
Potential Add-On Benefits of Dietary Intervention in the Treatment of Autosomal Dominant Polycystic Kidney Disease
 , , ,
, , ,
Abstract
1. Introduction
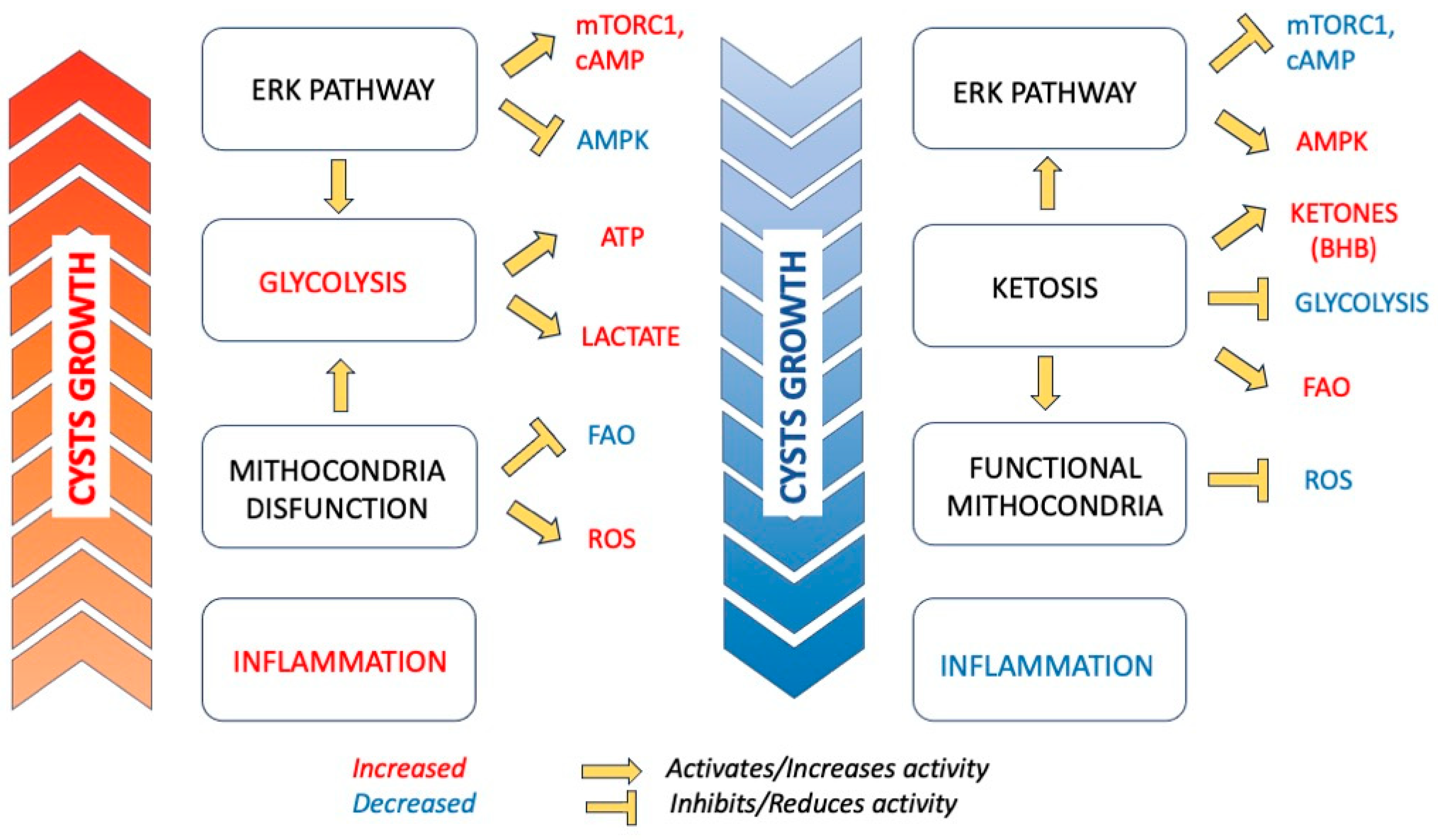
1.1. Metabolic Reprogramming in ADPKD
1.2. Pathophysiologic Rational
2. Preclinical Trials
3. Clinical Trials
4. Side Effects of Ketogenic Diet
4.1. Metabolic Acidosis
4.2. Kidney Stones and Hyperuricemia
4.3. Elevation of Low-Density Lipoprotein Cholesterol (LDL-C)
4.4. “Keto Flu” Symptoms
5. Ketosis-Inducing Therapies: SGLT2 Inhibitors
6. Dietary Counseling in ADPKD
6.1. Protein Intake
6.2. Salt Intake
6.3. Water Intake
6.4. Caffein Intake
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef] [PubMed]
- Gall, E.C.-L.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Gall, E.C.-L.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2018, 29, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.R.; Moore, B.S.; Luo, J.Z.; Sartori, G.; Fang, B.; Jacobs, S.; Abdalla, Y.; Taher, M.; Carey, D.J.; Triffo, W.J.; et al. Exome Sequencing of a Clinical Population for Autosomal Dominant Polycystic Kidney Disease. JAMA 2022, 328, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Mallawaarachchi, A.C.; Lundie, B.; Hort, Y.; Schonrock, N.; Senum, S.R.; Gayevskiy, V.; Minoche, A.E.; Hollway, G.; Ohnesorg, T.; Hinchcliffe, M.; et al. Genomic diagnostics in polycystic kidney disease: An assessment of real-world use of whole-genome sequencing. Eur. J. Hum. Genet. 2021, 29, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Gall, E.C.-L.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Gall, E.C.-L.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.-P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective Glucose Metabolism in Polycystic Kidney Disease Identifies A Novel Therapeutic Paradigm. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Padovano, V.; Podrini, C.; Boletta, A.; Caplan, M.J. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat. Rev. Nephrol. 2018, 14, 678–687. [Google Scholar] [CrossRef]
- Magistroni, R.; Boletta, A. Defective glycolysis and the use of 2-deoxy-D-glucose in polycystic kidney disease: From animal models to humans. J. Nephrol. 2017, 30, 511–519. [Google Scholar] [CrossRef]
- Pagliarini, R.; Podrini, C. Metabolic Reprogramming and Reconstruction: Integration of Experimental and Computational Studies to Set the Path forward in ADPKD. Front. Med. 2021, 8, 740087. [Google Scholar] [CrossRef] [PubMed]
- Seliger, S.L.; Abebe, K.Z.; Hallows, K.R.; Miskulin, D.C.; Perrone, R.D.; Watnick, T.; Bae, K.T. A Randomized Clinical Trial of Metformin to Treat Autosomal Dominant Polycystic Kidney Disease. Am. J. Nephrol. 2018, 47, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhao, J.; Wu, X.; Zhang, Y.; Li, Q.; Lin, S.; Bai, X.-Y.; Chen, X. The changes in glucose metabolism and cell proliferation in the kidneys of polycystic kidney disease mini-pig models. Biochem. Biophys. Res. Commun. 2017, 488, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of Aerobic Glycolysis Attenuates Disease Progression in Polycystic Kidney Disease. PLoS ONE 2016, 11, e0146654. [Google Scholar] [CrossRef] [PubMed]
- Menezes, L.F.; Lin, C.-C.; Zhou, F.; Germino, G.G. Fatty Acid Oxidation is Impaired in An Orthologous Mouse Model of Autosomal Dominant Polycystic Kidney Disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef]
- Menezes, L.F.; Zhou, F.; Patterson, A.D.; Piontek, K.B.; Krausz, K.W.; Gonzalez, F.J.; Germino, G.G. Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4α as a disease modifier. PLoS Genet. 2012, 8, 1003053. [Google Scholar] [CrossRef] [PubMed]
- Hajarnis, S.; Lakhia, R.; Yheskel, M.; Williams, D.; Sorourian, M.; Liu, X.; Aboudehen, K.; Zhang, S.; Kersjes, K.; Galasso, R.; et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat. Commun. 2017, 8, 14395. [Google Scholar] [CrossRef] [PubMed]
- Capelli, I.; Lerario, S.; Aiello, V.; Provenzano, M.; Di Costanzo, R.; Squadrani, A.; Vella, A.; Vicennati, V.; Poli, C.; La Manna, G.; et al. Diet and Physical Activity in Adult Dominant Polycystic Kidney Disease: A Review of the Literature. Nutrients 2023, 15, 2621. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell. Biol. 2017, 37, e00337-17. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Wang, W.; Farmer-Bailey, H.; Gitomer, B.; Malaczewski, M.; Klawitter, J.; Jovanovich, A.; Chonchol, M. Vascular Dysfunction, Oxidative Stress, and Inflammation in Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Meijer, E.; Boertien, W.E.; Nauta, F.L.; Bakker, S.J.; van Oeveren, W.; Rook, M.; van der Jagt, E.J.; van Goor, H.; Peters, D.J.; Navis, G.; et al. Association of urinary biomarkers with disease severity in patients with autosomal dominant polycystic kidney disease: A cross-sectional analysis. Am. J. Kidney Dis. 2010, 56, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Karihaloo, A.; Koraishy, F.; Huen, S.C.; Lee, Y.; Merrick, D.; Caplan, M.J.; Somlo, S.; Cantley, L.G. Macrophages promote cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhou, X.; Fan, L.X.; Yao, Y.; Swenson-Fields, K.I.; Gadjeva, M.; Wallace, D.P.; Peters, D.J.; Yu, A.; Grantham, J.J.; et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J. Clin. Investig. 2015, 125, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E. Pro: Tolvaptan delays the progression of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 30–34. [Google Scholar] [CrossRef]
