

Sulforaphane and Brain Health: From Pathways of Action to Effects on Specific Disorders


Abstract
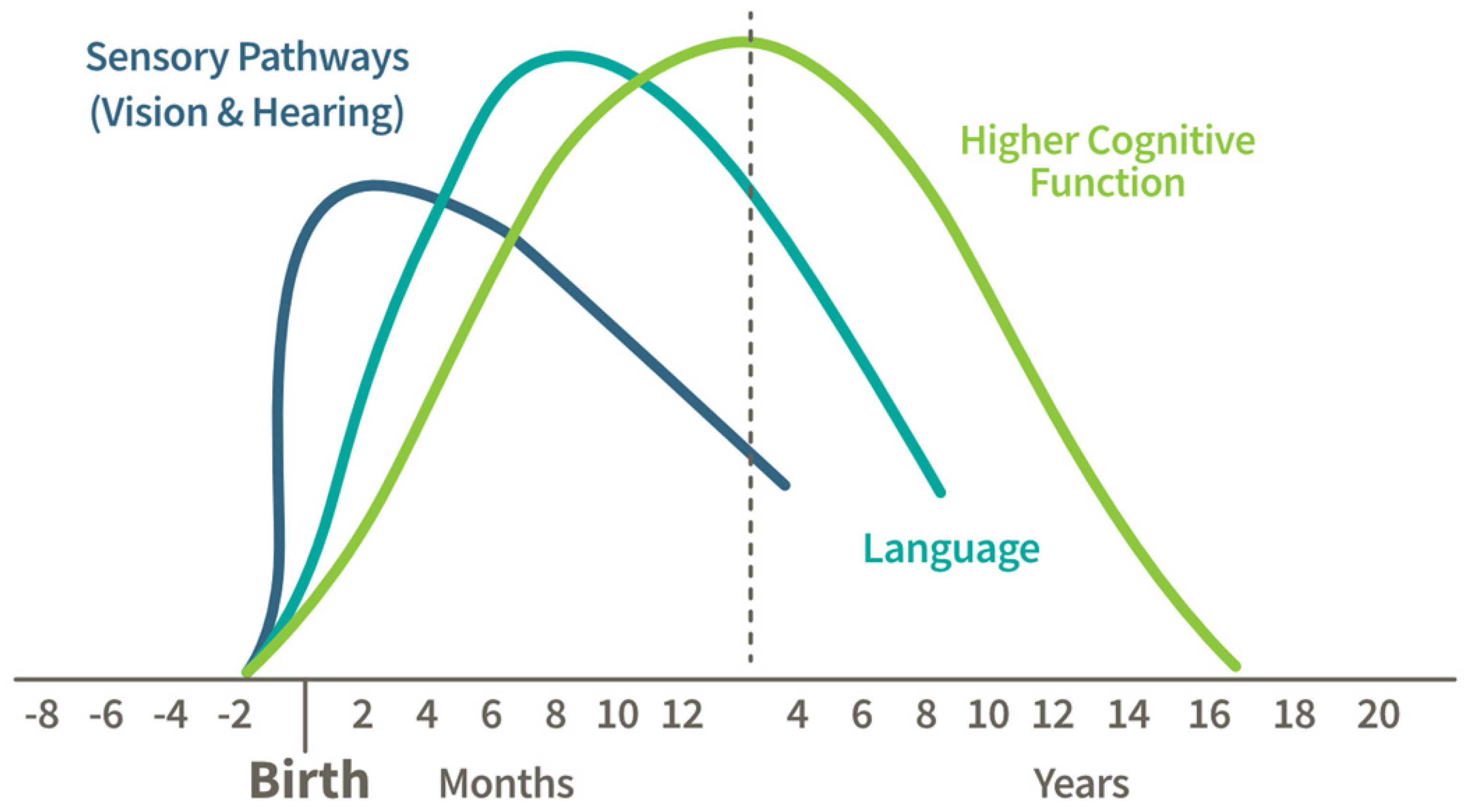
:1. Introduction
2. Pathways Through Which SF Has Been Shown to Impact Brain Health and Cognition
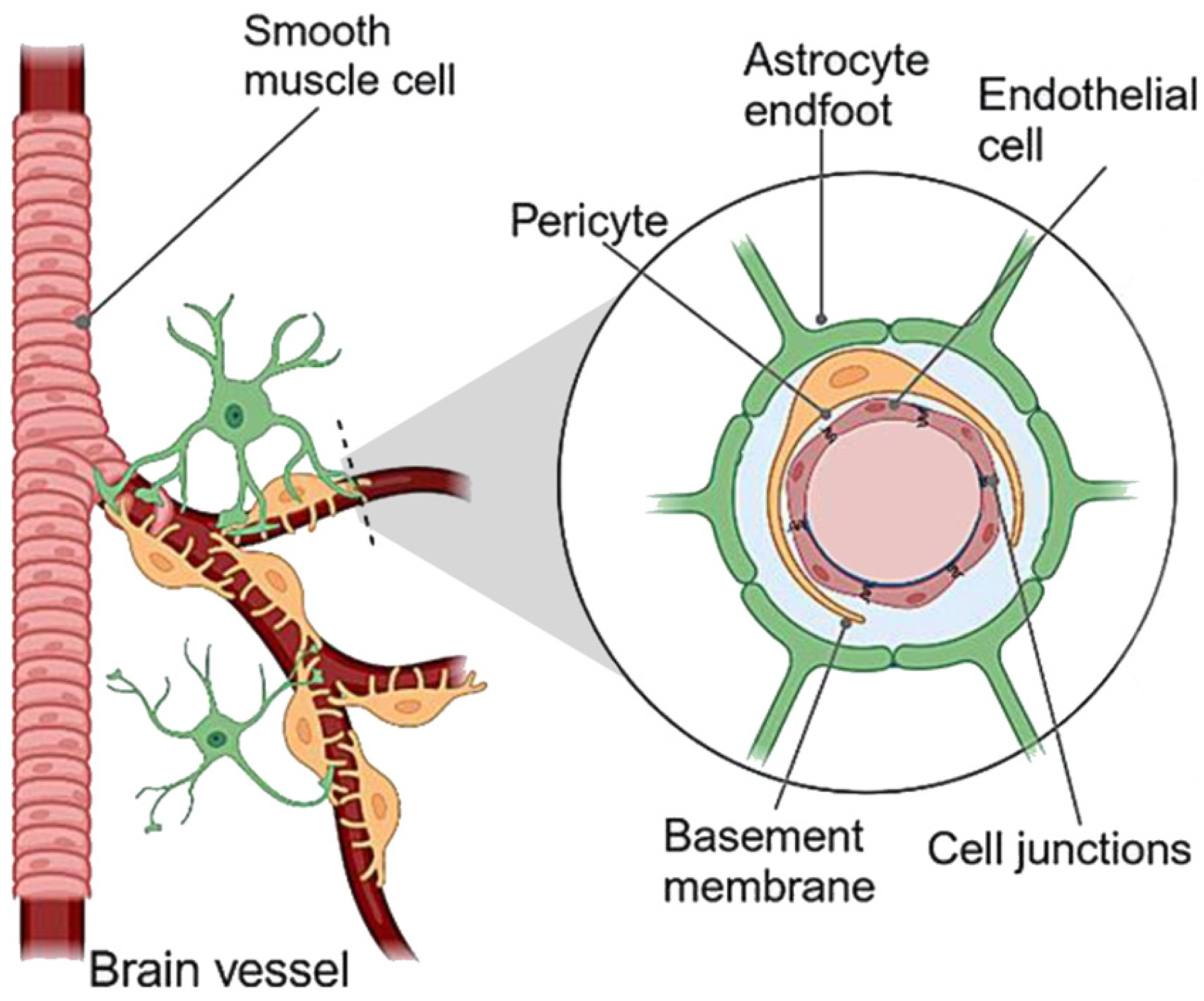
2.1. The Blood–Brain Barrier (BBB)
2.2. The Keap1/Nrf2 Pathway, Detoxication, and Oxidative Stress
2.2.1. Glutathione (GSH)
2.2.2. NAD(P)H:Quinone Oxidoreductase-1 (NQO1; Previously Called Quinone Reductase or QR)
2.2.3. Heme Oxygenase-1 (HO-1; HMOX; HSP32)
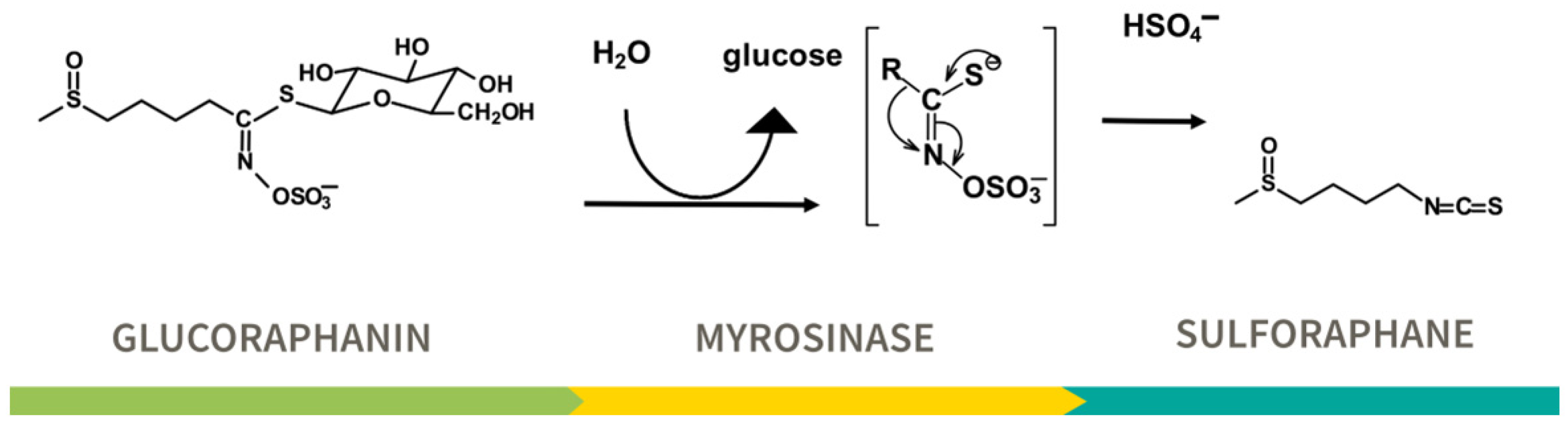
2.3. Detoxication

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Compound | Neurotoxin | Reference |
|---|---|---|
| advanced glycation endproducts (AGE) precursor | methylglyoxal | [49] |
| air pollutant | diesel exhaust | [93] |
| air pollutant | cigarette smoke | [94] |
| air pollutants from combustion and tobacco smoke | benzene, acrolein | [95,96,97,98] |
| antimuscarinic alkaloid | scopalamine | [99] |
| dinoflagellate-derived shellfish toxin | okadic acid | [100] |
| DNA alkylating antibiotic | streptozocin | [101] |
| fungicide | pyraclostrobin | [102] |
| fungicide | trifloxystrobin | [102] |
| fungicide | famoxadone | [102] |
| fungicide | fenamidone | [102] |
| heavy metal | Cadmium | [103] |
| heavy metal | Arsenic | [104] |
| heavy metal | lead | [105] |
| polycyclic aromatic hydrocarbons (PAH; air pollutants) | phenanthrene | [92] |
| pesticide | rotenone | [102,106] |
| pesticide | tributyltin | [107] |
| pesticide | paraquat | [108] |
| poisonous gas | carbon monoxide | [109] |
| spoiled grain contaminant | aflatoxin | [88] |
| street drug | methamphetamine | [110,111] |
| street drug | phencyclidine | [112,113] |
| street drug contaminant | MPTP | [114] |
| toxic pathogen-associated molecular patterns (PAMP) | bacterial lipopolysaccharide | [115] |
2.4. Mitochondrial Health and Function
2.5. Neuroinflammation
2.6. The Gut–Brain Axis
2.6.1. Gut Microbiota Modulation
2.6.2. Reduction in Gut Inflammation
2.6.3. Gut–Brain Signaling Pathways
2.6.4. Impact of SF-Induced Microbiome Changes on Neurological Disorders
2.7. Other Mechanisms
2.7.1. Neurogenesis and Synaptic Plasticity
2.7.2. The Heat Shock Response (HSR)
2.7.3. Apoptosis
2.7.4. Autophagy
2.7.5. Epigenetic Effects
3. Disorders
3.1. Mood, Cognitive, and Psychiatric Disorders (Affecting Mental Health and Cognition)
3.1.1. Mood, Depression, and Anxiety Disorders
3.1.2. Autism Spectrum Disorder (ASD)
3.1.3. Schizophrenia
3.1.4. Diabetes-Related Cognitive Decline
3.1.5. Cognitive Function and Memory Impairment
3.2. Neurodegenerative Diseases (Progressive Degeneration of Neurons)
3.2.1. Alzheimer’s Disease (AD)
3.2.2. Parkinson’s Disease (PD)
3.2.3. Huntington’s Disease
3.3. Brain Injuries and Acute Events (Brain Injuries or Events That Disrupt Normal Brain Function)
3.3.1. Traumatic Brain Injury (TBI) and Spinal Cord Injury (SCI)
3.3.2. Ischemia/Reperfusion-Induced Brain Injury
3.3.3. Stroke
3.3.4. Alcohol-Related Disorders
3.4. Epilepsy and Seizure Disorders (Affecting Motor Control and Electrical Activity in the Brain)
3.5. Sleep Disorders
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | Aberrant Behavior Checklist |
| Ach | acetylcholine |
| AchE | acetylcholineesterase |
| AD | Alzheimer’s Disease |
| ADH | alcohol dehydrogenase |
| AGE | advanced glycation endproducts |
| ALDH | aldehyde dehydrogenase |
| AQP4 | aquaporin 4 |
| ASD | autism spectrum disorder |
| Aβ | amyloid-beta |
| BBB | blood–brain barrier |
| Bcl | anti-apoptotic B-cell lymphoma-2 |
| HSP | heat shock protein |
| BDNF | brain-derived neutrophic factor |
| CHIP | carboxy-terminal Hsp70-interacting protein |
| CNS | central nervous system |
| COX-2 | cyclooxygenase-2 |
| DNMT | DNA methyl transferase |
| FAS | fetal alcohol syndrome |
| GCL | γ-glutamylcysteine ligase |
| GCLC | γ-glutamylcysteine ligase catalytic subunit |
| GCLM | γ-glutamylcysteine ligase modifier subunit |
| GGT | γ-glutamyl transferase |
| GPx | glutathione peroxidase(s) |
| GR | glucoraphanin |
| GS | glucosinolates |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| GST | glutathione S-transferase(s) |
| HDAC | histone deacetylase |
| HMOX | heme oxygenase |
| HO-1 | heme oxygenase-1 |
| HSR | heat shock response |
| ICH | intracerebral hemorrhage(s) |
| IL-1β | interleukin-1 beta |
| IL-6 | interleukin-6 |
| IP-10 | interferon-gamma inducible protein 10 |
| ITC | isothiocyanate(s) |
| Keap1 | Kelch-like ECH-associated protein 1 |
| KO | knock-out |
| MATRICS | Measurement and Treatment Research to Improve Cognition in Schizophrenia |
| MCCB | MATRICS Consensus Cognitive Battery |
| mHtt | mutant huntingtin |
| MIF | macrophage migration inhibitory factor |
| miRNA | microRNA |
| MPO | myeloperoxidase(s) |
| MPTP | methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NF-kB | nuclear factor kappa B |
| NMDA | N-methyl-D-aspartate |
| NQO1 | NAD(P)H quinone dehydrogenase 1 or NAD(P)H dehydrogenase, quinone 1 |
| Nrf2 | NF-E2 p45-related factor 2 |
| OARS | Ohio State University (OSU) Autism Rating Scale |
| PANSS | Positive and Negative Syndrome Scale |
| PCP | phencyclidine or phenylcyclohexyl piperidine |
| PD | Parkinson’s Disease |
| PGE2 | protaglandin E2 |
| Prx | peroxiredoxin |
| ROS | reactive oxygen species |
| SCFA | short-chain fatty acids |
| SCI | spinal cord injury |
| SF | sulforaphane |
| SOD | superoxide dismutase |
| SRS | Social Responsiveness Scales |
| TBI | traumatic brain injury |
| TCA | tricarboxylic acid cycle (also known as. Krebs cycle) |
| TNF-α | tumor necrosis factor alpha |
| Trx | thioredoxin |
References
- National Research Council, Institute of Medicine. From Neurons to Neighborhoods: The Science of Early Childhood Development; The National Academies Press: Washington, DC, USA, 2000; p. 608. [Google Scholar]
- Marzola, P.; Melzer, T.; Pavesi, E.; Gil-Mohapel, J.; Brocardo, P.S. Exploring the Role of Neuroplasticity in Development, Aging, and Neurodegeneration. Brain Sci. 2023, 13, 1610. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.M.; Schneider, D.C.; Mehta, N.K.; Myrskyla, M. Cognitive impairment in the U.S.: Lifetime risk, age at onset, and years impaired. SSM Popul. Health 2020, 11, 100577. [Google Scholar] [CrossRef] [PubMed]
- Graham, S. Results of case-control studies of diet and cancer in Buffalo, New York. Cancer Res. 1983, 43 (Suppl. S5), 2409–2413. [Google Scholar]
- Graham, S.; Dayal, H.; Swanson, M.; Mittelman, A.; Wilkinson, G. Diet in the Epidemiology of Cancer of the Colon and Rectum. J. Natl. Cancer Inst. 1978, 61, 709–714. [Google Scholar]
- Tuck, N.J.; Farrow, C.; Thomas, J.M. Assessing the effects of vegetable consumption on the psychological health of healthy adults: A systematic review of prospective research. Am. J. Clin. Nutr. 2019, 110, 196–211. [Google Scholar] [CrossRef]
- Radavelli-Bagatini, S.; Sim, M.; Blekkenhorst, L.C.; Bondonno, N.P.; Bondonno, C.P.; Woodman, R.; Dickson, J.M.; Magliano, D.J.; Shaw, J.E.; Daly, R.M.; et al. Associations of specific types of fruit and vegetables with perceived stress in adults: The AusDiab study. Eur. J. Nutr. 2022, 61, 2929–2938. [Google Scholar] [CrossRef]
- Haskell-Ramsay, C.F.; Docherty, S. Role of Fruit and Vegetables in Sustaining Healthy Cognitive Function: Evidence and Issues; Cambridge University Press: Cambridge, UK, 2023; pp. 305–314. [Google Scholar]
- Huang, L.; Zhao, C.; Gao, M.; Tao, Y.; Chen, X.; Chen, H.; Li, F.; Zheng, Y.; Lu, M.; Ma, Y.; et al. Associations of vegetable and fruit intake with cognitive function and its decline: Two longitudinal studies. J. Nutr. Health Aging 2024, 28, 100223. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, W.; Yang, Y.; Liu, H.; Liu, J.; Liu, Y. The association between dietary dark green vegetable intake and cognitive function in US older adults. Nutr. Bull. 2025, 50, 69–81. [Google Scholar] [CrossRef]
- Zhu, W.; Cremonini, E.; Mastaloudis, A.F.; Mitchell, A.E.; Bornhorst, G.M.; Oteiza, P.I. Optimization of sulforaphane bioavailability from a glucoraphanin-rich broccoli seed extract in a model of dynamic gastric digestion and absorption by Caco-2 cell monolayers. Food Funct. 2024, 16, 314–328. [Google Scholar] [CrossRef]
- Johnson, T.L.; Dinkova-Kostova, A.T.; Fahey, J.W. Glucosinolates from the Brassica Vegetables and Their Health Effects. In Encyclopedia of Food and Health; Elsevier: Amsterdam, The Netherlands, 2016; pp. 248–255. [Google Scholar]
