Q.; Yang, K.; Li, J.; Xiang, W.; Liu, H.; Zhu, X.; Chen, H. Safety and Efficacy of Pro-biotic Supplementation in 8 Types of Inflammatory Arthritis: A Systematic Review and Meta-Analysis of 34 Randomized Controlled Trials. Front. Immunol. 2022, 13, 961325. [Google Scholar] [CrossRef]
- Veronese, N.; Solmi, M.; Caruso, M.G.; Giannelli, G.; Osella, A.R.; Evangelou, E.; Maggi, S.; Fontana, L.; Stubbs, B.; Tzoulaki, I. Dietary Fiber and Health Outcomes: An Umbrella Review of Systematic Reviews and Meta-Analyses. Am. J. Clin. Nutr. 2018, 107, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xie, S. Dietary Fiber Intake Associated with Risk of Rheumatoid Arthritis among U.S. Adults: NHANES 2010–2020. Medicine 2023, 102, e33357. [Google Scholar] [CrossRef]
- Nguyen, H.D.T.; Le, T.M.; Lee, E.; Lee, D.; Choi, Y.; Cho, J.; Park, N.J.-Y.; Chong, G.O.; Seo, I.; Han, H.S. Relationship between Human Papillomavirus Status and the Cervicovaginal Microbiome in Cervical Cancer. Microorganisms 2023, 11, 1417. [Google Scholar] [CrossRef]
- Kyrgiou, M.; Mitra, A.; Moscicki, A.-B. Does the Vaginal Microbiota Play a Role in the Development of Cervical Cancer? Transl. Res. 2017, 179, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Sun, H.; Chu, J.; Gong, X.; Liu, X. Cervicovaginal Microbiota: A Promising Direction for Prevention and Treatment in Cervical Cancer. Infect. Agents Cancer 2024, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Bowden, S.J.; Doulgeraki, T.; Bouras, E.; Markozannes, G.; Athanasiou, A.; Grout-Smith, H.; Kechagias, K.S.; Ellis, L.B.; Zuber, V.; Chadeau-Hyam, M.; et al. Risk Factors for Human Papillomavirus Infection, Cervical Intraepithelial Neoplasia and Cervical Cancer: An Umbrella Review and Follow-up Mendelian Randomisation Studies. BMC Med. 2023, 21, 274. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, Z.E.; Łaniewski, P.; Thomas, N.; Roe, D.J.; Chase, D.M.; Herbst-Kralovetz, M.M. Deciphering the Complex Interplay between Microbiota, HPV, Inflammation and Cancer through Cervicovaginal Metabolic Profiling. EBioMedicine 2019, 44, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Ntuli, L.; Mtshali, A.; Mzobe, G.; Liebenberg, L.J.; Ngcapu, S. Role of Immunity and Vaginal Microbiome in Clearance and Persistence of Human Papillomavirus Infection. Front. Cell. Infect. Microbiol. 2022, 12, 927131. [Google Scholar] [CrossRef]
- Zhou, Z.-W.; Long, H.-Z.; Cheng, Y.; Luo, H.-Y.; Wen, D.-D.; Gao, L.-C. From Microbiome to Inflammation: The Key Drivers of Cervical Cancer. Front. Microbiol. 2021, 12, 767931. [Google Scholar] [CrossRef]
- Audirac-Chalifour, A.; Torres-Poveda, K.; Bahena-Román, M.; Téllez-Sosa, J.; Martínez-Barnetche, J.; Cortina-Ceballos, B.; López-Estrada, G.; Delgado-Romero, K.; Burguete-García, A.I.; Cantú, D.; et al. Cervical Microbiome and Cytokine Profile at Various Stages of Cervical Cancer: A Pilot Study. PLoS ONE 2016, 11, e0153274. [Google Scholar] [CrossRef] [PubMed]
- Głowienka-Stodolak, M.; Bagińska-Drabiuk, K.; Szubert, S.; Hennig, E.E.; Horala, A.; Dąbrowska, M.; Micek, M.; Ciebiera, M.; Zeber-Lubecka, N. Human Papillomavirus Infections and the Role Played by Cervical and Cervico-Vaginal Microbiota—Evidence from Next-Generation Sequencing Studies. Cancers 2024, 16, 399. [Google Scholar] [CrossRef] [PubMed]
- Ravilla, R.; Coleman, H.N.; Chow, C.-E.; Chan, L.; Fuhrman, B.J.; Greenfield, W.W.; Robeson, M.S.; Iver-son, K.; Spencer, H.; Nakagawa, M. Cervical Microbiome and Response to a Human Papillomavirus Therapeutic Vaccine for Treating High-Grade Cervical Squamous Intraepithelial Lesion. Integr. Cancer Ther. 2019, 18, 153473541989306. [Google Scholar] [CrossRef]
- Gutiérrez Salmeán, G.; Delgadillo González, M.; Rueda Escalona, A.A.; Leyva Islas, J.A.; Castro-Eguiluz, D. Effects of Prebiotics, Probiotics, and Synbiotics on the Prevention and Treatment of Cervical Cancer: Mexican Consensus and Recommendations. Front. Oncol. 2024, 14, 1383258. [Google Scholar] [CrossRef]