- Watkins, P.B.; Lewis, J.H.; Kaplowitz, N.; Alpers, D.H.; Blais, J.D.; Smotzer, D.M.; Krasa, H.; Ouyang, J.; Torres, V.E.; Czerwiec, F.S.; et al. Clinical Pattern of Tolvaptan-Associated Liver Injury in Subjects with Autosomal Dominant Polycystic Kidney Disease: Analysis of Clinical Trials Database. Drug Saf. 2015, 38, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, B.; Torra, R. Dietary Aspects and Drug-Related Side Effects in Autosomal Dominant Polycystic Kidney Disease Progression. Nutrients 2022, 14, 4651. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; You, Z.; Gitomer, B.; Brosnahan, G.; Torres, V.E.; Chapman, A.B.; Perrone, R.D.; Steinman, T.I.; Abebe, K.Z.; Rahbari-Oskoui, F.F.; et al. Overweight and Obesity Are Predictors of Progression in Early Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 571–578. [Google Scholar] [CrossRef]
- Nowak, K.L.; Steele, C.; Gitomer, B.; Wang, W.; Ouyang, J.; Chonchol, M.B. Overweight and Obesity and Progression of ADPKD. Clin. J. Am. Soc. Nephrol. 2021, 16, 908–915. [Google Scholar] [CrossRef]
- Nowak, K.L.; Hopp, K. Metabolic Reprogramming in Autosomal Dominant Polycystic Kidney Disease: Evidence and Therapeutic Potential. Clin. J. Am. Soc. Nephrol. 2020, 15, 577–584. [Google Scholar] [CrossRef]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Willett, W.C.; Volek, J.S.; Neuhouser, M.L. Dietary fat: From foe to friend? Science 2018, 362, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Huffman, J.; Kossoff, E.H. State of the ketogenic diet(s) in epilepsy. Curr. Neurol. Neurosci. Rep. 2006, 6, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T.; Saville, J.; Kalantar-Zadeh, K. Ketogenic metabolic therapy for chronic kidney disease—The pro part. Clin. Kidney J. 2024, 17, sfad273. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Morales, P.; Tapia, E.; Pedraza-Chaverri, J. β-Hydroxybutyrate: A signaling metabolite in starvation response? Cell. Signal. 2016, 28, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1437–1447. [Google Scholar] [CrossRef]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef]
- Torres, J.A.; Torres, J.A.; Kruger, S.L.; Kruger, S.L.; Broderick, C.; Broderick, C.; Amarlkhagva, T.; Amarlkhagva, T.; Agrawal, S.; Agrawal, S.; et al. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023.e5. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; Catenacci, V.A.; Dwivedi, N.; Kline, T.L.; Wang, W.; You, Z.; Nguyen, D.T.; Bing, K.; Poudyal, B.; Johnson, G.C.; et al. Weight loss and cystic disease progression in autosomal dominant polycystic kidney disease. iScience 2022, 25, 103697. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Sabatini, D.M. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009, 5, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T.; Olsan, E.E.; Talbot, J.J. Regulation of STATs by polycystin-1 and their role in polycystic kidney disease. JAKSTAT 2013, 2, e23650. [Google Scholar] [CrossRef] [PubMed]
- Bartmann, C.; Raman, S.R.J.; Flöter, J.; Schulze, A.; Bahlke, K.; Willingstorfer, J.; Strunz, M.; Wöckel, A.; Klement, R.J.; Kapp, M.; et al. Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer Metab. 2018, 6, 8. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Testa, F.; Marchiò, M.; Belli, M.; Giovanella, S.; Ligabue, G.; Cappelli, G.; Biagini, G.; Magistroni, R. A pilot study to evaluate tolerability and safety of a modified Atkins diet in ADPKD patients. PharmaNutrition 2019, 9, 100154. [Google Scholar] [CrossRef]
- Strubl, S.; Oehm, S.; Torres, J.A.; Grundmann, F.; Haratani, J.; Decker, M.; Vuong, S.; Bhandal, A.K.; Methot, N.; Haynie-Cion, R.; et al. Ketogenic dietary interventions in autosomal dominant polycystic kidney disease-a retrospective case series study: First insights into feasibility, safety and effects. Clin. Kidney J. 2022, 15, 1079–1092. [Google Scholar] [CrossRef]
- Bruen, D.M.; Kingaard, J.J.; Munits, M.; Paimanta, C.S.; Torres, J.A.; Saville, J.; Weimbs, T. Ren.Nu, a Dietary Program for Individuals with Autosomal-Dominant Polycystic Kidney Disease Implementing a Sustainable, Plant-Focused, Kidney-Safe, Ketogenic Approach with Avoidance of Renal Stressors. Kidney Dial. 2022, 2, 183–203. [Google Scholar] [CrossRef]
- Oehm, S.; Steinke, K.; Schmidt, J.; Arjune, S.; Todorova, P.; Lindemann, C.H.; Wöstmann, F.; Meyer, F.; Siedek, F.; Weimbs, T.; et al. RESET-PKD: A pilot trial on short-term ketogenic interventions in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2023, 38, 1623–1635. [Google Scholar] [CrossRef]
- Cukoski, S.; Lindemann, C.H.; Arjune, S.; Todorova, P.; Brecht, T.; Kühn, A.; Oehm, S.; Strubl, S.; Becker, I.; Kämmerer, U.; et al. Feasibility and impact of ketogenic dietary interventions in polycystic kidney disease: KETO-ADPKD-a randomized controlled trial. Cell Rep. Med. 2023, 4, 101283. [Google Scholar] [CrossRef] [PubMed]
- Testa, F.; Marchiò, M.; D’amico, R.; Giovanella, S.; Ligabue, G.; Fontana, F.; Alfano, G.; Cappelli, G.; Biagini, G.; Magistroni, R. GREASE II. A phase II randomized, 12-month, parallel-group, superiority study to evaluate the efficacy of a Modified Atkins Diet in Autosomal Dominant Polycystic Kidney Disease patients. PharmaNutrition 2020, 13, 100206. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Lockwood, M.B.; Rhee, C.M.; Tantisattamo, E.; Andreoli, S.; Balducci, A.; Laffin, P.; Harris, T.; Knight, R.; Kumaraswami, L.; et al. Patient-centred approaches for the management of unpleasant symptoms in kidney disease. Nat. Rev. Nephrol. 2022, 18, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Cukoski, S.; Kühn, A.; Lindemann, C.H.; Arjune, S.; Meyer, F.; Schömig, T.; Große-Hokamp, N.; Schmidt, J.; Antczak, P.; Weimbs, T.; et al. #2160 Ketosis moderates the effect on kidney volume in dietary interventions for ADPKD—More insights on the KETO ADPKD trial. Nephrol. Dial. Transplant. 2024, 39 (Suppl. S1), gfae069-0738-2160. [Google Scholar] [CrossRef]