- Barba, F.J.; Nikmaram, N.; Roohinejad, S.; Khelfa, A.; Zhu, Z.; Koubaa, M. Bioavailability of Glucosinolates and Their Breakdown Products: Impact of Processing. Front. Nutr. 2016, 3, 24. [Google Scholar] [CrossRef]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wehage, S.L.; Holtzclaw, W.D.; Kensler, T.W.; Egner, P.A.; Shapiro, T.A.; Talalay, P. Protection of humans by plant glucosinolates: Efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev. Res. 2012, 5, 603–611. [Google Scholar] [CrossRef]
- Elfoul, L.; Rabot, S.; Khelifa, N.; Quinsac, A.; Duguay, A.; Rimbault, A. Formation of allyl isothiocyanate from sinigrin in the digestive tract of rats monoassociated with a human colonic strain of Bacteroides thetaiotaomicron. FEMS Microbiol. Lett. 2001, 197, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.M.; Teran-Garcia, M.; Jeffery, E.H. Enhancing sulforaphane absorption and excretion in healthy men through the combined consumption of fresh broccoli sprouts and a glucoraphanin-rich powder. Br. J. Nutr. 2012, 107, 1333–1338. [Google Scholar] [CrossRef]
- Li, F.; Hullar, M.A.J.; Beresford, S.A.A.; Lampe, J.W. Variation of glucoraphanin metabolism in vivo and ex vivo by human gut bacteria. Br. J. Nutr. 2011, 106, 408–416. [Google Scholar] [CrossRef]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or sulforaphane: Is it the source or dose that matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef]
- Fahey, J.W.; Kensler, T.W. The Challenges of Designing and Implementing Clinical Trials With Broccoli Sprouts… and Turning Evidence Into Public Health Action. Front. Nutr. 2021, 8, 648788. [Google Scholar] [CrossRef]
- Liu, H.; Talalay, P.; Fahey, J.W. Biomarker-Guided Strategy for Treatment of Autism Spectrum Disorder (ASD). CNS Neurol. Disord. Drug Targets 2016, 15, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, T.; Mao, L.; Zhang, F. Sulforaphane Protects against Brain Diseases: Roles of Cytoprotective Enzymes. Austin J. Cerebrovasc. Dis. Stroke 2017, 4, 1054. [Google Scholar]
- Klomparens, E.A.; Ding, Y. The neuroprotective mechanisms and effects of sulforaphane. Brain Circ. 2019, 5, 74–83. [Google Scholar]
- Panjwani, A.A.; Liu, H.; Fahey, J.W. Crucifers and related vegetables and supplements for neurologic disorders: What is the evidence? Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.; Fahey, J.W.; Zimmerman, A.W.; Zhou, X.; Panjwani, A.A. The role of isothiocyanate-rich plants and supplements in neuropsychiatric disorders: A review and update. Front. Nutr. 2024, 11, 1448130. [Google Scholar] [CrossRef]
- Sá-Caputo, D.D.; Seixas, A.; Taiar, R.; Van der Zee, E.A.; Bernardo-Filho, M. Editorial: Non-pharmacological interventions in healthy and pathological aging: Facts and perspectives. Front. Aging Neurosci. 2023, 15, 1191281. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor NRf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef]
- Clarke, J.D.; Hsu, A.; Williams, D.E.; Dashwood, R.H.; Stevens, J.F.; Yamamoto, M.; Ho, E. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm. Res. 2011, 28, 3171–3179. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Wei, Z.; Niu, H.; Wu, L.; Chen, C.; Liu, H.; Cai, T.; Fan, H. Sulforaphane Wrapped in Self-Assembled Nanomicelle Enhances the Effect of Sonodynamic Therapy on Glioma. Pharmaceutics 2024, 17, 34. [Google Scholar] [CrossRef]
- Jaafaru, M.S.; Abd Karim, N.A.; Enas, M.E.; Rollin, P.; Mazzon, E.; Abdull Razis, A.F. Protective Effect of Glucosinolates Hydrolytic Products in Neurodegenerative Diseases (NDDs). Nutrients 2018, 10, 580. [Google Scholar] [CrossRef] [PubMed]
- Cazalla, E.; Cuadrado, A.; García-Yagüe, Á.J. Role of the transcription factor NRF2 in maintaining the integrity of the Blood-Brain Barrier. Fluids Barriers CNS 2024, 21, 1–29. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.M.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.R.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Mao, L.; Yang, T.; Li, X.; Lei, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Gao, Y.; Sun, B.; Zhang, F. Protective effects of sulforaphane in experimental vascular cognitive impairment: Contribution of the Nrf2 pathway. J. Cereb. Blood Flow. Metab. 2019, 39, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; Coman, D.; Jiang, L.; Rothman, D.L.; Hyder, F. Caloric restriction impedes age-related decline of mitochondrial function and neuronal activity. J. Cereb. Blood Flow. Metab. 2014, 34, 1440–1443. [Google Scholar] [CrossRef]
- Akbulut, K.G.; Gonül, B.; Akbulut, H. Exogenous melatonin decreases age-induced lipid peroxidation in the brain. Brain Res. 2008, 1238, 31–35. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Ruhee, R.T.; Suzuki, K. The integrative role of sulforaphane in preventing inflammation, oxidative stress and fatigue: A review of a potential protective phytochemical. Antioxidants 2020, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Kwak, M.K.; Pi, J. Nrf2 in host defense: Over the rainbow. Oxidative Med. Cell. Longev. 2013, 2013, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Joko, S.; Watanabe, M.; Fuda, H.; Takeda, S.; Furukawa, T.; Hui, S.-P.; Shrestha, R.; Chiba, H. Comparison of chemical structures and cytoprotection abilities between direct and indirect antioxidants. J. Funct. Foods 2017, 35, 245–255. [Google Scholar] [CrossRef]
- Fahey, J.W.; Talalay, P. Antioxidant functions of sulforaphane: A potent inducer of phase II detoxication enzymes. Food Chem. Toxicol. 1999, 37, 973–979. [Google Scholar] [CrossRef]
- Tsuji, P.A.; Stephenson, K.K.; Wade, K.L.; Liu, H.; Fahey, J.W. Structure-activity analysis of flavonoids: Direct and indirect antioxidant, and antiinflammatory potencies and toxicities. Nutr. Cancer 2013, 65, 1014–1025. [Google Scholar] [CrossRef]
- Lastra, D.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Perspectives on the Clinical Development of NRF2-Targeting Drugs. Handb. Exp. Pharmacol. 2021, 264, 93–141. [Google Scholar]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Cazalla, E.; Bach, A.; Bathish, B.; Naidu, S.D.; DeNicola, G.M.; Dinkova-Kostova, A.T.; Fernández-Ginés, R.; Grochot-Przeczek, A.; Hayes, J.D.; et al. Health position paper and redox perspectives—Bench to Bedside Transition for Pharmacological Regulation of NRF2 in Noncommunicable Diseases. Redox Biol. 2025, 81, 103569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Thirty years of NRF2: Advances and therapeutic challenges. Nat. Rev. Drug Discov. 2025, 1–24. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef]
- Krisanits, B.A.; Kaur, B.; Fahey, J.W.; Turner, D.P. The Anti-AGEing and RAGEing Potential of Isothiocyanates. Molecules 2024, 29, 5986. [Google Scholar] [CrossRef]
- Vina, J.; Hems, R.; Krebs, H.A. Maintenance of glutathione content in isolated hepatoyctes. Biochem. J. 1978, 170, 627–630. [Google Scholar] [CrossRef]
- Rae, C.D.; Williams, S.R. Glutathione in the human brain: Review of its roles and measurement by magnetic resonance spectroscopy. Anal. Biochem. 2017, 529, 127–143. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione is a crucial guardian of protein integrity in the brain upon nitric oxide imbalance. Commun. Integr. Biol. 2011, 4, 477–479. [Google Scholar] [CrossRef]
- Currais, A.; Maher, P. Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox Signal. 2013, 19, 813–822. [Google Scholar] [CrossRef]
- Mastaloudis, A.; Wood, S.M. Age-related changes in cellular protection, purification, and inflammation-related gene expression: Role of dietary phytonutrients. Ann. N. Y. Acad. Sci. 2012, 1259, 112–120. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Targeting transporters: Promoting blood-brain barrier repair in response to oxidative stress injury. Brain Res. 2015, 1623, 39–52. [Google Scholar] [CrossRef]
- Sotokawauchi, A.; Ishibashi, Y.; Matsui, T.; Yamagishi, S.I. Aqueous Extract of Glucoraphanin-Rich Broccoli Sprouts Inhibits Formation of Advanced Glycation End Products and Attenuates Inflammatory Reactions in Endothelial Cells. Evid.-Based Complement. Altern. Med. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Watanabe, K.; Ishibashi, M.; Saiki, R.; Kuni, K.; Nishimura, K.; Toida, T.; Kashiwagi, K.; Igarashi, K. Aggravation of brain infarction through an increase in acrolein production and a decrease in glutathione with aging. Biochem. Biophys. Res. Commun. 2016, 473, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Briviba, K.; Sies, H. Protection against peroxynitrite. FEBS Lett. 1999, 445, 226–230. [Google Scholar] [CrossRef]
- Cocco, T.; Sgobbo, P.; Clemente, M.; Lopriore, B.; Grattagliano, I.; Di Paola, M.; Villani, G. Tissue-specific changes of mitochondrial functions in aged rats: Effect of a long-term dietary treatment with N-acetylcysteine. Free Radic. Biol. Med. 2005, 38, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Garg, G.; Singh, S.; Singh, A.K.; Rizvi, S.I. Antiaging Effect of Metformin on Brain in Naturally Aged and Accelerated Senescence Model of Rat. Rejuvenation Res. 2017, 20, 173–182. [Google Scholar] [CrossRef]