- Mitra, A.; Gultekin, M.; Burney Ellis, L.; Bizzarri, N.; Bowden, S.; Taumberger, N.; Bracic, T.; Vieira-Baptista, P.; Sehouli, J.; Kyrgiou, M. Genital Tract Microbiota Composition Profiles and Use of Prebiotics and Probiotics in Gynaecological Cancer Prevention: Review of the Current Evidence, the European Society of Gynaecological Oncology Prevention Committee Statement. Lancet Microbe 2024, 5, e291–e300. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cao, C.; Ren, Y.; Weng, S.; Liu, L.; Guo, C.; Wang, L.; Han, X.; Ren, J.; Liu, Z. Antitumor Effects of Fecal Microbiota Transplantation: Implications for Microbiome Modulation in Cancer Treatment. Front. Immunol. 2022, 13, 949490. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; An, Y.; Dong, Y.; Chu, Q.; Wei, J.; Wang, B.; Cao, H. Fecal Microbiota Transplantation: No Long-er Cinderella in Tumour Immunotherapy. EBioMedicine 2024, 100, 104967. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Sun, J.; Zhang, G. A Viable Remedy for Overcoming Resistance to Anti-PD-1 Immunotherapy: Fecal Microbiota Transplantation. Crit. Rev. Oncol. Hematol. 2024, 200, 104403. [Google Scholar] [CrossRef]
- Meng, Y.; Sun, J.; Zhang, G. Vaginal Microbiota Transplantation Is a Truly Opulent and Promising Edge: Fully Grasp Its Potential. Front. Cell. Infect. Microbiol. 2024, 14, 1280636. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Peng, L.; Zheng, W.; Huang, F.; Zhang, N.; Wu, D.; Yang, Y. Fecal Microbiota Transplantation for Rheumatoid Arthritis: A Case Report. Clin. Case Rep. 2021, 9, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chu, J.; Feng, S.; Guo, C.; Xue, B.; He, K.; Li, L. Immunological Mechanisms of Inflammatory Diseases Caused by Gut Microbiota Dysbiosis: A Review. Biomed. Pharmacother. 2023, 164, 114985. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid Arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Falconer, J.; Murphy, A.N.; Young, S.P.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. The Immunology of Rheumatoid Arthritis. Nat. Immunol. 2021, 22, 10–18. [Google Scholar] [CrossRef]
- Ding, Q.; Hu, W.; Wang, R.; Yang, Q.; Zhu, M.; Li, M.; Cai, J.; Rose, P.; Mao, J.; Zhu, Y.Z. Signaling Path-ways in Rheumatoid Arthritis: Implications for Targeted Therapy. Signal Transduct. Target. Ther. 2023, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Liu, N.; Sigdel, K.R.; Duan, L. Role of NLRP3 Inflammasome in Rheumatoid Arthritis. Front. Immu. 2022, 13, 931690. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lin, W.; Kuang, Y.; Wang, J.; Xu, S.; Shen, C.; Qiu, Q.; Shi, M.; Xiao, Y.; Liang, L.; et al. CGAS/STING Signaling in the Regulation of Rheumatoid Synovial Aggression. Ann. Transl. Med. 2022, 10, 431. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; Maini, R.N.; Feldmann, M. Role of Pro-Inflammatory Cytokines in Rheumatoid Arthritis. In Springer Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 1998; Volume 20, pp. 133–147. [Google Scholar] [CrossRef]
- Livshits, G.; Kalinkovich, A. Hierarchical, Imbalanced pro-Inflammatory Cytokine Networks Govern the Pathogenesis of Chronic Arthropathies. Osteoarthr. Cartil. 2018, 26, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.V.; De Medeiros Fernandes, T.A.A.; De Azevedo, J.C.V.; Cobucci, R.N.O.; De Carvalho, M.G.F.; Andrade, V.S.; De Araújo, J.M.G. Link between Chronic Inflammation and Human Papillomavirus-Induced Carcinogenesis (Review). Oncol. Lett. 2015, 9, 1015–1026. [Google Scholar] [CrossRef]
- Parida, S.; Mandal, M. Inflammation Induced by Human Papillomavirus in Cervical Cancer and Its Implication in Prevention. Eur. J. Cancer Prev. 2014, 23, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Sales, K.J.; Katz, A.A.; Howard, B.; Soeters, R.P.; Millar, R.P.; Jabbour, H.N. Cyclooxygenase-1 is up-regulated in cervical carcinomas: Autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin e receptors, and angiogenic factors by cyclooxygenase-1. Cancer Res. 2002, 62, 424–432. [Google Scholar] [PubMed]
- García-Quiroz, J.; Vázquez-Almazán, B.; García-Becerra, R.; Díaz, L.; Avila, E. The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics. Cells 2022, 11, 2528. [Google Scholar] [CrossRef]
- Trujillo-Cirilo, L.; Weiss-Steider, B.; Vargas-Angeles, C.A.; Corona-Ortega, M.T.; Rangel-Corona, R. Im-mune Microenvironment of Cervical Cancer and the Role of IL-2 in Tumor Promotion. Cytokine 2023, 170, 156334. [Google Scholar] [CrossRef]