- University of Colorado Denver. Daily Caloric Restriction and Intermittent Fasting in Overweight and Obese Adults with Autosomal Dominant Polycystic Kidney Disease. Clinical Trial Registration NCT03342742. 2022. Available online: https://clinicaltrials.gov/study/NCT03342742 (accessed on 1 January 2024).
- University of Colorado Denver. Time Restricted Feeding in Overweight and Obese Adults with Autosomal Dominant Polycystic Kidney Disease. Clinical Trial Registration NCT04534985. 2023. Available online: https://clinicaltrials.gov/study/NCT04534985 (accessed on 1 January 2024).
- University of Colorado Denver. Daily Caloric Restriction in Overweight and Obese Adults with ADPKD. Clinical Trial Registration NCT04907799. 2024. Available online: https://clinicaltrials.gov/study/NCT04907799 (accessed on 1 January 2024).
- Universitair Ziekenhuis Brussel. Treat Autosomal Dominant Polycystic Kidney Disease with Oral Ketone Ester? Clinical Trial Registration NCT06100133. 2023. Available online: https://clinicaltrials.gov/study/NCT06100133 (accessed on 1 January 2024).
- Phinney, S.D.; Bistrian, B.R.; Wolfe, R.R.; Blackburn, G.L. The human metabolic response to chronic ketosis without caloric restriction: Physical and biochemical adaptation. Metabolism 1983, 32, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.J.; Sharma, A.P.; Ross, M.L.; Welvaert, M.; Slater, G.J.; Burke, L.M. Chronic Ketogenic Low Carbohydrate High Fat Diet Has Minimal Effects on Acid-Base Status in Elite Athletes. Nutrients 2018, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arbelaez, D.; Crujeiras, A.B.; Castro, A.I.; Goday, A.; Mas-Lorenzo, A.; Bellon, A.; Tejera, C.; Bellido, D.; Galban, C.; Sajoux, I.; et al. Acid-base safety during the course of a very low-calorie-ketogenic diet. Endocrine 2017, 58, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, S.D.; Zoungas, S.; Caramori, M.L.; Chan, J.C.; Heerspink, H.J.; Hurst, C.; Liew, A.; Michos, E.D.; Olowu, W.A.; Sadusky, T.; et al. Diabetes Management in Chronic Kidney Disease: Synopsis of the KDIGO 2022 Clinical Practice Guideline Update. Ann. Intern. Med. 2023, 176, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L. Metabolic Acidosis in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 263–275. [Google Scholar] [CrossRef]
- Acharya, P.; Acharya, C.; Thongprayoon, C.; Hansrivijit, P.; Kanduri, S.R.; Kovvuru, K.; Medaura, J.; Vaitla, P.; Anton, D.F.G.; Mekraksakit, P.; et al. Incidence and Characteristics of Kidney Stones in Patients on Ketogenic Diet: A Systematic Review and Meta-Analysis. Diseases 2021, 9, 39. [Google Scholar] [CrossRef]
- Kocyigit, I.; Yilmaz, M.I.; Orscelik, O.; Sipahioglu, M.H.; Unal, A.; Eroglu, E.; Kalay, N.; Tokgoz, B.; Axelsson, J.; Oymak, O. Serum uric acid levels and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Nephron Clin. Pract. 2013, 123, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, M.; Dhayat, N.A.; Anderegg, M.; Semmo, M.; Huynh-Do, U.; Vogt, B.; Ferraro, P.M.; Fuster, D.G. Urinary Lithogenic Risk Profile in ADPKD Patients Treated with Tolvaptan. Clin. J. Am. Soc. Nephrol. 2020, 15, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Erickson, S.B.; Smith, L.H.; Wilson, D.M.; Hattery, R.R.; Segura, J.W. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 1988, 11, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Daudon, M.; Cohen-Solal, F.; Lacour, B.; Jungers, P. Urinary stones and urinary tract abnormalities. Is the stone composition independent of the anatomical abnormality? Prog. Urol. 2003, 13, 1320–1329. [Google Scholar]
- Kielb, S.; Koo, H.P.; Bloom, D.A.; Faerber, G.J. Nephrolithiasis associated with the ketogenic diet. J. Urol. 2000, 164, 464–466. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Shi, R.; Patel, J. Risks of the ketogenic diet in CKD—The con part. Clin. Kidney J. 2024, 17, sfad274. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.; Raggi, P. The ketogenic diet: Pros and cons. Atherosclerosis 2020, 292, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Kwiterovich, P.O.; Vining, E.P.G.; Pyzik, P.; Skolasky, R.; Freeman, J.M. Effect of a high-fat ketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteins in children. JAMA 2003, 290, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Groesbeck, D.K.; Bluml, R.M.; Kossoff, E.H. Long-term use of the ketogenic diet in the treatment of epilepsy. Dev. Med. Child. Neurol. 2006, 48, 978–981. [Google Scholar] [CrossRef]
- Falkenhain, K.; Roach, L.A.; McCreary, S.; McArthur, E.; Weiss, E.J.; Francois, M.E.; Little, J.P. Effect of carbohydrate-restricted dietary interventions on LDL particle size and number in adults in the context of weight loss or weight maintenance: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2021, 114, 1455–1466. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Horne, B.; Muhlestein, J.; Lappé, D.; May, H.; Carlquist, J.; Galenko, O.; Brunisholz, K.; Anderson, J. Randomized cross-over trial of short-term water-only fasting: Metabolic and cardiovascular consequences. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Bueno, N.B.; de Melo, I.S.V.; de Oliveira, S.L.; da Rocha Ataide, T. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, J.Z.; Day, A.; Brinkworth, G.D.; Sato, J.; Yamada, S.; Jönsson, T.; Beardsley, J.; Johnson, J.A.; Thabane, L.; Johnston, B.C. Efficacy and safety of low and very low carbohydrate diets for type 2 diabetes remission: Systematic review and meta-analysis of published and unpublished randomized trial data. BMJ 2021, 372, m4743. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, C.F.; Bolick, J.P.; Kris-Etherton, P.M.; Sikand, G.; Aspry, K.E.; Soffer, D.E.; Willard, K.-E.; Maki, K.C. Review of current evidence and clinical recommendations on the effects of low-carbohydrate and very-low-carbohydrate (including ketogenic) diets for the management of body weight and other cardiometabolic risk factors: A scientific statement from the National Lipid Association Nutrition and Lifestyle Task Force. J. Clin. Lipidol. 2019, 13, 689–711.e1. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Hattori, Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: Proposed role of ketone utilization. Heart Fail. Rev. 2021, 26, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-M.; Feng, S.-T.; Wen, Y.; Tang, T.-T.; Wang, B.; Liu, B.-C. Cardiorenal protection of SGLT2 inhibitors-Perspectives from metabolic reprogramming. EBioMedicine 2022, 83, 104215. [Google Scholar] [CrossRef] [PubMed]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419.e6. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, S.; Liu, Y.; Spichtig, D.; Kapoor, S.; Koepsell, H.; Mohebbi, N.; Segerer, S.; Serra, A.L.; Rodriguez, D.; et al. Targeting of sodium-glucose cotransporters with phlorizin inhibits polycystic kidney disease progression in Han:SPRD rats. Kidney Int. 2013, 84, 962–968. [Google Scholar] [CrossRef]
- Rodriguez, D.; Kapoor, S.; Edenhofer, I.; Segerer, S.; Riwanto, M.; Kipar, A.; Yang, M.; Mei, C.; Wüthrich, R.P. Inhibition of Sodium-Glucose Cotransporter 2 with Dapagliflozin in Han: SPRD Rats with Polycystic Kidney Disease. Kidney Blood Press. Res. 2015, 40, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Rodriguez, D.; Riwanto, M.; Edenhofer, I.; Segerer, S.; Mitchell, K.; Wüthrich, R.P. Effect of Sodium-Glucose Cotransport Inhibition on Polycystic Kidney Disease Progression in PCK Rats. PLoS ONE 2015, 10, 0125603. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, W.N.; Song, X.; Kanhai, A.A.; Iliuta, I.-A.; Bozovic, A.; Steinberg, G.R.; Peters, D.J.; Pei, Y. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine 2019, 47, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- The EMPA-KIDNEY Collaborative Group. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Bankir, L.; Roussel, R.; Bouby, N. Protein- and diabetes-induced glomerular hyperfiltration: Role of glucagon, vasopressin, and urea. Am. J. Physiol. Renal Physiol. 2015, 309, F2–F23. [Google Scholar] [CrossRef] [PubMed]
- Aukema, H.M.; Ogborn, M.R.; Tomobe, K.; Takahashi, H.; Hibino, T.; Holub, B.J. Effects of dietary protein restriction and oil type on the early progression of murine polycystic kidney disease. Kidney Int. 1992, 42, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Klahr, S.; Breyer, J.A.; Beck, G.J.; Dennis, V.W.; Hartman, J.A.; Roth, D.; Steinman, T.I.; Wang, S.R.; Yamamoto, M.E. Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. Modification of Diet in Renal Disease Study Group. J. Am. Soc. Nephrol. 1995, 5, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Kramers, B.J.; Koorevaar, I.W.; Drenth, J.P.; de Fijter, J.W.; Neto, A.G.; Peters, D.J.; Vart, P.; Wetzels, J.F.; Zietse, R.; Gansevoort, R.T.; et al. Salt, but not protein intake, is associated with accelerated disease progression in autosomal dominant polycystic kidney disease. Kidney Int. 2020, 98, 989–998. [Google Scholar] [CrossRef]
- Torres, V.E.; Grantham, J.J.; Chapman, A.B.; Mrug, M.; Bae, K.T.; King, B.F.; Wetzel, L.H.; Martin, D.; Lockhart, M.E.; Bennett, W.M.; et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 640–647. [Google Scholar] [CrossRef]
- Heida, J.E.; Gansevoort, R.T.; Messchendorp, A.L.; Meijer, E.; Casteleijn, N.F.; Boertien, W.E.; Zittema, D.; on behalf of the DIPAK Consortium. Use of the Urine-to-Plasma Urea Ratio to Predict ADPKD Progression. Clin. J. Am. Soc. Nephrol. 2021, 16, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Graudal, N.; Jürgens, G.; Baslund, B.; Alderman, M.H. Compared with usual sodium intake, low- and excessive-sodium diets are associated with increased mortality: A meta-analysis. Am. J. Hypertens. 2014, 27, 1129–1137. [Google Scholar] [CrossRef]
- Cogswell, M.E.; Mugavero, K.; Bowman, B.A.; Frieden, T.R. Dietary Sodium and Cardiovascular Disease Risk--Measurement Matters. N. Engl. J. Med. 2016, 375, 580–586. [Google Scholar] [CrossRef]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R.; Simons-Morton, D.G.; et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef]
- McMahon, E.J.; Campbell, K.L.; Bauer, J.D.; Mudge, D.W. Altered dietary salt intake for people with chronic kidney disease. Cochrane Database Syst. Rev. 2015, 2, CD010070. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Holtkamp, F.A.; Parving, H.-H.; Navis, G.J.; Lewis, J.B.; Ritz, E.; de Graeff, P.A.; de Zeeuw, D. Moderation of dietary sodium potentiates the renal and cardiovascular protective effects of angiotensin receptor blockers. Kidney Int. 2012, 82, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Vegter, S.; Perna, A.; Postma, M.J.; Navis, G.; Remuzzi, G.; Ruggenenti, P. Sodium intake, ACE inhibition, and progression to ESRD. J. Am. Soc. Nephrol. 2012, 23, 165–173. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. Blood pressure in early autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2015, 372, 976–977. [Google Scholar] [CrossRef]