- Yoshida, M.; Mikami, T.; Higashi, K.; Saiki, R.; Mizoi, M.; Fukuda, K.; Nakamura, T.; Ishii, I.; Nishimura, K.; Toida, T.; et al. Inverse correlation between stroke and urinary 3-hydroxypropyl mercapturic acid, an acrolein-glutathione metabolite. Clin. Chim. Acta 2012, 413, 753–759. [Google Scholar] [CrossRef]
- Javed, Z.; Qamar, U.; Sathyapalan, T. Pituitary and/or hypothalamic dysfunction following moderate to severe traumatic brain injury: Current perspectives. Indian J. Endocrinol. Metab. 2015, 19, 753–763. [Google Scholar] [CrossRef]
- Liu, H.; Wang, H.; Shenvi, S.; Hagen, T.M.; Liu, R.M. Glutathione metabolism during aging and in Alzheimer disease. Ann. N. Y. Acad. Sci. 2004, 1019, 346–349. [Google Scholar] [CrossRef]
- Liu, R.M. Down-regulation of γ-glutamylcysteine synthetase regulatory subunit gene expression in rat brain tissue during aging. J. Neurosci. Res. 2002, 68, 344–351. [Google Scholar] [CrossRef]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Rebrin, I.; Forster, M.J.; Sohal, R.S. Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice. Brain Res. 2007, 1127, 10–18. [Google Scholar] [CrossRef]
- Emir, U.E.; Raatz, S.; McPherson, S.; Hodges, J.S.; Torkelson, C.; Tawfik, P.; White, T.; Terpstra, M. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 2011, 24, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Dean, O.; Bush, A.I.; Berk, M.; Copolov, D.L.; van den Buuse, M. Glutathione depletion in the brain disrupts short-term spatial memory in the Y-maze in rats and mice. Behav. Brain Res. 2009, 198, 258–262. [Google Scholar] [CrossRef] [PubMed]
- González-Fraguela, M.E.; Blanco, L.; Fernández, C.I.; Lorigados, L.; Serrano, T.; Fernández, J.L. Glutathione depletion: Starting point of brain metabolic stress, neuroinflammation and cognitive impairment in rats. Brain Res. Bull. 2018, 137, 120–131. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Nucifora, L.G.; Koga, M.; Shaffer, L.S.; Higgs, C.; Tanaka, T.; Wang, A.M.; Coughlin, J.M.; Barker, P.B.; Fahey, J.W.; et al. Sulforaphane Augments Glutathione and Influences Brain Metabolites in Human Subjects: A Clinical Pilot Study. Mol. Neuropsychiatry 2018, 3, 214–222. [Google Scholar] [CrossRef]
- Zimmerman, A.W.; Singh, K.; Connors, S.L.; Liu, H.; Panjwani, A.A.; Lee, L.C.; Diggins, E.; Foley, A.; Melnyk, S.; Singh, I.N.; et al. Randomized controlled trial of sulforaphane and metabolite discovery in children with Autism Spectrum Disorder. Mol. Autism 2021, 12, 38. [Google Scholar] [CrossRef]
- Mizuno, K.; Kume, T.; Muto, C.; Takada-Takatori, Y.; Izumi, Y.; Sugimoto, H.; Akaike, A. Glutathione biosynthesis via activation of the nuclear factor E2-related factor 2 (Nrf2)--antioxidant-response element (ARE) pathway is essential for neuroprotective effects of sulforaphane and 6-(methylsulfinyl) hexyl isothiocyanate. J. Pharmacol. Sci. 2011, 115, 320–328. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ(10) Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Y. NAD(P)H: Quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc. Toxicol. 2012, 12, 39–45. [Google Scholar] [CrossRef]
- Talalay, P.; Dinkova-Kostova, A.T. Role of Nicotinamide Quinone Oxidoreductase 1 (NQO1) in Protection against Toxicity of Electrophiles and Reactive Oxygen Intermediates. Methods Enzymol. 2004, 382, 355–364. [Google Scholar]
- Ushida, Y.; Suganuma, H.; Yanaka, A. Low-Dose of the Sulforaphane Precursor Glucoraphanin as a Dietary Supplement Induces Chemoprotective Enzymes in Humans. Food Nutr. Sci. 2015, 6, 1603–1612. [Google Scholar] [CrossRef]
- Soane, L.; Li Dai, W.; Fiskum, G.; Bambrick, L.L. Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J. Neurosci. Res. 2010, 88, 1355–1363. [Google Scholar] [CrossRef]
- Talalay, P. Chemoprotection against cancer by induction of Phase 2 enzymes. BioFactors 2000, 12, 5–11. [Google Scholar] [CrossRef]
- Fahey, J.W.; Dinkova-Kostova, A.T.; Stephenson, K.K.; Talalay, P. The “Prochaska” Microtiter Plate Bioassay for Inducers of NQO1. Methods Enzymol. 2004, 382, 243–258. [Google Scholar]
- Liu, H.; Zimmerman, A.W.; Singh, K.; Connors, S.L.; Diggins, E.; Stephenson, K.K.; Dinkova-Kostova, A.T.; Fahey, J.W. Biomarker Exploration in Human Peripheral Blood Mononuclear Cells for Monitoring Sulforaphane Treatment Responses in Autism Spectrum Disorder. Sci. Rep. 2020, 10, 5822. [Google Scholar] [CrossRef]
- Brown, R.H.; Reynolds, C.; Brooker, A.; Talalay, P.; Fahey, J.W. Sulforaphane improves the bronchoprotective response in asthmatics through Nrf2-mediated gene pathways. Respir. Res. 2015, 16, 106. [Google Scholar] [CrossRef]
- Prestera, T.; Talalay, P.; Alam, J.; Ahn, Y.I.; Lee, P.J.; Choi, A.M. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: Regulation by upstream antioxidant-responsive elements (ARE). Mol. Med. 1995, 1, 827–837. [Google Scholar] [CrossRef]
- Wu, Y.H.; Hsieh, H.L. Roles of Heme Oxygenase-1 in Neuroinflammation and Brain Disorders. Antioxidants 2022, 11, 923. [Google Scholar] [CrossRef]
- Shimizu, S.; Kasai, S.; Yamazaki, H.; Tatara, Y.; Mimura, J.; Engler, M.J.; Tanji, K.; Nikaido, Y.; Inoue, T.; Suganuma, H.; et al. Sulforaphane Increase Mitochondrial Biogenesis-Related Gene Expression in the Hippocampus and Suppresses Age-Related Cognitive Decline in Mice. Int. J. Mol. Sci. 2022, 23, 8433. [Google Scholar] [CrossRef]
- Nihart, A.J.; Garcia, M.A.; El Hayek, E.; Liu, R.; Olewine, M.; Kingston, J.D.; Castillo, E.F.; Gullapalli, R.R.; Howard, T.; Bleske, B.; et al. Bioaccumulation of microplastics in decedent human brains. Nat. Med. 2025, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fabiano, N.; Luu, B.; Puder, D. Human microplastic removal: What does the evidence tell us? Brain Med. 2025, 1, 1–2. [Google Scholar] [CrossRef]
- Cascajosa-Lira, A.; Prieto, A.I.; Pichardo, S.; Jos, A.; Cameán, A.M. Protective effects of sulforaphane against toxic substances and contaminants: A systematic review. Phytomedicine 2024, 130, 155731. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Eaton, D.L. 65 Years on—Aflatoxin Biomarkers Blossoming: Whither Next? Toxins 2024, 16, 496. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.G.; Zhu, Y.R.; Qian, G.S.; Wang, J.B.; Lu, J.H.; Kensler, T.W.; Jacobson, L.P.; Munoz, A.; Groopman, J.D. Fifty Years of Aflatoxin Research in Qidong, China: A Celebration of Team Science to Improve Public Health. Toxins 2025, 17, 79. [Google Scholar] [CrossRef]
- Chen, J.G.; Kensler, T.W.; Zhu, J.; Zhu, Y.R.; Wang, J.B.; Lu, J.H.; Muñoz, A.; Groopman, J.D. Profound primary prevention of liver cancer following a natural experiment in China: A 50-year perspective and public health implications. Int. J. Cancer 2025, 156, 756–763. [Google Scholar] [CrossRef]
- Song, C.; Wang, Z.; Cao, J.; Dong, Y.; Chen, Y. Neurotoxic mechanisms of mycotoxins: Focus on aflatoxin B1 and T-2 toxin. Env. Pollut. 2024, 356, 124359. [Google Scholar] [CrossRef]
- Kensler, T.W.; Chen, J.G.; Egner, P.A.; Fahey, J.W.; Jacobson, L.P.; Stephenson, K.K.; Ye, L.; Coady, J.L.; Wang, J.B.; Wu, Y.; et al. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo Township, Qidong, People’s Republic of China. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2605–2613. [Google Scholar] [CrossRef]
- Heber, D.; Li, Z.; Garcia-Lloret, M.; Wong, A.M.; Lee, T.Y.; Thames, G.; Krak, M.; Zhang, Y.; Nel, A. Sulforaphane-rich broccoli sprout extract attenuates nasal allergic response to diesel exhaust particles. Food Funct. 2014, 5, 35–41. [Google Scholar] [CrossRef]
- Sidhaye, V.K.; Holbrook, J.T.; Burke, A.; Sudini, K.R.; Sethi, S.; Criner, G.J.; Fahey, J.W.; Berenson, C.S.; Jacobs, M.R.; Thimmulappa, R.; et al. Compartmentalization of anti-oxidant and anti-inflammatory gene expression in current and former smokers with COPD. Respir. Res. 2019, 20, 190. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Ng, D.; Carmella, S.G.; Chen, M.; Jacobson, L.P.; Munoz, A.; Egner, P.A.; Chen, J.G.; Qian, G.S.; Chen, T.Y.; et al. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 2012, 33, 101–107. [Google Scholar] [CrossRef]
- Chen, J.G.; Johnson, J.; Egner, P.; Ng, D.; Zhu, J.; Wang, J.B.; Xue, X.F.; Sun, Y.; Zhang, Y.H.; Lu, L.L.; et al. Dose-dependent detoxication of the airborne pollutant benzene in a randomized trial of broccoli sprout beverage in Qidong, China. Am. J. Clin. Nutr. 2019, 110, 675–684. [Google Scholar] [CrossRef]