- Vitkauskaite, A.; Urboniene, D.; Celiesiute, J.; Jariene, K.; Skrodeniene, E.; Nadisauskiene, R.J.; Vaitkiene, D. Circulating Inflammatory Markers in Cervical Cancer Patients and Healthy Controls. J. Immunotoxicol. 2020, 17, 105–109. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Caseley, E.A.; Poulter, J.A.; Rodrigues, F.; Caseley, E.A.; Poulter, J.A.; McDermott, M.F. Inflammasome Inhibition under Physiological and Pharmacological Conditions. Genes Immun. 2020, 21, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, X.; Huang, E.; Wang, Q.; Wen, C.; Yang, G.; Lu, L.; Cui, D. Inflammasome and Its Therapeutic Targeting in Rheumatoid Arthritis. Front. Immunol. 2022, 12, 816839. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Spano, M.; Di Matteo, G.; Ingallina, C.; Ambroselli, D.; Carradori, S.; Gallorini, M.; Giusti, A.M.; Salvo, A.; Grosso, M.; Mannina, L. Modulatory Properties of Food and Nutraceutical Components Targeting NLRP3 Inflammasome Activation. Nutrients 2022, 14, 490. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Zhao, C.; Gu, Y.; Zeng, X.; Wang, J. NLRP3 Inflammasome Regulates Th17 Differentiation in Rheumatoid Arthritis. Clin. Immunol. 2018, 197, 154–160. [Google Scholar] [CrossRef]
- Tenshin, H.; Teramachi, J.; Ashtar, M.; Hiasa, M.; Inoue, Y.; Oda, A.; Tanimoto, K.; Shimizu, S.; Higa, Y.; Harada, T.; et al. TGF-β-activated Kinase-1 Inhibitor LL-Z1640-2 Reduces Joint Inflammation and Bone Destruction in Mouse Models of Rheumatoid Arthritis by Inhibiting NLRP3 Inflammasome, TACE, TNF-α and RANKL Expression. Clin. Transl. Immunol. 2022, 11, e1371. [Google Scholar] [CrossRef]
- Mathews, R.J.; Robinson, J.I.; Battellino, M.; Wong, C.; Taylor, J.C.; Eyre, S.; Churchman, S.M.; Wilson, A.G.; Isaacs, J.D.; Hyrich, K.; et al. Evidence of NLRP3-Inflammasome Activation in Rheumatoid Arthritis (RA); Genetic Variants within the NLRP3-Inflammasome Complex in Relation to Susceptibility to RA and Response to Anti-TNF Treatment. Ann. Rheum. Dis. 2014, 73, 1202–1210. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, P.; Zhang, Y.; Wei, M.; Tian, T.; Zhu, D.; Guan, Y.; Wei, W.; Ma, Y. The Research Progression of Direct NLRP3 Inhibitors to Treat Inflammatory Disorders. Cell Immunol. 2024, 397–398, 104810. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Zeiser, R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 2020, 11, 01444. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Bricher, P.; Leal, V.N.C.; Lima, S.; Souza, P.R.E.; Crovella, S. Role of Inflammasome Genetics in Susceptibility to HPV Infection and Cervical Cancer Development. J. Med. Virol. 2016, 88, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Reinholz, M.; Kawakami, Y.; Salzer, S.; Kreuter, A.; Dombrowski, Y.; Koglin, S.; Kresse, S.; Ruzicka, T.; Schauber, J. HPV16 Activates the AIM2 Inflammasome in Keratinocytes. Arch. Dermatol. Res. 2013, 305, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, J.A.; da Silva, M.I.F.; de Moura, P.M.M.F.; Leitão, M.d.C.G.; Coimbra, E.C. Reduced Expression of IL-1β and IL-18 Proinflammatory Interleukins Increases the Risk of Developing Cervical Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 2715–2721. [Google Scholar] [CrossRef] [PubMed]
- So, D.; Shin, H.-W.; Kim, J.; Lee, M.; Myeong, J.; Chun, Y.-S.; Park, J.-W. Cervical Cancer Is Addicted to SIRT1 Disarming the AIM2 Antiviral Defense. Oncogene 2018, 37, 5191–5204. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, X.; Xu, Y.; Zhu, J.; Li, J.; Zou, Z.; Chen, L.; Zhang, B.; Hua, C.; Rui, H.; et al. HPV E7 Inhibits Cell Pyroptosis by Promoting TRIM21-Mediated Degradation and Ubiquitination of the IFI16 Inflammasome. Int. J. Biol. Sci. 2020, 16, 2924–2937. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-S.; Kang, J.-W.; Cho, M.; Cho, C.-W.; Lee, S.; Choe, Y.-K.; Kim, Y.; Choi, I.; Park, S.-N.; Kim, S.; et al. Down Modulation of IL-18 Expression by Human Papillomavirus Type 16 E6 Oncogene via Binding to IL-18. FEBS Lett. 2001, 501, 139–145. [Google Scholar] [CrossRef]
- García-Closas, R.; Castellsagué, X.; Bosch, X.; González, C.A. The Role of Diet and Nutrition in Cervical Carcinogenesis: A Review of Recent Evidence. Int. J. Cancer 2005, 117, 629–637. [Google Scholar] [CrossRef]