- Amro, O.W.; Paulus, J.K.; Noubary, F.; Perrone, R.D. Low-Osmolar Diet and Adjusted Water Intake for Vasopressin Reduction in Autosomal Dominant Polycystic Kidney Disease: A Pilot Randomized Controlled Trial. Am. J. Kidney Dis. 2016, 68, 882–891. [Google Scholar] [CrossRef]
- Taylor, J.M.; Ptomey, L.; Hamilton-Reeves, J.M.; Sullivan, D.K.; Creed, C.; Carlson, S.E.; Wesson, D.E.; Grantham, J.J.; Gibson, C.A. Experiences and Perspectives of Polycystic Kidney Disease Patients following a Diet of Reduced Osmoles, Protein, and Acid Precursors Supplemented with Water: A Qualitative Study. PLoS ONE 2016, 11, 0161043. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.B.; Torres, V.E.; Perrone, R.D.; Steinman, T.I.; Bae, K.T.; Miller, J.P.; Miskulin, D.C.; Oskoui, F.R.; Masoumi, A.; Hogan, M.C.; et al. The HALT polycystic kidney disease trials: Design and implementation. Clin. J. Am. Soc. Nephrol. 2010, 5, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Abebe, K.Z.; Schrier, R.W.; Perrone, R.D.; Chapman, A.B.; Yu, A.S.; Braun, W.E.; Steinman, T.I.; Brosnahan, G.; Hogan, M.C.; et al. Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease. Kidney Int. 2017, 91, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.-U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Burrowes, J.D.; Byham-Gray, L.D.; Campbell, K.L.; Carrero, J.-J.; Chan, W.; Fouque, D.; Friedman, A.N.; Ghaddar, S.; Goldstein-Fuchs, D.J.; et al. KDOQI Clinical Practice Guideline for Nutrition in CKD: 2020 Update. Am. J. Kidney Dis. 2020, 76 (Suppl. S1), S1–S107. [Google Scholar] [CrossRef] [PubMed]
- Zittema, D.; Boertien, W.E.; van Beek, A.P.; Dullaart, R.P.; Franssen, C.F.; de Jong, P.E.; Meijer, E.; Gansevoort, R.T. Vasopressin, copeptin, and renal concentrating capacity in patients with autosomal dominant polycystic kidney disease without renal impairment. Clin. J. Am. Soc. Nephrol. 2012, 7, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Wang, X.; Qian, Q.; Somlo, S.; Harris, P.C.; Gattone, V.H. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat. Med. 2004, 10, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Nagao, S.; Wallace, D.P.; Belibi, F.A.; Cowley, B.D.; Pelling, J.C.; Grantham, J.J. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003, 63, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Wallace, D.P.; Magenheimer, B.S.; Hempson, S.J.; Grantham, J.J.; Calvet, J.P. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 2004, 279, 40419–40430. [Google Scholar] [CrossRef]
- Grantham, J.J. Therapy for polycystic kidney disease? It’s water, stupid! J. Am. Soc. Nephrol. 2008, 19, 1–7. [Google Scholar] [CrossRef]
- Hopp, K.; Wang, X.; Ye, H.; Irazabal, M.V.; Harris, P.C.; Torres, V.E. Effects of hydration in rats and mice with polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2015, 308, F261–F266. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Nishii, K.; Katsuyama, M.; Kurahashi, H.; Marunouchi, T.; Takahashi, H.; Wallace, D.P. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J. Am. Soc. Nephrol. 2006, 17, 2220–2227. [Google Scholar] [CrossRef] [PubMed]
- Gattone, V.H.; Wang, X.; Harris, P.C.; Torres, V.E. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003, 9, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Nagao, S.; Kasahara, M.; Takahashi, H.; Grantham, J.J. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am. J. Kidney Dis. 1997, 30, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Creed, C.; Winklhofer, F.T.; Grantham, J.J. Water prescription in autosomal dominant polycystic kidney disease: A pilot study. Clin. J. Am. Soc. Nephrol. 2011, 6, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Hamilton-Reeves, J.M.; Sullivan, D.K.; Gibson, C.A.; Creed, C.; Carlson, S.E.; Wesson, D.E.; Grantham, J.J. Diet and polycystic kidney disease: A pilot intervention study. Clin. Nutr. 2017, 36, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Morgenthaler, N.G.; Struck, J.; Alonso, C.; Bergmann, A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin. Chem. 2006, 52, 112–119. [Google Scholar] [CrossRef] [PubMed]
- El-Damanawi, R.; Lee, M.; Harris, T.; Cowley, L.B.; Bond, S.; Pavey, H.; Sandford, R.N.; Wilkinson, I.B.; Frankl, F.E.K.; Hiemstra, T.F. High water vs. ad libitum water intake for autosomal dominant polycystic kidney disease: A randomized controlled feasibility trial. QJM 2020, 113, 258–265. [Google Scholar] [CrossRef]
- Higashihara, E.; Nutahara, K.; Tanbo, M.; Hara, H.; Miyazaki, I.; Kobayashi, K.; Nitatori, T. Does increased water intake prevent disease progression in autosomal dominant polycystic kidney disease? Nephrol. Dial. Transplant. 2014, 29, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- El-Damanawi, R.; Lee, M.; Harris, T.; Mader, L.B.; Bond, S.; Pavey, H.; Sandford, R.N.; Wilkinson, I.B.; Burrows, A.; Woznowski, P.; et al. Randomised controlled trial of high versus ad libitum water intake in patients with autosomal dominant polycystic kidney disease: Rationale and design of the DRINK feasibility trial. BMJ Open 2018, 8, e022859. [Google Scholar]
- Wong, A.T.Y.; Mannix, C.; Grantham, J.J.; Allman-Farinelli, M.; Badve, S.V.; Boudville, N.; Byth, K.; Chan, J.; Coulshed, S.; Edwards, M.E.; et al. Randomised controlled trial to determine the efficacy and safety of prescribed water intake to prevent kidney failure due to autosomal dominant polycystic kidney disease (PREVENT-ADPKD). BMJ Open 2018, 8, e018794. [Google Scholar] [CrossRef]
- A Clinical Trial of Water Therapy for ADPKD. PKD Clinical Studies. Available online: https://clinicalstudies.pkdcure.org/study/a-clinical-trial-of-water-therapy-for-adpkd/ (accessed on 1 January 2024).