- Egner, P.A.; Chen, J.G.; Zarth, A.T.; Ng, D.K.; Wang, J.B.; Kensler, K.H.; Jacobson, L.P.; Munoz, A.; Johnson, J.L.; Groopman, J.D.; et al. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: Results of a randomized clinical trial in China. Cancer Prev. Res. 2014, 7, 813–823. [Google Scholar] [CrossRef]
- Bauman, J.E.; Hsu, C.H.; Centuori, S.; Guillen-Rodriguez, J.; Garland, L.L.; Ho, E.; Padi, M.; Bageerathan, V.; Bengtson, L.; Wojtowicz, M.; et al. Randomized Crossover Trial Evaluating Detoxification of Tobacco Carcinogens by Broccoli Seed and Sprout Extract in Current Smokers. Cancers 2022, 14, 2129. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.; Seo, S.G.; Choi, B.R.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane alleviates scopolamine-induced memory impairment in mice. Pharmacol. Res. 2014, 85, 23–32. [Google Scholar] [CrossRef]
- Dwivedi, S.; Rajasekar, N.; Hanif, K.; Nath, C.; Shukla, R. Sulforaphane Ameliorates Okadaic Acid-Induced Memory Impairment in Rats by Activating the Nrf2/HO-1 Antioxidant Pathway. Mol. Neurobiol. 2016, 53, 5310–5323. [Google Scholar] [CrossRef]
- Wang, G.; Fang, H.; Zhen, Y.; Xu, G.; Tian, J.; Zhang, Y.; Zhang, D.; Zhang, G.; Xu, J.; Zhang, Z.; et al. Sulforaphane Prevents Neuronal Apoptosis and Memory Impairment in Diabetic Rats. Cell Physiol. Biochem. 2016, 39, 901–907. [Google Scholar] [CrossRef]
- Pearson, B.L.; Simon, J.M.; McCoy, E.S.; Salazar, G.; Fragola, G.; Zylka, M.J. Identification of chemicals that mimic transcriptional changes associated with autism, brain aging and neurodegeneration. Nat. Commun. 2016, 7, 11173. [Google Scholar] [CrossRef]
- Wang, L.; Gallagher, E.P. Role of Nrf2 antioxidant defense in mitigating cadmium-induced oxidative stress in the olfactory system of zebrafish. Toxicol. Appl. Pharmacol. 2013, 266, 177–186. [Google Scholar] [CrossRef]
- Shavali, S.; Sens, D.A. Synergistic neurotoxic effects of arsenic and dopamine in human dopaminergic neuroblastoma SH-SY5Y cells. Toxicol. Sci. 2008, 102, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, X.; Yin, Y.; Sun, H.; Ge, H.; Li, W. Effects of sulforaphane and vitamin E on cognitive disorder and oxidative damage in lead-exposed mice hippocampus at lactation. J. Trace Elem. Med. Biol. 2017, 44, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Kawami, T.; Ishida, A.; Yamazaki, T. Tributyltin induces oxidative stress and neuronal injury by inhibiting glutathione S-transferase in rat organotypic hippocampal slice cultures. Neurochem. Int. 2012, 60, 782–790. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.; Yang, L.; Chu, B.; Shi, J.; Xie, W.; Zhao, Y.; Zhang, P. Sulforaphane inhibits paraquat-induced oxidative stress by activating the nuclear factor erythroid 2-related factor 2 signaling pathway. Lat. Am. J. Pharm. 2018, 37, 1772–1779. [Google Scholar]
- Bi, M.; Li, Q.; Guo, D.; Ding, X.; Bi, W.; Zhang, Y.; Zou, Y. Sulphoraphane Improves Neuronal Mitochondrial Function in Brain Tissue in Acute Carbon Monoxide Poisoning Rats. Basic. Clin. Pharmacol. Toxicol. 2017, 120, 541–549. [Google Scholar] [CrossRef]
- Chen, H.; Wu, J.; Zhang, J.; Fujita, Y.; Ishima, T.; Iyo, M.; Hashimoto, K. Protective effects of the antioxidant sulforaphane on behavioral changes and neurotoxicity in mice after the administration of methamphetamine. Psychopharmacol. 2012, 222, 37–45. [Google Scholar] [CrossRef]
- Chen, L.; Ru, Q.; Xiong, Q.; Yang, J.; Xu, G.; Wu, Y. Potential Effects of Nrf2 in Exercise Intervention of Neurotoxicity Caused by Methamphetamine Oxidative Stress. Oxid. Med. Cell Longev. 2022, 2022, 4445734. [Google Scholar] [CrossRef]
- Shirai, Y.; Fujita, Y.; Hashimoto, K. Effects of the antioxidant sulforaphane on hyperlocomotion and prepulse inhibition deficits in mice after phencyclidine administration. Clin. Psychopharmacol. Neurosci. 2012, 10, 94–98. [Google Scholar] [CrossRef]
- Shirai, Y.; Fujita, Y.; Hashimoto, R.; Ohi, K.; Yamamori, H.; Yasuda, Y.; Ishima, T.; Suganuma, H.; Ushida, Y.; Takeda, M.; et al. Dietary intake of sulforaphane-rich broccoli sprout extracts during juvenile and adolescence can prevent phencyclidine-induced cognitive deficits at adulthood. PLoS ONE 2015, 10, e0127244. [Google Scholar] [CrossRef]
- Pu, Y.; Qu, Y.; Chang, L.; Wang, S.M.; Zhang, K.; Ushida, Y.; Suganuma, H.; Hashimoto, K. Dietary intake of glucoraphanin prevents the reduction of dopamine transporter in the mouse striatum after repeated administration of MPTP. Neuropsychopharmacol. Rep. 2019, 39, 247–251. [Google Scholar] [CrossRef]
- Townsend, B.E.; Johnson, R.W. Sulforaphane reduces lipopolysaccharide-induced proinflammatory markers in hippocampus and liver but does not improve sickness behavior. Nutr. Neurosci. 2017, 20, 195–202. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, X.; Zhang, X. The impact of exposure to air pollution on cognitive performance. Proc. Natl. Acad. Sci. USA 2018, 115, 9193–9197. [Google Scholar] [CrossRef]
- Hajdusianek, W.; Żórawik, A.; Waliszewska-Prosół, M.; Poręba, R.; Gać, P. Tobacco and nervous system development and function—New findings 2015–2020. Brain Sci. 2021, 11, 797. [Google Scholar] [CrossRef]
- Barnhill, L.M.; Khuansuwan, S.; Juarez, D.; Murata, H.; Araujo, J.A.; Bronstein, J.M. Diesel exhaust extract exposure induces neuronal toxicity by disrupting autophagy. Toxicol. Sci. 2020, 176, 193–202. [Google Scholar] [CrossRef]
- Gawryluk, J.R.; Polombo, D.J.; Curran, J.; Parker, A.; Carlsten, C. Brief diesel exhaust exposure acutely impairs functional brain connectivity in humans: A randomized controlled crossover study. Environ. Health Glob. Access Sci. Sour. 2023, 22, 1–7. [Google Scholar]
- Noah, T.L.; Zhang, H.; Zhou, H.; Glista-Baker, E.; Müller, L.; Bauer, R.N.; Meyer, M.; Murphy, P.C.; Jones, S.; Letang, B.; et al. Effect of broccoli sprouts on nasal response to live attenuated influenza virus in smokers: A randomized, double-blind study. PLoS ONE 2014, 9, e98671. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Hrelia, S. Antiglycative activity of sulforaphane: A new avenue to counteract neurodegeneration? Neural. Regen. Res. 2015, 10, 1750–1751. [Google Scholar]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective Effect of Sulforaphane against Methylglyoxal Cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef]
- Reddam, A.; McLarnan, S.; Kupsco, A. Environmental Chemical Exposures and Mitochondrial Dysfunction: A Review of Recent Literature. Curr. Env. Health Rep. 2022, 9, 631–649. [Google Scholar] [CrossRef]
- Amigo, I.; Kowaltowski, A.J. Dietary restriction in cerebral bioenergetics and redox state. Redox Biol. 2014, 2, 296–304. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Administration of the Nrf2-ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radic. Biol. Med. 2013, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Negrette-Guzmán, M.; Huerta-Yepez, S.; Tapia, E.; Pedraza-Chaverri, J. Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: A seemingly contradictory dual role and an integrative hypothesis. Free Radic. Biol. Med. 2013, 65, 1078–1089. [Google Scholar] [CrossRef]
- Brose, R.D.; Shin, G.; McGuinness, M.C.; Schneidereith, T.; Purvis, S.; Dong, G.X.; Keefer, J.; Spencer, F.; Smith, K.D. Activation of the stress proteome as a mechanism for small molecule therapeutics. Hum. Mol. Genet. 2012, 21, 4237–4252. [Google Scholar] [CrossRef]
- Townsend, B.E.; Johnson, R.W. Sulforaphane induces Nrf2 target genes and attenuates inflammatory gene expression in microglia from brain of young adult and aged mice. Exp. Gerontol. 2016, 73, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Healy, Z.R.; Liu, H.; Holtzclaw, W.D.; Talalay, P. Inactivation of tautomerase activity of macrophage migration inhibitory factor by sulforaphane: A potential biomarker for anti-inflammatory intervention. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1516–1523. [Google Scholar] [CrossRef]
- Wise, R.A.; Holbrook, J.T.; Criner, G.; Sethi, S.; Rayapudi, S.; Sudini, K.R.; Sugar, E.A.; Burke, A.; Thimmulappa, R.; Singh, A.; et al. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: A randomized, double-blind, placebo controlled trial. PLoS ONE 2016, 11, e0163716. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L. Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta Pharmacol. Sin. 2007, 28, 1343–1354. [Google Scholar] [CrossRef]