- Medina-Contreras, O.; Luvián-Morales, J.; Valdez-Palomares, F.; Flores-Cisneros, L.; Sánchez-López, M.; Soto-Lugo, J.H.; Castro-Eguiluz, D. Immunonutrition in Cervical Cancer: Immune Response Modulation by Diet. Rev. Investig. Clínica 2020, 72, 219–230. [Google Scholar] [CrossRef]
- Naresh, A.; Hagensee, M.; Myers, L.; Cameron, J. Association of Diet Quality and Dietary Components with Clinical Resolution of HPV. Nutr. Cancer 2021, 73, 2579–2588. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Fu, Q.; Tseng, T.; Zhu, X.; Reiss, K.; Joseph Su, L.; Hagensee, M.E. Impact of Dietary Quality on Genital Oncogenic Human Papillomavirus Infection in Women. J. Infect. Dis. 2023, 228, 1385–1393. [Google Scholar] [CrossRef]
- Maugeri, A.; Barchitta, M.; Magnano San Lio, R.; Scalisi, A.; Agodi, A. Antioxidant and Inflammatory Potential of Diet among Women at Risk of Cervical Cancer: Findings from a Cross-Sectional Study in Italy. Public Health Nutr. 2022, 25, 1577–1585. [Google Scholar] [CrossRef]
- Frega, A.; Gentili, C.; Proietti, S.; Lepore, E.; Unfer, V.; Fuso, A. Epigallocatechin Gallate, Folic Acid, Vitamin B12, and Hyaluronic Acid Significantly Increase Apoptosis and P53 Expression in HeLa Cells. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 5240–5245. [Google Scholar] [PubMed]
- Hewavisenti, R.V.; Arena, J.; Ahlenstiel, C.L.; Sasson, S.C. Human Papillomavirus in the Setting of Immunodeficiency: Pathogenesis and the Emergence of next-Generation Therapies to Reduce the High As-sociated Cancer Risk. Front. Immunol. 2023, 14, 1112513. [Google Scholar] [CrossRef]
- Tomita, L.Y.; Horta, B.L.; da Silva, L.L.S.; Malta, M.B.; Franco, E.L.; Cardoso, M.A. Fruits and Vegetables and Cervical Cancer: A Systematic Review and Meta-Analysis. Nutr. Cancer 2021, 73, 62–74. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Zhao, K.; Zhou, Y.; Li, W.; Pan, C.; Qiang, L.; Li, Z.; Lu, N. Small Molecule GL-V9 Protects against Colitis-Associated Colorectal Cancer by Limiting NLRP3 Inflammasome through Autophagy. Oncoimmunology 2018, 7, e1375640. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of CGAS–STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Gong, J.; Gao, X.; Ge, S.; Li, H.; Wang, R.; Zhao, L. The Role of CGAS-STING Signalling in Metabolic Diseases: From Signalling Networks to Targeted Intervention. Int. J. Biol. Sci. 2024, 20, 152–174. [Google Scholar] [CrossRef]
- Mao, Y.; Luo, W.; Zhang, L.; Wu, W.; Yuan, L.; Xu, H.; Song, J.; Fujiwara, K.; Abe, J.; LeMaire, S.A.; et al. STING–IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arter. Thromb. Vasc. Biol. 2017, 37, 920–929. [Google Scholar] [CrossRef]
- An, C.; Li, Z.; Chen, Y.; Huang, S.; Yang, F.; Hu, Y.; Xu, T.; Zhang, C.; Ge, S. The CGAS-STING Pathway in Cardiovascular Diseases: From Basic Research to Clinical Perspectives. Cell Biosci. 2024, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cervantes, C.; He, S.; He, J.; Plasko, G.R.; Wen, J.; Li, Z.; Yin, D.; Zhang, C.; Liu, M.; et al. Mitochondrial Stress-Activated CGAS-STING Pathway Inhibits Thermogenic Program and Contributes to Over-nutrition-Induced Obesity in Mice. Commun. Biol. 2020, 3, 257. [Google Scholar] [CrossRef] [PubMed]
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Sfikakis, P.P. DNA Damage Accumulation, Defective Chromatin Organization and Deficient DNA Repair Capacity in Patients with Rheumatoid Arthritis. Clin. Immunol. 2019, 203, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Shao, L. DNA Damage Response Signals Transduce Stress From Rheumatoid Arthritis Risk Factors Into T Cell Dysfunction. Front. Immunol. 2018, 9, 3055. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Goronzy, J.J.; Weyand, C.M. DNA Damage, Metabolism and Aging in pro-Inflammatory T Cells. Exp. Gerontol. 2018, 105, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, R.; Lin, H.; Qiu, Q.; Lao, M.; Zeng, S.; Wang, C.; Xu, S.; Zou, Y.; Shi, M.; et al. Accumulation of Cytosolic DsDNA Contributes to Fibroblast-like Synoviocytes-Mediated Rheumatoid Arthritis Synovial Inflammation. Int. Immunopharmacol. 2019, 76, 105791. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Yoshida, K.; Hashiramoto, A.; Matsui, K. Cell-Free DNA in Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 8941. [Google Scholar] [CrossRef] [PubMed]