- Butcher, R.W.; Sutherland, E.W. Adenosine 3′,5′-phosphate in biological materials. I. Purification and properties of cyclic 3′,5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′,5′-phosphate in human urine. J. Biol. Chem. 1962, 237, 1244–1250. [Google Scholar]
- Belibi, F.A.; Wallace, D.P.; Yamaguchi, T.; Christensen, M.; Reif, G.; Grantham, J.J. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Belibi, F.A.; Reif, G.; Wallace, D.P.; Yamaguchi, T.; Olsen, L.; Li, H.; Helmkamp, G.M.; Grantham, J.J. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004, 66, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Tanner, G.A.; Tanner, J.A. Chronic caffeine consumption exacerbates hypertension in rats with polycystic kidney disease. Am. J. Kidney Dis. 2001, 38, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, K.A.; El Ters, M.; Torres, V.E.; Harris, P.C.; Chapman, A.B.; Mrug, M.; Rahbari-Oskoui, F.F.; Bae, K.T.; Landsittel, D.P.; Bennett, W.M.; et al. Relationship between caffeine intake and autosomal dominant polycystic kidney disease progression: A retrospective analysis using the CRISP cohort. BMC Nephrol. 2018, 19, 378. [Google Scholar] [CrossRef] [PubMed]
- Girardat-Rotar, L.; Puhan, M.A.; Braun, J.; Serra, A.L. Long-term effect of coffee consumption on autosomal dominant polycystic kidneys disease progression: Results from the Suisse ADPKD, a Prospective Longitudinal Cohort Study. J. Nephrol. 2018, 31, 87–94. [Google Scholar] [CrossRef]
- Chieng, D.; Kistler, P.M. Coffee and tea on cardiovascular disease (CVD) prevention. Trends Cardiovasc. Med. 2022, 32, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F. Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics 2008, 5, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Caprio, M.; Watanabe, M.; Cammarata, G.; Feraco, A.; Muscogiuri, G.; Verde, L.; Colao, A.; Savastano, S. Could very low-calorie ketogenic diets turn off low grade inflammation in obesity? Emerging evidence. Crit. Rev. Food Sci. Nutr. 2023, 63, 8320–8336. [Google Scholar] [CrossRef]
- Saslow, L.R.; Jones, L.M.; Sen, A.; Wolfson, J.A.; Diez, H.L.; O’brien, A.; Leung, C.W.; Bayandorian, H.; Daubenmier, J.; Missel, A.L.; et al. Comparing Very Low-Carbohydrate vs DASH Diets for Overweight or Obese Adults with Hypertension and Prediabetes or Type 2 Diabetes: A Randomized Trial. Ann. Fam. Med. 2023, 21, 256–263. [Google Scholar] [CrossRef]
- Saudek, C.D.; Boulter, P.R.; Arky, R.A. The natriuretic effect of glucagon and its role in starvation. J. Clin. Endocrinol. Metab. 1973, 36, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Starvation Ketosis and the Kidney. Am. J. Nephrol. 2021, 52, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Golan, R.; Harman-Boehm, I.; Henkin, Y.; Schwarzfuchs, D.; Rudich, A.; Kovsan, J.; Fiedler, G.M.; Blüher, M.; Stumvoll, M.; et al. Renal function following three distinct weight loss dietary strategies during 2 years of a randomized controlled trial. Diabetes Care 2013, 36, 2225–2232. [Google Scholar] [CrossRef]
- Friedman, A.N.; Chambers, M.; Kamendulis, L.M.; Temmerman, J. Short-term changes after a weight reduction intervention in advanced diabetic nephropathy. Clin. J. Am. Soc. Nephrol. 2013, 8, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Bruci, A.; Tuccinardi, D.; Tozzi, R.; Balena, A.; Santucci, S.; Frontani, R.; Mariani, S.; Basciani, S.; Spera, G.; Gnessi, L.; et al. Very Low-Calorie Ketogenic Diet: A Safe and Effective Tool for Weight Loss in Patients with Obesity and Mild Kidney Failure. Nutrients 2020, 12, 333. [Google Scholar] [CrossRef] [PubMed]
- Athinarayanan, S.J.; Roberts, C.G.; Adams, R.N.; Volk, B.M.; Phinney, S.D.; Volek, J.; Mckenzie, A.L. 410-P: Two-Year (2y) eGFR Slope in People with Type 2 Diabetes (T2D) Receiving a Very Low Carbohydrate Diet (VLCD) Intervention. Diabetes 2023, 72 (Suppl. S1), 410. [Google Scholar] [CrossRef]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 5. Facilitating Positive Health Behaviors and Well-being to Improve Health Outcomes: Standards of Care in Diabetes-2023. Diabetes Care 2023, 46 (Suppl. S1), S68–S96. [Google Scholar] [CrossRef]
- Yancy, W.S.; Olsen, M.K.; Guyton, J.R.; Bakst, R.P.; Westman, E.C. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: A randomized, controlled trial. Ann. Intern. Med. 2004, 140, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Stern, L.; Iqbal, N.; Seshadri, P.; Chicano, K.L.; Daily, D.A.; McGrory, J.; Williams, M.; Gracely, E.J.; Samaha, F.F. The effects of low-carbohydrate versus conventional weight loss diets in severely obese adults: One-year follow-up of a randomized trial. Ann. Intern. Med. 2004, 140, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Westman, E.C.; Yancy, W.S.; Mavropoulos, J.C.; Marquart, M.; McDuffie, J.R. The effect of a low-carbohydrate, ketogenic diet versus a low-glycemic index diet on glycemic control in type 2 diabetes mellitus. Nutr. Metab. 2008, 5, 36. [Google Scholar] [CrossRef]
- Jhee, J.H.; Kee, Y.K.; Park, S.; Kim, H.; Park, J.T.; Han, S.H.; Kang, S.-W.; Yoo, T.-H. High-protein diet with renal hyperfiltration is associated with rapid decline rate of renal function: A community-based prospective cohort study. Nephrol. Dial. Transplant. 2020, 35, 98–106. [Google Scholar] [CrossRef]