- Nair, S.; Doh, S.T.; Chan, J.Y.; Kong, A.N.; Cai, L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer 2008, 99, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Alam, J.; Venkatesan, M.I.; Eiguren-Fernandez, A.; Schmitz, D.; Di Stefano, E.; Slaughter, N.; Killeen, E.; Wang, X.; Huang, A.; et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: Protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 2004, 173, 3467–3481. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Yao, C.A. Potential role of NRF2 activators with dual antiviral and anti-inflammatory properties in the management of viral pneumonia. Infect. Drug Resist. 2020, 13, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhäuser, C. Nuclear Factor κB Is a Molecular Target for Sulforaphane-mediated Anti-inflammatory Mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef]
- Suganuma, H.; Fahey, J.W.; Bryan, K.E.; Healy, Z.R.; Talalay, P. Stimulation of phagocytosis by sulforaphane. Biochem. Biophys. Res. Commun. 2011, 405, 146–151. [Google Scholar] [CrossRef]
- Nagata, N.; Xu, L.; Kohno, S.; Ushida, Y.; Aoki, Y.; Umeda, R.; Fuke, N.; Zhuge, F.; Ni, Y.; Nagashimada, M.; et al. Glucoraphanin ameliorates obesity and insulin resistance through adipose tissue browning and reduction of metabolic endotoxemia in mice. Diabetes 2017, 66, 1222–1236. [Google Scholar] [CrossRef]
- Huang, C.; Wu, J.; Chen, D.; Jin, J.; Wu, Y.; Chen, Z. Effects of sulforaphane in the central nervous system. Eur. J. Pharmacol. 2019, 853, 153–168. [Google Scholar] [CrossRef]
- Gao, J.; Xiong, B.; Zhang, B.; Li, S.; Huang, N.; Zhan, G.; Jiang, R.; Yang, L.; Wu, Y.; Miao, L.; et al. Sulforaphane Alleviates Lipopolysaccharide-induced Spatial Learning and Memory Dysfunction in Mice: The Role of BDNF-mTOR Signaling Pathway. Neuroscience 2018, 388, 357–366. [Google Scholar] [CrossRef]
- Chen, S.; Chen, H.; Du, Q.; Shen, J. Targeting Myeloperoxidase (MPO) Mediated Oxidative Stress and Inflammation for Reducing Brain Ischemia Injury: Potential Application of Natural Compounds. Front. Physiol. 2020, 11, 433. [Google Scholar] [CrossRef]
- Zhao, H.D.; Zhang, F.; Shen, G.; Li, Y.B.; Li, Y.H.; Jing, H.R.; Ma, L.F.; Yao, J.H.; Tian, X.F. Sulforaphane protects liver injury induced by intestinal ischemia reperfusion through Nrf2-ARE pathway. World J. Gastroenterol. 2010, 16, 3002–3010. [Google Scholar] [CrossRef]
- Yu, C.; He, Q.; Zheng, J.; Li, L.Y.; Hou, Y.H.; Song, F.Z. Sulforaphane improves outcomes and slows cerebral ischemic/reperfusion injury via inhibition of NLRP3 inflammasome activation in rats. Int. Immunopharmacol. 2017, 45, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mohr, A.; Heitplatz, B.; Hansen, U.; Pascher, A.; Brockmann, J.G.; Becker, F. Sulforaphane elicits protective effects in intestinal ischemia reperfusion injury. Int. J. Mol. Sci. 2020, 21, 5189. [Google Scholar] [CrossRef]
- Yao, W.; Zhang, J.C.; Ishima, T.; Dong, C.; Yang, C.; Ren, Q.; Ma, M.; Han, M.; Wu, J.; Suganuma, H.; et al. Role of Keap1-Nrf2 signaling in depression and dietary intake of glucoraphanin confers stress resilience in mice. Sci. Rep. 2016, 6, 30659. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The microbiota-gut-brain axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Holman, J.; Hurd, M.; Moses, P.L.; Mawe, G.M.; Zhang, T.; Ishaq, S.L.; Li, Y. Interplay of broccoli/broccoli sprout bioactives with gut microbiota in reducing inflammation in inflammatory bowel diseases. J. Nutr. Biochem. 2023, 113, 109238. [Google Scholar] [CrossRef]
- Holman, J.M.; Colucci, L.; Baudewyns, D.; Balkan, J.; Hunt, T.; Hunt, B.; Kinney, M.; Holcomb, L.; Stratigakis, A.; Chen, G.; et al. Steamed broccoli sprouts alleviate DSS-induced inflammation and retain gut microbial biogeography in mice. mSystems 2023, 8, e0053223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Holman, J.; McKinstry, D.; Trindade, B.C.; Eaton, K.A.; Mendoza-Castrejon, J.; Ho, S.; Wells, E.; Yuan, H.; Wen, B.; et al. A steamed broccoli sprout diet preparation that reduces colitis via the gut microbiota: Broccoli sprouts reduce colitis via gut microbiota. J. Nutr. Biochem. 2023, 112, 109215. [Google Scholar] [CrossRef]
- Bouranis, J.A.; Beaver, L.M.; Choi, J.; Wong, C.P.; Jiang, D.; Sharpton, T.J.; Stevens, J.F.; Ho, E. Article composition of the gut microbiome influences production of sulforaphane-nitrile and iberin-nitrile from glucosinolates in broccoli sprouts. Nutrients 2021, 13, 3013. [Google Scholar] [CrossRef]
- Bouranis, J.A.; Beaver, L.M.; Ho, E. Metabolic Fate of Dietary Glucosinolates and Their Metabolites: A Role for the Microbiome. Front. Nutr. 2021, 8, 748433. [Google Scholar] [CrossRef]
- Kaczmarek, J.L.; Liu, X.; Charron, C.S.; Novotny, J.A.; Jeffery, E.H.; Seifried, H.E.; Ross, S.A.; Miller, M.J.; Swanson, K.S.; Holscher, H.D. Broccoli consumption affects the human gastrointestinal microbiota. J. Nutr. Biochem. 2019, 63, 27–34. [Google Scholar] [CrossRef]
- Kellingray, L.; Tapp, H.S.; Saha, S.; Doleman, J.F.; Narbad, A.; Mithen, R.F. Consumption of a diet rich in Brassica vegetables is associated with a reduced abundance of sulphate-reducing bacteria: A randomised crossover study. Mol. Nutr. Food Res. 2017, 61, 1600992. [Google Scholar] [CrossRef] [PubMed]
- Bouranis, J.A.; Beaver, L.M.; Wong, C.P.; Choi, J.; Hamer, S.; Davis, E.W.; Brown, K.S.; Jiang, D.; Sharpton, T.J.; Stevens, J.F.; et al. Sulforaphane and Sulforaphane-Nitrile Metabolism in Humans Following Broccoli Sprout Consumption: Inter-individual Variation, Association with Gut Microbiome Composition, and Differential Bioactivity. Mol. Nutr. Food Res. 2024, 68, 2300286. [Google Scholar] [CrossRef] [PubMed]
- Bouranis, J.A.; Beaver, L.M.; Jiang, D.; Choi, J.; Wong, C.P.; Davis, E.W.; Williams, D.E.; Sharpton, T.J.; Stevens, J.F.; Ho, E. Interplay between Cruciferous Vegetables and the Gut Microbiome: A Multi-Omic Approach. Nutrients 2023, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Bouranis, J.A.; Wong, C.P.; Beaver, L.M.; Uesugi, S.L.; Papenhausen, E.M.; Choi, J.; Davis, E.W.; Da Silva, A.N.; Kalengamaliro, N.; Chaudhary, R.; et al. Sulforaphane Bioavailability in Healthy Subjects Fed a Single Serving of Fresh Broccoli Microgreens. Foods 2023, 12, 3784. [Google Scholar] [CrossRef]
- Holcomb, L.; Holman, J.M.; Hurd, M.; Lavoie, B.; Colucci, L.; Hunt, B.; Hunt, T.; Kinney, M.; Pathak, J.; Mawe, G.M.; et al. Early life exposure to broccoli sprouts confers stronger protection against enterocolitis development in an immunological mouse model of inflammatory bowel disease. mSystems 2023, 8, e0068823. [Google Scholar] [CrossRef]
- Zhu, W.; Cremonini, E.; Mastaloudis, A.; Oteiza, P.I. Glucoraphanin and sulforaphane mitigate TNFα-induced Caco-2 monolayers permeabilization and inflammation. Redox Biol. 2024, 76, 103359. [Google Scholar] [CrossRef]
- Lista, S.; Munafo, A.; Caraci, F.; Imbimbo, C.; Emanuele, E.; Minoretti, P.; Pinto-Fraga, J.; Merino-Pais, M.; Crespo-Escobar, P.; Lopez-Ortiz, S.; et al. Gut microbiota in Alzheimer’s disease: Understanding molecular pathways and potential therapeutic perspectives. Ageing Res. Rev. 2025, 104, 102659. [Google Scholar] [CrossRef]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef]
- Han, Z.; Xu, Q.; Li, C.; Zhao, H. Effects of sulforaphane on neural stem cell proliferation and differentiation. Genesis 2017, 55, e23022. [Google Scholar] [CrossRef]
- Grieco, S.F.; Holmes, T.C.; Xu, X. Neuregulin directed molecular mechanisms of visual cortical plasticity. J. Comp. Neurol. 2019, 527, 668–678. [Google Scholar] [CrossRef]
- Jonas, E.A. Contributions of Bcl-xL to acute and long term changes in bioenergetics during neuronal plasticity. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Dukay, B.; Csoboz, B.; Tóth, M.E. Heat-shock proteins in neuroinflammation. Front. Pharmacol. 2019, 10, 920. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Kaji, H. Thermal Effect on Heat Shock Protein 70 Family to Prevent Atherosclerotic Cardiovascular Disease. Biomolecules 2023, 13, 867. [Google Scholar] [CrossRef]
- Zheng, Z.; Kim, J.Y.; Ma, H.; Lee, J.E.; Yenari, M.A. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J. Cereb. Blood Flow. Metab. 2008, 28, 53–63. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, J.; Yu, S.; Chen, Y.; Wu, J.; Zhao, Y. Sulforaphane protects primary cultures of cortical neurons against injury induced by oxygen-glucose deprivation/reoxygenation via antiapoptosis. Neurosci. Bull. 2012, 28, 509–516. [Google Scholar] [CrossRef]
- Lee, C.; Park, G.H.; Lee, S.R.; Jang, J.H. Attenuation of beta-amyloid-induced oxidative cell death by sulforaphane via activation of NF-E2-related factor 2. Oxid. Med. Cell Longev. 2013, 2013, 313510. [Google Scholar] [CrossRef]
- Giordano, S.; Dodson, M.; Ravi, S.; Redmann, M.; Ouyang, X.; Usmar, V.M.D.; Zhang, J. Bioenergetic adaptation in response to autophagy regulators during rotenone exposure. J. Neurochem. 2014, 131, 625–633. [Google Scholar] [CrossRef]