- Skopelja-Gardner, S.; An, J.; Elkon, K.B. Role of the CGAS–STING Pathway in Systemic and Organ-Specific Diseases. Nat. Rev. Nephrol. 2022, 18, 558–572. [Google Scholar] [CrossRef]
- Chen, Z.; Bozec, A.; Ramming, A.; Schett, G. Anti-Inflammatory and Immune-Regulatory Cytokines in Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2019, 15, 9–17. [Google Scholar] [CrossRef]
- van Holten, J.; Reedquist, K.; Sattonet-Roche, P.; Smeets, T.J.; Plater-Zyberk, C.; Vervoordeldonk, M.J.; Tak, P.P. Treatment with Recombinant Interferon-β Reduces Inflammation and Slows Cartilage Destruction in the Collagen-Induced Arthritis Model of Rheumatoid Arthritis. Arthritis Res. Ther. 2004, 6, R239. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Delgado, J.E.; Bastián-Hernandez, Y.; Macias-Segura, N.; Santiago-Algarra, D.; Castillo-Ortiz, J.D.; Alemán-Navarro, A.L.; Martínez-Tejada, P.; Enciso-Moreno, L.; Garcia-De Lira, Y.; Olguín-Calderón, D.; et al. Type I Interferon Gene Response Is Increased in Early and Established Rheumatoid Arthritis and Correlates with Autoantibody Production. Front. Immunol. 2017, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.M.A.; Isaacs, J.D.; Cooles, F.A.H. Role of IFN-α in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2024, 26, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Lübbers, J.; Brink, M.; van de Stadt, L.A.; Vosslamber, S.; Wesseling, J.G.; van Schaardenburg, D.; Rantapää-Dahlqvist, S.; Verweij, C.L. The Type I IFN Signature as a Biomarker of Preclinical Rheumatoid Arthritis. Ann. Rheum. Dis. 2013, 72, 776–780. [Google Scholar] [CrossRef]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon Target-Gene Expression and Epigenomic Signatures in Health and Disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.S.; Taylor, P.C.; Choy, E.H.; Sebba, A.; Quebe, A.; Knopp, K.L.; Porreca, F. The Jak/STAT Pathway: A Focus on Pain in Rheumatoid Arthritis. Semin. Arthritis Rheum. 2021, 51, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Wampler Muskardin, T.; Vashisht, P.; Dorschner, J.M.; Jensen, M.A.; Chrabot, B.S.; Kern, M.; Curtis, J.R.; Danila, M.I.; Cofield, S.S.; Shadick, N.; et al. Increased Pretreatment Serum IFN-β/α Ratio Predicts Non-Response to Tumour Necrosis Factor α Inhibition in Rheumatoid Arthritis. Ann. Rheum. Dis. 2016, 75, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Yarilina, A.; Ivashkiv, L.B. Type I Interferon: A New Player in TNF Signaling. TNF Pathophysiol. 2010, 11, 94–104. [Google Scholar]
- Shen, H.; Jin, L.; Zheng, Q.; Ye, Z.; Cheng, L.; Wu, Y.; Wu, H.; Jon, T.G.; Liu, W.; Pan, Z.; et al. Synergisti-ally Targeting Synovium STING Pathway for Rheumatoid Arthritis Treatment. Bioact. Mater. 2023, 24, 37–53. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, L.; Pang, Y. CGAS-STING Pathway in Pathogenesis and Treatment of Osteoarthritis and Rheumatoid Arthritis. Front. Immunol. 2024, 15, 1384372. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.F.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A Critical Function for Type I Interferons in Cancer Immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing CGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Huo, L.; Xiao, B.; Ouyang, Y.; Chen, F.; Li, J.; Zheng, X.; Wei, D.; Wu, Y.; Zhang, R.; et al. Activating STING/TBK1 Suppresses Tumor Growth via Degrading HPV16/18 E7 Oncoproteins in Cervical Cancer. Cell Death Differ. 2024, 31, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Kol, A.; Lubbers, J.M.; Terwindt, A.L.J.; Workel, H.H.; Plat, A.; Wisman, G.B.A.; Bart, J.; Nijman, H.W.; De Bruyn, M. Combined STING Levels and CD103+ T Cell Infiltration Have Significant Prognostic Implications for Patients with Cervical Cancer. Oncoimmunology 2021, 10, 1936391. [Google Scholar] [CrossRef]
- Luo, K.; Li, N.; Ye, W.; Gao, H.; Luo, X.; Cheng, B. Activation of Stimulation of Interferon Genes (STING) Signal and Cancer Immunotherapy. Molecules 2022, 27, 4638. [Google Scholar] [CrossRef]
- Huang, C.; Shao, N.; Huang, Y.; Chen, J.; Wang, D.; Hu, G.; Zhang, H.; Luo, L.; Xiao, Z. Overcoming Challenges in the Delivery of STING Agonists for Cancer Immunotherapy: A Comprehensive Review of Strategies and Future Perspectives. Mater. Today Bio 2023, 23, 100839. [Google Scholar] [CrossRef]