- Menon, V.; Kopple, J.D.; Wang, X.; Beck, G.J.; Collins, A.J.; Kusek, J.W.; Greene, T.; Levey, A.S.; Sarnak, M.J. Effect of a very low-protein diet on outcomes: Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) Study. Am. J. Kidney Dis. 2009, 53, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Joshi, S.; Schlueter, R.; Cooke, J.; Brown-Tortorici, A.; Donnelly, M.; Schulman, S.; Lau, W.-L.; Rhee, C.M.; Streja, E.; et al. Plant-Dominant Low-Protein Diet for Conservative Management of Chronic Kidney Disease. Nutrients 2020, 12, 1931. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; Torres, V.E. Recent Advances in the Management of Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-Y.; Ma, T.-L.; Hung, C.-C.; Tian, Y.-C.; Chen, Y.-C.; Yang, C.-W.; Cheng, Y.-C. Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency. Sci. Rep. 2017, 7, 7161. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Soler, N.M.; Li, H.; Pham, J.; Rivera, D.; Ho, P.-Y.; Mancino, V.; Saitta, B.; Hallows, K.R. Metformin improves relevant disease parameters in an autosomal dominant polycystic kidney disease mouse model. Am. J. Physiol. Renal Physiol. 2022, 322, F27–F41. [Google Scholar] [CrossRef] [PubMed]
- Takiar, V.; Nishio, S.; Seo-Mayer, P.; King, J.D., Jr.; Li, H.; Zhang, L.; Karihaloo, A.; Hallows, K.R.; Somlo, S.; Caplan, M.J. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2462–2467. [Google Scholar] [CrossRef]
- McCarty, M.F.; Barroso-Aranda, J.; Contreras, F. Activation of AMP-activated kinase as a strategy for managing autosomal dominant polycystic kidney disease. Med. Hypotheses 2009, 73, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- Perrone, R.D.; Abebe, K.Z.; Watnick, T.J.; Althouse, A.D.; Hallows, K.R.; Lalama, C.M.; Miskulin, D.C.; Seliger, S.L.; Tao, C.; Harris, P.C.; et al. Primary results of the randomized trial of metformin administration in polycystic kidney disease (TAME PKD). Kidney Int. 2021, 100, 684–696. [Google Scholar] [CrossRef]
- Brosnahan, G.M.; Wang, W.; Gitomer, B.; Struemph, T.; George, D.; You, Z.; Nowak, K.L.; Klawitter, J.; Chonchol, M.B. Metformin Therapy in Autosomal Dominant Polycystic Kidney Disease: A Feasibility Study. Am. J. Kidney Dis. 2022, 79, 518–526. [Google Scholar] [CrossRef]

{kind=link}
| Authors, Year of Publication | Intervention | Animal Model | Duration | Findings |
|---|---|---|---|---|
| Warner et al., 2016 [39] | 10–40% DCR versus ad libitum | Pkd1RC/RC mice; Pkd2. WS25/− mice | From 6 wk to 7.5 mo | Reduced cyst area, kidney fibrosis, inflammation, and injury; Improved kidney function; Increased activation of LKB1-AMPK and reduced activation of mTOR-S6K. |
| Kipp et al., 2016 [40] | 23% DCR versus ad libitum | Pkd1cond/cond:Nescre mice | Postnatal wk 5–12 | Reduced cyst growth, proliferation and fibrosis; Maintained renal function; Reduced mTOR activation, not increased activation of LKB1-AMPK. |
| Hopp et al., 2021 [42] | 30% DCR versus ad libitum | Pkd1RC/RC mice | From 3 to 6 mo of age | Reduced cyst area, reduced kidney weight and fibrosis; Increased p-AMPK/AMPK, not reduced p-S6/S6. |
| Hopp et al., 2021 [42] | IMF (80% food restriction 3 d/wk, ad libitum other days) versus ad libitum every day | Pkd1RC/RC mice | From 3 to 6 mo of age | No reduction in cyst area; No reduction in kidney weight and fibrosis. |
| Hopp et al., 2021 [42] | TRF (8 h during the 12 h dark cycle) versus ad libitum | Pkd1RC/RC mice | From 3 to 6 mo of age | No reduction in cyst area; No reduction in kidney weight and fibrosis. |
| Torres et al., 2019 [41] | TRF (8 h during the 12 h dark cycle) versus ad libitum | Han:SPRD rat | Postnatal weeks 3—8 | Reduced cystogenesis and cyst growth; Reduced fibrosis; Reduced activation of mTOR-S6K and STAT3. |
| Torres et al., 2019 [41] | Ketogenic diet | Han:SPRD rat, juvenile and adult and male versus female | 5 wk (age 3–8 wk); (age 8–12 wk) | Reduced kidney weight and cystic indices; Reduced increase in serum creatinine; Reduced activation of mTOR-S6K and STAT3, increased p-AMPK (in the juvenile model); Reduced kidney weight and cystic indices; renal function not affected (in the adult model). |
| Torres et al., 2019 [41] | Acute fasting | Han:SPRD rat; Pkd1cond/cond:Nescre mice, Feline PKD1 models | 48 h fasting with free access to water (Han:SPRD rat); 24 h fasting with free access to water (Pkd1 cond/cond:Nes cre mice); 72 h fasting with free access to water (Feline PKD1 models) | Reduced cystic area and kidney mass (Han:SPRD rat, Feline PKD1 models); No change in kidney mass (Pkd1cond/cond:Nescre mice). |
| Torres et al., 2019 [41] | Ad libitum feed + oral BHB | Han:SPRD rat, juvenile and adult and male versus female | 5 wk | Reduced kidney mass and cystic area; Inhibition of proliferation; Reduced fibrosis; Improved kidney function. |
| Authors, Year of Publication | Intervention | Study Design | Population (N) | Duration | Findings |
|---|---|---|---|---|---|
| Hopp et al., 2021 [42] | 34% DCR versus IMF (80% restriction every other day) | Pilot clinical trial | Adult overweight or obese ADPKD patients (28) | 1 year | Higher adherence with fewer side effects in DCR compared to IMF; Higher weight loss in DCR compared to IMF; Annual change in htTKV qualitatively low versus historical control and correlated with weight loss in both DCR and IMF; No annual change in eGFR in both DCR and IMF; Improved lipid profile only in DCR. |
| Testa et al., 2019 [47] | Modified Atkins diet | Pilot clinical trial | Adult patients with rapidly progressive ADPKD (3) | 3 mo | High rate of overall satisfaction; Increase in total cholesterol levels. |
| Strubl et al., 2022 [48] | Self-initiated KD and/or TRD | Retrospective case series study of self-reported observation and medical data | Adult ADPKD patients (121) | ≥6 mo | Improvement of overall health with both KD or TRD; Amelioration of ADPKD-related symptoms and weight loss more pronounced in KD compared to TRD; Stabilization of eGFR. |