- Bertova, A.; Kontar, S.; Ksinanova, M.; Vergara, A.Y.; Sulova, Z.; Breier, A.; Imrichova, D. Sulforaphane and Benzyl Isothiocyanate Suppress Cell Proliferation and Trigger Cell Cycle Arrest, Autophagy, and Apoptosis in Human AML Cell Line. Int. J. Mol. Sci. 2024, 25, 13511. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; D’Mello, S.R. Complex neuroprotective and neurotoxic effects of histone deacetylases. J. Neurochem. 2018, 145, 96–110. [Google Scholar] [CrossRef]
- Shankar, S.; Kumar, D.; Srivastava, R.K. Epigenetic modifications by dietary phytochemicals: Implications for personalized nutrition. Pharmacol. Ther. 2013, 138, 1–17. [Google Scholar] [CrossRef]
- Santin-Marquez, R.; Alarcon-Aguilar, A.; Lopez-Diazguerrero, N.E.; Chondrogianni, N.; Konigsberg, M. Sulforaphane—Role in aging and neurodegeneration. Geroscience 2019, 41, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef]
- Ho, E.; Clarke, J.D.; Dashwood, R.H. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J. Nutr. 2009, 139, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Choi, B.R.; Yang, H.; Hwang, Y.; Park, J.H.Y.; LaFerla, F.M.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane epigenetically enhances neuronal BDNF expression and TrkB signaling pathways. Mol. Nutr. Food Res. 2017, 61, 1600194. [Google Scholar] [CrossRef]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenetics 2011, 3, 3. [Google Scholar] [CrossRef]
- Yang, A.Y.; Kim, H.; Li, W.; Kong, A.N.T. Natural compound-derived epigenetic regulators targeting epigenetic readers, writers and erasers. Curr. Top. Med. Chem. 2016, 16, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhang, J.; Chang, N. Epigenetic modification of Nrf2 by sulforaphane increases the antioxidative and anti-inflammatory capacity in a cellular model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 824, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, A.; Ishima, T.; Hirai, A.; Suzuki, S.; Suganuma, H.; Hashimoto, K. Dietary intake of glucoraphanin during pregnancy and lactation prevents the behavioral abnormalities in the offspring after maternal immune activation. Neuropsychopharmacol. Rep. 2020, 40, 268–274. [Google Scholar] [CrossRef]
- Kasai, S.; Kokubu, D.; Mizukami, H.; Itoh, K. Mitochondrial Reactive Oxygen Species, Insulin Resistance, and Nrf2-Mediated Oxidative Stress Response—Toward an Actionable Strategy for Anti-Aging. Biomolecules 2023, 13, 1544. [Google Scholar] [CrossRef]
- Grillon, C.; Robinson, O.J.; Cornwell, B.; Ernst, M. Modeling anxiety in healthy humans: A key intermediate bridge between basic and clinical sciences. Neuropsychopharmacology 2019, 44, 1999–2010. [Google Scholar] [CrossRef]
- Wu, S.; Gao, Q.; Zhao, P.; Gao, Y.; Xi, Y.; Wang, X.; Liang, Y.; Shi, H.; Ma, Y. Sulforaphane produces antidepressant- and anxiolytic-like effects in adult mice. Behav. Brain Res. 2016, 301, 55–62. [Google Scholar] [CrossRef]
- Zhang, J.C.; Yao, W.; Dong, C.; Yang, C.; Ren, Q.; Ma, M.; Han, M.; Wu, J.; Ushida, Y.; Suganuma, H.; et al. Prophylactic effects of sulforaphane on depression-like behavior and dendritic changes in mice after inflammation. J. Nutr. Biochem. 2017, 39, 134–144. [Google Scholar] [CrossRef]
- Ghazizadeh-Hashemi, F.; Bagheri, S.; Ashraf-Ganjouei, A.; Moradi, K.; Shahmansouri, N.; Mehrpooya, M.; Noorbala, A.A.; Akhondzadeh, S. Efficacy and safety of sulforaphane for treatment of mild to moderate depression in patients with history of cardiac interventions: A randomized, double-blind, placebo-controlled clinical trial. Psychiatry Clin. Neurosci. 2021, 75, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Chamorro, P.; Redondo, A.; Riego, G.; Leanez, S.; Pol, O. Sulforaphane Inhibited the Nociceptive Responses, Anxiety- and Depressive-Like Behaviors Associated With Neuropathic Pain and Improved the Anti-allodynic Effects of Morphine in Mice. Front. Pharmacol. 2018, 9, 1332. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Warren, Z.; Williams, A.R.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Fitzgerald, R.T.; Furnier, S.M.; Hughes, M.M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. MMWR Surveill. Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef]
- Iffland, M.; Livingstone, N.; Jorgensen, M.; Hazell, P.; Gillies, D. Pharmacological intervention for irritability, aggression, and self-injury in autism spectrum disorder (ASD). Cochrane Database Syst. Rev. 2023, 2023, CD011769. [Google Scholar]
- Henneberry, E.; Lamy, M.; Dominick, K.C.; Erickson, C.A. Decades of Progress in the Psychopharmacology of Autism Spectrum Disorder. J. Autism Dev. Disord. 2021, 51, 4370–4394. [Google Scholar] [CrossRef]
- Gupta, N.; Gupta, M. Off-label psychopharmacological interventions for autism spectrum disorders: Strategic pathways for clinicians. CNS Spectr. 2024, 29, 10–25. [Google Scholar] [CrossRef]
- Persico, A.M.; Asta, L.; Chehbani, F.; Mirabelli, S.; Parlatini, V.; Cortese, S.; Arango, C.; Vitiello, B. The pediatric psychopharmacology of autism spectrum disorder: A systematic review—Part II: The future. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2025, 136, 111176. [Google Scholar] [CrossRef]
- Suchitra, M.R.; Yogitha, P.S. Unraveling Neurodevelopmental Disorders: Signalling Pathways as Potential Targets for Therapeutic Intervention in Autism Spectrum Disorder. Biomed. Pharmacol. J. 2024, 17, 1383–1394. [Google Scholar] [CrossRef]
- Magner, M.; Thorová, K.; Župová, V.; Houška, M.; Švandová, I.; Novotná, P.; Tříska, J.; Vrchotová, N.; Soural, I.; Jílek, L. Sulforaphane Treatment in Children with Autism: A Prospective Randomized Double-Blind Study. Nutrients 2023, 15, 718. [Google Scholar] [CrossRef] [PubMed]
- Bent, S.; Lawton, B.; Warren, T.; Widjaja, F.; Dang, K.; Fahey, J.W.; Cornblatt, B.; Kinchen, J.M.; Delucchi, K.; Hendren, R.L. Identification of urinary metabolites that correlate with clinical improvements in children with autism treated with sulforaphane from broccoli. Mol. Autism 2018, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Lynch, R.; Diggins, E.L.; Connors, S.L.; Zimmerman, A.W.; Singh, K.; Liu, H.; Talalay, P.; Fahey, J.W. Sulforaphane from Broccoli Reduces Symptoms of Autism: A Follow-up Case Series from a Randomized Double-blind Study. Glob. Adv. Health Med. 2017, 6, 2164957X17735826. [Google Scholar] [CrossRef]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef]
- Ou, J.; Smith, R.C.; Tobe, R.H.; Lin, J.; Arriaza, J.; Fahey, J.W.; Liu, R.; Zeng, Y.; Liu, Y.; Huang, L.; et al. Efficacy of Sulforaphane in Treatment of Children with Autism Spectrum Disorder: A Randomized Double-Blind Placebo-Controlled Multi-center Trial. J. Autism Dev. Disord. 2024, 54, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Momtazmanesh, S.; Amirimoghaddam-Yazdi, Z.; Moghaddam, H.S.; Mohammadi, M.R.; Akhondzadeh, S. Sulforaphane as an adjunctive treatment for irritability in children with autism spectrum disorder: A randomized, double-blind, placebo-controlled clinical trial. Psychiatry Clin. Neurosci. 2020, 74, 398–405. [Google Scholar] [CrossRef]
- Yang, J.; He, L.; Dai, S.; Zheng, H.; Cui, X.; Ou, J.; Zhang, X. Therapeutic efficacy of sulforaphane in autism spectrum disorders and its association with gut microbiota: Animal model and human longitudinal studies. Front. Nutr. 2023, 10, 1294057. [Google Scholar] [CrossRef]
- Hamoud, A.F.; Al-Saadi, N.H. Sphingomyelin, Plasminogen, and Docosahexaenoic in Sera of Autism Spectrum Disorder Children. Adv. Life Sci. 2024, 11, 226–231. [Google Scholar]
- Dai, S.; Hou, Y.; Lin, J.; Shen, Y.; Cheng, D.; Wu, R.; Ou, J. Metabolomics mechanism of sulforaphane in the treatment of autism spectrum disorders. Chin. J. Psychiatry 2024, 57, 337–344. [Google Scholar]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S.F.; Al-Ayadhi, L.Y.; Attia, S.M.; Al-Harbi, N.O.; Alzahrani, K.S.; Bakheet, S.A. Differential regulation of Nrf2 is linked to elevated inflammation and nitrative stress in monocytes of children with autism. Psychoneuroendocrinology 2020, 113, 104554. [Google Scholar] [CrossRef]
- Pecorelli, A.; Ferrara, F.; Messano, N.; Cordone, V.; Schiavone, M.L.; Cervellati, F.; Woodby, B.; Cervellati, C.; Hayek, J.; Valacchi, G. Alterations of mitochondrial bioenergetics, dynamics, and morphology support the theory of oxidative damage involvement in autism spectrum disorder. FASEB J. 2020, 34, 6521–6538. [Google Scholar] [CrossRef]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From subclinical condition to pathological biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 2, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and Done? Targeting the NRF2 Pathway with Sulforaphane. Trends Food Sci. Technol. 2017, 69 Pt B, 257–269. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar] [CrossRef]