- Garland, K.M.; Sheehy, T.L.; Wilson, J.T. Chemical and Biomolecular Strategies for STING Pathway Activation in Cancer Immunotherapy. Chem. Rev. 2022, 122, 5977–6039. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Structural Elucidation and Physiologic Functions of Specialized Pro-Resolving Mediators and Their Receptors. Mol. Asp. Med. 2017, 58, 114–129. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of Inflammation: A New Therapeutic Frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Perretti, M.; Cooper, D.; Dalli, J.; Norling, L.V. Immune Resolution Mechanisms in Inflammatory Arthritis. Nat. Rev. Rheumatol. 2017, 13, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Resolution of Inflammation in Arthritis. Semin. Immunopathol. 2019, 41, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Arnardottir, H.H.; Dalli, J.; Norling, L.V.; Colas, R.A.; Perretti, M.; Serhan, C.N. Resolvin D3 Is Dysregulated in Arthritis and Reduces Arthritic Inflammation. J. Immunol. 2016, 197, 2362–2368. [Google Scholar] [CrossRef]
- Zaninelli, T.H.; Fattori, V.; Verri, W.A. Harnessing Inflammation Resolution in Arthritis: Current Under-standing of Specialized Pro-Resolving Lipid Mediators’ Contribution to Arthritis Physiopathology and Future Perspectives. Front. Physiol. 2021, 12, 729134. [Google Scholar] [CrossRef]
- Sigaux, J.; Bellicha, A.; Buscail, C.; Julia, C.; Flipo, R.-M.; Cantagrel, A.; Laporte, F.; Beal, C.; Boissier, M.-C.; Semerano, L. Serum Fatty Acid Profiles Are Associated with Disease Activity in Early Rheumatoid Arthritis: Results from the ESPOIR Cohort. Nutrients 2022, 14, 2947. [Google Scholar] [CrossRef] [PubMed]
- Murata, M. Inflammation and Cancer. Environ. Health Prev. Med. 2018, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and Tumor Progression: Sig-naling Pathways and Targeted Intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Lavy, M.; Gauttier, V.; Poirier, N.; Barillé-Nion, S.; Blanquart, C. Specialized Pro-Resolving Mediators Mitigate Cancer-Related Inflammation: Role of Tumor-Associated Macrophages and Therapeutic Opportunities. Front. Immunol. 2021, 12, 702785. [Google Scholar] [CrossRef]
- Martínez-Micaelo, N.; González-Abuín, N.; Pinent, M.; Ardévol, A.; Blay, M. Dietary Fatty Acid Compo-sition Is Sensed by the NLRP3 Inflammasome: Omega-3 Fatty Acid (DHA) Prevents NLRP3 Activation in Human Macrophages. Food Funct. 2016, 7, 3480–3487. [Google Scholar] [CrossRef]
- Shen, L.; Yang, Y.; Ou, T.; Key, C.-C.C.; Tong, S.H.; Sequeira, R.C.; Nelson, J.M.; Nie, Y.; Wang, Z.; Boudyguina, E.; et al. Dietary PUFAs Attenuate NLRP3 Inflammasome Activation via Enhancing Macrophage Autophagy. J. Lipid Res. 2017, 58, 1808–1821. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.; Kumar, A.; Pawar, S.V. A Review on Rheumatoid Arthritis Interventions and Current Developments. Curr. Drug Targets 2021, 22, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Sulciner, M.L. Resolution Medicine in Cancer, Infection, Pain and Inflammation: Are We on Track to Address the next Pandemic? Cancer Metastasis Rev. 2023, 42, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Zhou, J.; Zang, Y. Effects of Omega-3 Supplementation on Lipid Metabolism, Inflammation, and Disease Activity in Rheumatoid Arthritis: A Meta-Analysis of Randomized Controlled Trials. Clin. Rheumatol. 2024, 43, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H. Pro-Resolving and Anti-Inflammatory Effects of Resolvins and Protectins in Rheumatoid Arthritis. Inflammopharmacology 2023, 31, 2995–3004. [Google Scholar] [CrossRef] [PubMed]
- Zaninelli, T.H.; Fattori, V.; Saraiva-Santos, T.; Badaro-Garcia, S.; Staurengo-Ferrari, L.; Andrade, K.C.; Artero, N.A.; Ferraz, C.R.; Bertozzi, M.M.; Rasquel-Oliveira, F.; et al. RvD1 Disrupts Nociceptor Neuron and Macrophage Activation and Neuroimmune Communication, Reducing Pain and Inflammation in Gouty Arthritis in Mice. Br. J. Pharmacol. 2022, 179, 4500–4515. [Google Scholar] [CrossRef]