| Bruen et al., 2022 [49] | PFKD including KetoCitra® | Real-life experience clinical trial | Adult ADPKD patients (24) | 12 wk | High rate of adherence and satisfaction; 50% of pts refer amelioration of ADPKD-related symptoms; Weight loss; Improved renal function. |
| Oehm et al., 2023 [50] | 3-day WF or a 14-day KD | Prospective interventional short-term clinical trial | Adult patients with rapidly progressive ADPKD (10) | 3 days (WF) 14 days (KD) | High rate of satisfaction; No change in TKV; Weight loss; Increased levels of total and LDL cholesterol in KD; Increased uric acid levels. |
| Cukoski et al., 2023 [51] | KD versus WF (3 of 14 d) versus ad libitum diet | Exploratory, randomized, open, single-center, three arm dietary intervention study | Adult ADPKD patients (63); randomized to KD (23), WF (21), and ad libitum diet (19) | 12 wk | Increased eGFR in KD group; Significant weight loss only in KD group; Decrease ht-TKV in KD group (p = 0.08); Increased eGFR in KD group (p = 0.00); Increased cholesterol and uric acid levels in KD group. |
| Dietary Intervention | Proposed Mechanism | Available Studies | Conclusions | Status of Clinical Trials |
|---|---|---|---|---|
| Daily caloric restriction | Inhibition of mTOR pathway and activation of AMPK, preventing cell proliferation, fibrosis, and cyst growth. | Preclinical trial [40,41,43] Pilot clinical trial [42] | Difficult long-term adherence or feasibility; Possible recommendation in overweight or obese patients, under medical monitoring. | No published data yet (NCT03342742); Ongoing clinical trial (NCT04907799). |
| Intermittent fasting | Inhibition of mTOR pathway and activation of AMPK, preventing cell proliferation, fibrosis, and cyst growth. | Preclinical trial [42] Pilot clinical trial [42] | Not enough evidence to specifically recommend in ADPKD patients. | No published data yet (NCT03342742). |
| Time-restricted feeding | Reduction in mTOR and STAT3 signaling, preventing cell proliferation, fibrosis, and cyst growth. | Preclinical trial [41,42] | Not enough evidence to specifically recommend in ADPKD patients. | Ongoing clinical trials (NCT04534985). |
| Ketogenic diet | Inhibition of mTOR pathway and activation of AMPK, preventing cell proliferation, fibrosis, and cyst growth. | Preclinical trial [41] Pilot clinical trial [47] Retrospective case series study [48] Real-life clinical trial [49] Prospective interventional short-term clinical trial [50] RCT [51] | Not enough evidence to specifically recommend in ADPKD patients. | No planned clinical trials. |
| BHB | Inhibition of mTOR pathway and activation of AMPK, preventing cell proliferation, fibrosis, and cyst growth. Anti-inflammatory effects. Improvement of mitochondrial efficiency. | Preclinical trial [41] | Not enough evidence to specifically recommend in ADPKD patients. | Not recruiting clinical trial yet (NCT06100133). |
| Reduced protein intake | Reduction in hyperfiltration and vasopressin levels. | Preclinical trial [91], Prospective clinical trial [92], Observational cohort study [93,94], Post hoc analysis of the DIPAK observational data [95] | No specific evidence in ADPKD patients, but in accordance with the current management of CKD. | No planned clinical trials. |
| Reduced salt intake | Decrease in vasopressin secretion, RAAS activation, and blood pressure. | Observational cohort study [93,94], Pilot RCT [104], Qualitative study [105], Post hoc analysis of the HALT-PKD data [107] | No specific evidence in ADPKD patients, but in accordance with the current management of CKD. | No planned clinical trials. |
| High water intake | Reduction in V2 receptor activation and cAMP production. | Preclinical trial [115,116,117,118] Pilot study [119,120], Pilot RCT [104], Prospective clinical trial [123] | No specific evidence in ADPKD patients. Recommendation to reduce water intake as eGFR declines, in line with current CKD management. | Ongoing clinical trials (ANZCTR12614001216606; NCT02933268; NCT03102632). |
| Low caffeine intake | Activation of phosphodiesterases, enhanced hydrolysis of cAMP. | Preclinical trial [130], Retrospective, post hoc analysis of the CRISP cohort [131], Prospective analysis of the Suisse PKD cohort [132] | No evidence in animal models or ADPKD patients. | No planned clinical trials. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosati, E.; Condello, G.; Tacente, C.; Mariani, I.; Tommolini, V.; Calvaruso, L.; Fulignati, P.; Grandaliano, G.; Pesce, F. Potential Add-On Benefits of Dietary Intervention in the Treatment of Autosomal Dominant Polycystic Kidney Disease. Nutrients 2024, 16, 2582. https://doi.org/10.3390/nu16162582
Rosati E, Condello G, Tacente C, Mariani I, Tommolini V, Calvaruso L, Fulignati P, Grandaliano G, Pesce F. Potential Add-On Benefits of Dietary Intervention in the Treatment of Autosomal Dominant Polycystic Kidney Disease. Nutrients. 2024; 16(16):2582. https://doi.org/10.3390/nu16162582
Chicago/Turabian StyleRosati, Erica, Giulia Condello, Chiara Tacente, Ilaria Mariani, Valeria Tommolini, Luca Calvaruso, Pierluigi Fulignati, Giuseppe Grandaliano, and Francesco Pesce. 2024. "Potential Add-On Benefits of Dietary Intervention in the Treatment of Autosomal Dominant Polycystic Kidney Disease" Nutrients 16, no. 16: 2582. https://doi.org/10.3390/nu16162582
APA StyleRosati, E., Condello, G., Tacente, C., Mariani, I., Tommolini, V., Calvaruso, L., Fulignati, P., Grandaliano, G., & Pesce, F. (2024). Potential Add-On Benefits of Dietary Intervention in the Treatment of Autosomal Dominant Polycystic Kidney Disease. Nutrients, 16(16), 2582. https://doi.org/10.3390/nu16162582