- Matsuura, A.; Ishima, T.; Fujita, Y.; Iwayama, Y.; Hasegawa, S.; Kawahara-Miki, R.; Maekawa, M.; Toyoshima, M.; Ushida, Y.; Suganuma, H.; et al. Dietary glucoraphanin prevents the onset of psychosis in the adult offspring after maternal immune activation. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Shiina, A.; Kanahara, N.; Sasaki, T.; Oda, Y.; Hashimoto, T.; Hasegawa, T.; Yoshida, T.; Iyo, M.; Hashimoto, K. An open study of sulforaphane-rich broccoli sprout extract in patients with schizophrenia. Clin. Psychopharmacol. Neurosci. 2015, 13, 62–67. [Google Scholar] [CrossRef]
- Hei, G.; Smith, R.C.; Li, R.; Ou, J.; Song, X.; Zheng, Y.; He, Y.; Arriaza, J.; Fahey, J.W.; Cornblatt, B.; et al. Sulforaphane Effects on Cognition and Symptoms in First and Early Episode Schizophrenia: A Randomized Double-Blind Trial. Schizophr. Bull. Open 2022, 3, sgac024. [Google Scholar] [CrossRef]
- Dickerson, F.; Origoni, A.; Katsafanas, E.; Squire, A.; Newman, T.; Fahey, J.; Xiao, J.C.; Stallings, C.; Goga, J.; Khushalani, S.; et al. Randomized controlled trial of an adjunctive sulforaphane nutraceutical in schizophrenia. Schizophr. Res. 2021, 231, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F. Efficacy of Add-On Sulforaphane for Improving Symptoms and Cognition in Schizophrenia: A Randomized Double-Blind Study. In Schizophrenia International Research Society; Springer Nature: London, UK, 2021. [Google Scholar]
- Huang, J.; Chen, A.; Jin, H.; Liu, F.; Hei, G.; Teng, Z.; Xiao, J.; Wu, R.; Zhao, J.; Davis, J.M.; et al. Efficacy and Safety of Sulforaphane Added to Antipsychotics for the Treatment of Negative Symptoms of Schizophrenia: A Randomized Controlled Trial. J. Clin. Psychiatry 2025, 86, 24m15272. [Google Scholar] [CrossRef] [PubMed]
- Faust, T.E.; Xin, W.; Lee, B.; Agarwal, A.; Saha, S.; Palen, T.; Cash-Padgett, T.; Wood, D.; Bonci, A.; Fahey, J.W.; et al. Astrocyte redox dysregulation causes prefrontal hypoactivity: Sulforaphane treats non-ictal pathophysiology in ALDH7A1-mediated epilepsy. bioRxiv 2019. [Google Scholar] [CrossRef]
- Riederer, P.; Korczyn, A.D.; Ali, S.S.; Bajenaru, O.; Choi, M.S.; Chopp, M.; Dermanovic-Dobrota, V.; Grünblatt, E.; Jellinger, K.A.; Kamal, M.A.; et al. The diabetic brain and cognition. J. Neural Transm. 2017, 124, 1431–1454. [Google Scholar] [CrossRef]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9, eaah4477. [Google Scholar] [CrossRef]
- Dwibedi, C.; Axelsson, A.S.; Abrahamsson, B.; Fahey, J.W.; Asplund, O.; Hansson, O.; Ahlqvist, E.; Tremaroli, V.; Backhed, F.; Rosengren, A.H. Effect of broccoli sprout extract and baseline gut microbiota on fasting blood glucose in prediabetes: A randomized, placebo-controlled trial. Nat. Microbiol. 2025, 10, 681–693. [Google Scholar] [CrossRef]
- Dash, P.K.; Zhao, J.; Orsi, S.A.; Zhang, M.; Moore, A.N. Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci. Lett. 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Park, H.S.; Hwang, E.S.; Choi, G.Y.; Kim, H.B.; Park, K.S.; Sul, J.Y.; Hwang, Y.; Choi, G.W.; Kim, B.I.; Park, H.; et al. Sulforaphane enhances long-term potentiation and ameliorate scopolamine-induced memory impairment. Physiol. Behav. 2021, 238, 113467. [Google Scholar] [CrossRef]
- Nouchi, R.; Hu, Q.; Saito, T.; Kawata, N.Y.D.S.; Nouchi, H.; Kawashima, R. Brain training and sulforaphane intake interventions separately improve cognitive performance in healthy older adults, whereas a combination of these interventions does not have more beneficial effects: Evidence from a randomized controlled trial. Nutrients 2021, 13, 352. [Google Scholar] [CrossRef]
- Nouchi, R.; Hu, Q.; Ushida, Y.; Suganuma, H.; Kawashima, R. Effects of sulforaphane intake on processing speed and negative moods in healthy older adults: Evidence from a randomized controlled trial. Front. Aging Neurosci. 2022, 14, 929628. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimers Dement. 2023, 19, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, B.R.; Kim, J.; LaFerla, F.M.; Park, J.H.Y.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane Upregulates the Heat Shock Protein Co-Chaperone CHIP and Clears Amyloid-β and Tau in a Mouse Model of Alzheimer’s Disease. Mol. Nutr. Food Res. 2018, 62, e1800240. [Google Scholar] [CrossRef]
- Zhang, R.; Miao, Q.W.; Zhu, C.X.; Zhao, Y.; Liu, L.; Yang, J.; An, L. Sulforaphane ameliorates neurobehavioral deficits and protects the brain from amyloid β deposits and peroxidation in mice with Alzheimer-like lesions. Am. J. Alzheimer’s Dis. Other Dem. 2015, 30, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, R.; Zhan, Z.; Li, X.; Zhou, F.; Xing, A.; Jiang, C.; Chen, Y.; An, L. Beneficial Effects of Sulforaphane Treatment in Alzheimer’s Disease May Be Mediated through Reduced HDAC1/3 and Increased P75NTR Expression. Front. Aging Neurosci. 2017, 9, 121. [Google Scholar] [CrossRef]
- Hou, T.T.; Yang, H.Y.; Wang, W.; Wu, Q.Q.; Tian, Y.R.; Jia, J.P. Sulforaphane Inhibits the Generation of Amyloid-β Oligomer and Promotes Spatial Learning and Memory in Alzheimer’s Disease (PS1V97L) Transgenic Mice. J. Alzheimer’s Dis. 2018, 62, 1803–1813. [Google Scholar] [CrossRef]
- Sunkaria, A.; Bhardwaj, S.; Yadav, A.; Halder, A.; Sandhir, R. Sulforaphane attenuates postnatal proteasome inhibition and improves spatial learning in adult mice. J. Nutr. Biochem. 2018, 51, 69–79. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, J.; Fang, L.; Li, X.; Zhao, Y.; Shi, W.; An, L. Neuroprotective effects of sulforaphane on cholinergic neurons in mice with Alzheimer’s disease-like lesions. Int. J. Mol. Sci. 2014, 15, 14396–14410. [Google Scholar] [CrossRef]
- Park, H.M.; Kim, J.A.; Kwak, M.K. Protection against amyloid beta cytotoxicity by sulforaphane: Role of the proteasome. Arch. Pharmacal Res. 2009, 32, 109–115. [Google Scholar] [CrossRef]
- Wang, W.; Wei, C.; Quan, M.; Li, T.; Jia, J. Sulforaphane Reverses the Amyloid-beta Oligomers Induced Depressive-Like Behavior. J. Alzheimers Dis. 2020, 78, 127–137. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Djemil, A.; D’Amico, M.; Pruccoli, L.; Cantelli-Forti, G.; Hrelia, P.; Tarozzi, A. Comparison of Adaptive Neuroprotective Mechanisms of Sulforaphane and its Interconversion Product Erucin in in Vitro and in Vivo Models of Parkinson’s Disease. J. Agric. Food Chem. 2018, 66, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. NeuroToxicology 2013, 36, 63–71. [Google Scholar] [CrossRef]
- Hacet, F.B.E.; Vatansever, H.S.; Yücecan, S. Investigation of Neuroprotective Effects of Sulforaphane and Allyl Isothiocyanate in an in vitro Alzheimer’s Disease Model. Pharmacogn. Mag. 2023, 19, 822–830. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Baird, L.; Holmström, K.M.; Meyer, C.J.; Abramov, A.Y. The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem. Soc. Trans. 2015, 43, 602–610. [Google Scholar] [CrossRef]
- Wright, A.F. The Redox Stress Test: A novel technique reveals oxidative stress in Parkinson’s disease. Med. Res. Arch. 2024, 12. [Google Scholar] [CrossRef]
- Islam, M.R.; Jony, M.H.; Thufa, G.K.; Akash, S.; Dhar, P.S.; Rahman, M.M.; Afroz, T.; Ahmed, M.; Hemeg, H.A.; Rauf, A.; et al. A clinical study and future prospects for bioactive compounds and semi-synthetic molecules in the therapies for Huntington’s disease. Mol. Neurobiol. 2024, 61, 1237–1270. [Google Scholar] [CrossRef] [PubMed]
- Luis-García, E.R.; Limón-Pacheco, J.H.; Serrano-García, N.; Hernández-Pérez, A.D.; Pedraza-Chaverri, J.; Orozco-Ibarra, M. Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J. Biochem. Mol. Toxicol. 2017, 31, e21837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef]
- Brokowska, J.; Herman-Antosiewicz, A.; Hać, A. Isothiocyanates induce autophagy and inhibit protein synthesis in primary cells via modulation of AMPK-mTORC1-S6K1 signaling pathway, and protect against mutant huntingtin aggregation. Eur. J. Nutr. 2025, 64, 1–13. [Google Scholar] [CrossRef]
- Zhao, J.; Moore, A.N.; Clifton, G.L.; Dash, P.K. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J. Neurosci. Res. 2005, 82, 499–506. [Google Scholar] [CrossRef]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood-brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Dash, P.K.; Kan, Y.W.; Grotta, J.C.; Aronowski, J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 2007, 38, 3280–3286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Hong, Y.; Yan, W.; Chen, S.; Sun, C.R. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol. Sin. 2010, 31, 1421–1430. [Google Scholar]