- Allen, B.L.; Montague-Cardoso, K.; Simeoli, R.; Colas, R.A.; Oggero, S.; Vilar, B.; McNaughton, P.A.; Dalli, J.; Perretti, M.; Sher, E.; et al. Imbalance of Proresolving Lipid Mediators in Persistent Allodynia Dissoci-ated from Signs of Clinical Arthritis. Pain 2020, 161, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Battista, M.J.; Schmidt, M.; Garcia, M.; Siepmann, T.; Hasenburg, A.; Anic, K. Efficacy and Safety of Immunotherapy for Cervical Cancer—A Systematic Review of Clinical Trials. Cancers 2022, 14, 441. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, C.A.; Khan, S.F.; Schäfer, G.; Mbatani, N.; Adams, T.; Moodley, J.; Prince, S. Cervical Cancer Therapies: Current Challenges and Future Perspectives. Tumour Virus Res. 2022, 13, 200238. [Google Scholar] [CrossRef]
- Tsuda, N.; Watari, H.; Ushijima, K. Chemotherapy and Molecular Targeting Therapy for Recurrent Cer-vical Cancer. Chin. J. Cancer Res. 2016, 28, 241–253. [Google Scholar] [CrossRef]
- Áyen, Á.; Jiménez Martínez, Y.; Boulaiz, H. Targeted Gene Delivery Therapies for Cervical Cancer. Cancers 2020, 12, 1301. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, M.; Valsecchi, A.A.; Tuninetti, V.; Scotto, G.; Borella, F.; Valabrega, G. Immunotherapy for Cervical Cancer: Are We Ready for Prime Time? Int. J. Mol. Sci. 2022, 23, 3559. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lanqing, G.; Huang, Z.; Xin, X.; Minglin, L.; Fa-hui, L.; Zou, H.; Min, J. T Cell Immunotherapy for Cervical Cancer: Challenges and Opportunities. Front. Immunol. 2023, 14, 1105265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, K.; Long, T.; Long, J.; Li, Y.; Li, J.; Cheng, L. Dietary Fish and Omega-3 Polyunsaturated Fatty Acids Intake and Cancer Survival: A Systematic Review and Meta-Analysis. Crit. Rev. Food Sci. Nutr. 2023, 63, 6235–6251. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins Suppress Tumor Growth and Enhance Cancer Therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Torres, W.; Pérez, J.L.; Díaz, M.P.; D’Marco, L.; Checa-Ros, A.; Carrasquero, R.; Angarita, L.; Gómez, Y.; Chacín, M.; Ramírez, P.; et al. The Role of Specialized Pro-Resolving Lipid Mediators in Inflammation-Induced Carcinogenesis. Int. J. Mol. Sci. 2023, 24, 12623. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Gartung, A.; Yang, J.; Yang, H.; Gilligan, M.M.; Sulciner, M.L.; Bhasin, S.S.; Bielenberg, D.R.; Chang, J.; Schmidt, B.A.; et al. Preoperative Stimulation of Resolution and Inflammation Blockade Eradicates Micrometastases. J. Clin. Investig. 2019, 129, 2964–2979. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Hua, X.; Zhou, J.; Yang, R. Resolvin D1 and E1 Alleviate the Progress of Hepatitis to-ward Liver Cancer in Long-Term Concanavalin A-Induced Mice through Inhibition of NF-ΚB Activity. Oncol. Rep. 2016, 35, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Shao, J.; Zhou, S.; Zhao, Z.; Li, F.; Xiang, R.; Zhao, A.Z.; Pan, J. Inhibition of Lung Cancer Growth and Metastasis by DHA and Its Metabolite, RvD1, through MiR-138-5p/FOXC1 Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 479. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xu, Q.; Yin, G.; Xu, W.; Jiang, H. Resolvin D1 Inhibits the Proliferation of Lipopolysaccha-ride treated HepG2 Hepatoblastoma and PLC/PRF/5 Hepatocellular Carcinoma Cells by Targeting the MAPK Pathway. Exp. Ther. Med. 2018, 16, 3603–3610. [Google Scholar] [CrossRef]
- Shan, K.; Feng, N.; Cui, J.; Wang, S.; Qu, H.; Fu, G.; Li, J.; Chen, H.; Wang, X.; Wang, R.; et al. Resolvin D1 and D2 Inhibit Tumour Growth and Inflammation via Modulating Macrophage Polarization. J. Cell Mol. Med. 2020, 24, 8045–8056. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Wang, L.; Yao, B.; Chen, T.; Li, Q.; Liu, Z.; Liu, R.; Niu, Y.; Song, T.; et al. Resolvin D1 Prevents Epithelial-Mesenchymal Transition and Reduces the Stemness Features of Hepatocellular Carcinoma by Inhibiting Paracrine of Cancer-Associated Fibroblast-Derived COMP. J. Exp. Clin. Cancer Res. 2019, 38, 170. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Lee, H.-N.; Surh, Y.-J. RvD1 Inhibits TNFα-Induced c-Myc Expression in Normal Intestinal Epithelial Cells and Destabilizes Hyper-Expressed c-Myc in Colon Cancer Cells. Biochem. Biophys. Res. Commun. 2018, 496, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Mattoscio, D.; Isopi, E.; Lamolinara, A.; Patruno, S.; Medda, A.; De Cecco, F.; Chiocca, S.; Iezzi, M.; Ro-mano, M.; Recchiuti, A. Resolvin D1 Reduces Cancer Growth Stimulating a Protective Neutrophil-Dependent Recruitment of Anti-Tumor Monocytes. J. Exp. Clin. Cancer Res. 2021, 40, 129. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Specialized Pro-Resolving Mediator Network: An Update on Production and Actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Wu, P.; Li, H.; Jin, S.; Zhou, X.; Li, Y.; Ye, D.; Chen, B.; Wan, J. BML-111, a Lipoxin Receptor Agonist, Modulates the Immune Response and Reduces the Severity of Collagen-Induced Arthritis. Inflamm. Res. 2008, 57, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Norling, L.V.; Headland, S.E.; Dalli, J.; Arnardottir, H.H.; Haworth, O.; Jones, H.R.; Irimia, D.; Serhan, C.N.; Perretti, M. Proresolving and Cartilage-Protective Actions of Resolvin D1 in Inflammatory Arthritis. JCI Insight 2016, 1, e85922. [Google Scholar] [CrossRef]
- Hao, H.; Liu, M.; Wu, P.; Cai, L.; Tang, K.; Yi, P.; Li, Y.; Chen, Y.; Ye, D. Lipoxin A4 and Its Analog Sup-press Hepatocellular Carcinoma via Remodeling Tumor Microenvironment. Cancer Lett. 2011, 309, 85–94. [Google Scholar] [CrossRef]
- Du, Y.; Yang, J.; Su, T.; Shen, Z.; Li, J. Lipid Mediator Lipoxin A4 and Its Analog BML-111 Exert Anti-tumor Effects in Melanoma. Ann. Transl. Med. 2021, 9, 802. [Google Scholar] [CrossRef]
- Dong, T.; Dave, P.; Yoo, E.; Ebright, B.; Ahluwalia, K.; Zhou, E.; Asante, I.; Salimova, M.; Pei, H.; Lin, T.; et al. NAP1051, a Lipoxin A4 Biomimetic Analogue, Demonstrates Antitumor Activity Against the Tumor Microenvironment. Mol. Cancer Ther. 2021, 20, 2384–2397. [Google Scholar] [CrossRef]
- Khanna, S.; Jaiswal, K.S.; Gupta, B. Managing Rheumatoid Arthritis with Dietary Interventions. Front. Nutr. 2017, 4, 301603. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.A.; Michelsen, M.K.; Carpenter, C.L. Modern Diets and the Health of Our Planet: An Investigation into the Environmental Impacts of Food Choices. Nutrients 2023, 15, 692. [Google Scholar] [CrossRef] [PubMed]
- Trilleaud, C.; Gauttier, V.; Biteau, K.; Girault, I.; Belarif, L.; Mary, C.; Pengam, S.; Teppaz, G.; Thepenier, V.; Danger, R.; et al. Agonist Anti-ChemR23 MAb Reduces Tissue Neutrophil Accumulation and Triggers Chronic Inflammation Resolution. Sci. Adv. 2021, 7, eabd1453. [Google Scholar] [CrossRef] [PubMed]
- Lavy, M.; Gauttier, V.; Dumont, A.; Chocteau, F.; Deshayes, S.; Fresquet, J.; Dehame, V.; Girault, I.; Tril-leaud, C.; Neyton, S.; et al. ChemR23 Activation Reprograms Macrophages toward a Less Inflammatory Phenotype and Dampens Carcinoma Progression. Front. Immunol. 2023, 14, 1196731. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source * | N/n | Statistical Indicator |
|---|---|---|
| E. Foster et al., 2020 [39] | 1246/25 (CIN 1) | AHR 1.23 (95% CI 1.05–1.42) |
| H. Wadström et al., 2016 [4] | 44,613/212 (CIN 2+) | HR 1.39 (95: CI 1.16–1.66) |
| P. Dugué et al., 2015 [38] | 56,142/140 (CC) | SIR 1.1 (95% CI 0.9–1.2) |
| M. Beydon et al., 2023 [1] | 189,335/332 (CC) | SIR 1.80 (95% CI 1.62–2.01) |
| S. Kim et al., 2014 [3] | 58,979/818 (CIN 2 + CC) | HR 1.49 (95% CI 1.11–2.01) |
| H. Lee et al., 2019 [5] | 1586/9 (CC) | SIR 3.65 (95% CI 1.65–6.42) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gusakov, K.; Kalinkovich, A.; Ashkenazi, S.; Livshits, G. Nature of the Association between Rheumatoid Arthritis and Cervical Cancer and Its Potential Therapeutic Implications. Nutrients 2024, 16, 2569. https://doi.org/10.3390/nu16152569
Gusakov K, Kalinkovich A, Ashkenazi S, Livshits G. Nature of the Association between Rheumatoid Arthritis and Cervical Cancer and Its Potential Therapeutic Implications. Nutrients. 2024; 16(15):2569. https://doi.org/10.3390/nu16152569
Chicago/Turabian StyleGusakov, Kirill, Alexander Kalinkovich, Shai Ashkenazi, and Gregory Livshits. 2024. "Nature of the Association between Rheumatoid Arthritis and Cervical Cancer and Its Potential Therapeutic Implications" Nutrients 16, no. 15: 2569. https://doi.org/10.3390/nu16152569
APA StyleGusakov, K., Kalinkovich, A., Ashkenazi, S., & Livshits, G. (2024). Nature of the Association between Rheumatoid Arthritis and Cervical Cancer and Its Potential Therapeutic Implications. Nutrients, 16(15), 2569. https://doi.org/10.3390/nu16152569