- Benedict, A.L.; Mountney, A.; Hurtado, A.; Bryan, K.E.; Schnaar, R.L.; Dinkova-Kostova, A.T.; Talalay, P. Neuroprotective effects of sulforaphane after contusive spinal cord injury. J. Neurotrauma 2012, 29, 2576–2586. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; De Rivero Vaccari, J.P.; Wang, H.; Diaz, P.; German, R.; Marcillo, A.E.; Keane, R.W. Activation of the nuclear factor E2-related factor 2/antioxidant response element pathway is neuroprotective after spinal cord injury. J. Neurotrauma 2012, 29, 936–945. [Google Scholar] [CrossRef]
- Mao, L.; Wang, H.; Wang, X.; Liao, H.; Zhao, X. Transcription factor Nrf2 protects the spinal cord from inflammation produced by spinal cord injury. J. Surg. Res. 2011, 170, e105–e115. [Google Scholar] [CrossRef]
- Mao, L.; Wang, H.D.; Wang, X.L.; Qiao, L.; Yin, H.X. Sulforaphane attenuates matrix metalloproteinase-9 expression following spinal cord injury in mice. Ann. Clin. Lab. Sci. 2010, 40, 354–360. [Google Scholar]
- Liu, F.; Huang, J.; Hei, G.; Wu, R.; Liu, Z.; Liu, Z. Effects of sulforaphane on cognitive function in patients with frontal brain damage: Study protocol for a randomised controlled trial. BMJ Open 2020, 10, e037543. [Google Scholar] [CrossRef]
- Zhao, J.; Kobori, N.; Aronowski, J.; Dash, P.K. Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci. Lett. 2006, 393, 108–112. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Ting, S.M.; Song, S.; Zhang, J.; Edwards, N.J.; Aronowski, J. Cleaning up after ICH: The role of Nrf2 in modulating microglia function and hematoma clearance. J. Neurochem. 2015, 133, 144–152. [Google Scholar] [CrossRef]
- Yin, X.P.; Chen, Z.Y.; Zhou, J.; Wu, D.; Bao, B. Mechanisms underlying the perifocal neuroprotective effect of the Nrf2–ARE signaling pathway after intracranial hemorrhage. Drug Des. Dev. Ther. 2015, 9, 5973–5986. [Google Scholar]
- Zhao, X.; Wen, L.; Dong, M.; Lu, X. Sulforaphane activates the cerebral vascular Nrf2–ARE pathway and suppresses inflammation to attenuate cerebral vasospasm in rat with subarachnoid hemorrhage. Brain Res. 2016, 1653, 1–7. [Google Scholar] [CrossRef]
- Ping, Z.; Liu, W.; Kang, Z.; Cai, J.; Wang, Q.; Cheng, N.; Wang, S.; Wang, S.; Zhang, J.H.; Sun, X. Sulforaphane protects brains against hypoxic-ischemic injury through induction of Nrf2-dependent phase 2 enzyme. Brain Res. 2010, 1343, 178–185. [Google Scholar] [CrossRef]
- Nutt, D.; Hayes, A.; Fonville, L.; Zafar, R.; Palmer, E.O.C.; Paterson, L.; Lingford-Hughes, A. Alcohol and the brain. Nutrients 2021, 13, 3938. [Google Scholar] [CrossRef]
- Rusyn, I.; Bataller, R. Alcohol and toxicity. J. Hepatol. 2013, 59, 387–388. [Google Scholar] [CrossRef]
- Crabb, D.W.; Matsumoto, M.; Chang, D.; You, M. In Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 2004, 63, 49–63. [Google Scholar] [CrossRef]
- Ushida, Y.; Talalay, P. Sulforaphane accelerates acetaldehyde metabolism by inducing aldehyde dehydrogenases: Relevance to ethanol intolerance. Alcohol. Alcohol. 2013, 48, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Wehage, S.L.; Talalay, P. Quantification of skin erythema response to topical alcohol in alcohol-intolerant East Asians. Ski. Res. Technol. 2017, 23, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Popova, S.; Charness, M.E.; Burd, L.; Crawford, A.; Hoyme, H.E.; Mukherjee, R.A.S.; Riley, E.P.; Elliott, E.J. Fetal alcohol spectrum disorders. Nat. Rev. Dis. Prim. 2023, 9, 11. [Google Scholar] [CrossRef]
- Yuan, F.; Chen, X.; Liu, J.; Feng, W.; Cai, L.; Wu, X.; Chen, S.Y. Sulforaphane restores acetyl-histone H3 binding to Bcl-2 promoter and prevents apoptosis in ethanol-exposed neural crest cells and mouse embryos. Exp. Neurol. 2018, 300, 60–66. [Google Scholar] [CrossRef]
- Beghi, E.; Giussani, G.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Adib, M.G.; Agrawal, S.; Alahdab, F.; Awasthi, A.; et al. Global, regional, and national burden of epilepsy, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 357–375. [Google Scholar] [CrossRef]
- Zack, M.M.; Kobau, R. National and state estimates of the numbers of adults and children with active epilepsy—United States, 2015. Morb. Mortal. Wkly. Rep. 2017, 66, 821–825. [Google Scholar] [CrossRef]
- Sandouka, S.; Shekh-Ahmad, T. Induction of the nrf2 pathway by sulforaphane is neuroprotective in a rat temporal lobe epilepsy model. Antioxidants 2021, 10, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.H. Status epilepticus: An overview of the clinical problem. Epilepsia 1999, 40, s3–s8. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pozo, C.; Tan, K.N.; Borges, K. Sulforaphane is anticonvulsant and improves mitochondrial function. J. Neurochem. 2015, 135, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Folbergrova, J.; Jesina, P.; Otahal, J. Protective Effect of Sulforaphane on Oxidative Stress and Mitochondrial Dysfunction Associated with Status Epilepticus in Immature Rats. Mol. Neurobiol. 2023, 60, 2024–2035. [Google Scholar] [CrossRef]
- Daněk, J.; Danačíková, Š.; Kala, D.; Svoboda, J.; Kapoor, S.; Pošusta, A.; Folbergrová, J.; Tauchmannová, K.; Mráček, T.; Otáhal, J. Sulforaphane Ameliorates Metabolic Changes Associated With Status Epilepticus in Immature Rats. Front. Cell. Neurosci. 2022, 16, 855161. [Google Scholar] [CrossRef]
- Wennberg, A.M.V.; Wu, M.N.; Rosenberg, P.B.; Spira, A.P. Sleep Disturbance, Cognitive Decline, and Dementia: A Review. Semin. Neurol. 2017, 37, 395–406. [Google Scholar]
- Kikuchi, M.; Aoki, Y.; Kishimoto, N.; Masuda, Y.; Suzuki, N.; Takashimizu, S.; Yoshida, K.; Aizawa, K.; Suganuma, H.; Nishizaki, Y. Effects of glucoraphanin-rich broccoli sprout extracts on sleep quality in healthy adults: An exploratory study. J. Funct. Foods 2021, 84, 104574. [Google Scholar] [CrossRef]
- Li, X.; Ying, H.; Zhang, Z.; Yang, Z.; You, C.; Cai, X.; Lin, Z.; Xiao, Y. Sulforaphane Attenuates Chronic Intermittent Hypoxia-Induced Brain Damage in Mice via Augmenting Nrf2 Nuclear Translocation and Autophagy. Front. Cell Neurosci. 2022, 16, 827527. [Google Scholar] [CrossRef]
- Braley, T.J.; Huber, A.K.; Segal, B.M.; Kaplish, N.; Saban, R.; Washnock-Schmid, J.M.; Chervin, R.D. A randomized, subject and rater-blinded, placebo-controlled trial of dimethyl fumarate for obstructive sleep apnea. Sleep. 2018, 41, zsy109. [Google Scholar] [CrossRef]
- Qiu, X. Biosynthesis of docosahexaenoic acid (DHA, 22:6-4, 7,10,13,16,19): Two distinct pathways. Prostaglandins Leukot. Essent. Fat. Acids 2003, 68, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Pekovic-Vaughan, V.; Gibbs, J.; Yoshitane, H.; Yang, N.; Pathiranage, D.; Guo, B.; Sagami, A.; Taguchi, K.; Bechtold, D.; Loudon, A.; et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes. Dev. 2014, 28, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Raphaely, M. The Impact of Sulforaphane on Sex-Specific Conditions and Hormone Balance: A Comprehensive Review. Appl. Sci. 2025, 15, 522. [Google Scholar] [CrossRef]




| Representative Proteins | Half-Life |
|---|---|
| Biotransformation and detoxification including: AHR, CYP1B1, UGT1A1, GSTMQ, ABCB6, ABCC1, CBR1, EPHX1, ALDH3A2, ADH7, NQO1 | 8–87 h |
| Antioxidant enzymes, including: GPX4, GSR1, TXN1, PRDX1, SRXN1 | 9–54 h |
| Carbohydrate metabolism, including: G6PD, PGD, TALD01, TKT, ME1, UGDH | 37–363 h |
| Lipid Metabolism, including: ACO7, ACOX1, SCD2, NFE2L2 | 0.3–149 h |
| Haem and iron metabolism, including: HMOX1 (HO-1), BLVRA, BLVRB, FECH, FTH1, FTL1 | 0.3–149 h |
| Mediators of inflammation, including: PLA2G7, PTGR1, CEBPB | 6–91 h |
| Protein recycling and turnover, including: PLAMP2, SQSTM1, PSMB1 | 6–133 h |
| Regulation of GSH synthesis, including: GR, GPO, GST, GCL | 18–45 h |
| Disorder |
|---|
| Discussed in this review |
| alcohol-related disorders (including frank toxicity and FAS) |
| Alzheimer’s disease (AD) |
| autism spectrum disorder (ASD) |
| cognitive function and memory impairment |
| diabetes-related cognitive decline |
| epilepsy |
| Huntington’s disease |
| ischemia/reperfusion-induced brain injury |
| mood disorders, depression, and anxiety |
| Parkinson’s disease (PD) |
| pathologies from environmental and food-borne toxins |
| schizophrenia |
| sleep/circadian disorder(s) |
| stroke |
| traumatic brain injury (TBI) and spinal cord injury (SCI) |
| Not further discussed herein |
| amyotrophic lateral sclerosis |
| cerebral palsy |
| Friedreich’s ataxia |
| Herpes-encephalitis-induced neurotoxicity |
| hyperammonemia-induced hepatic encephalopathy |
| multiple sclerosis |
| vascular dementia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahey, J.W.; Liu, H.; Batt, H.; Panjwani, A.A.; Tsuji, P. Sulforaphane and Brain Health: From Pathways of Action to Effects on Specific Disorders. Nutrients 2025, 17, 1353. https://doi.org/10.3390/nu17081353
Fahey JW, Liu H, Batt H, Panjwani AA, Tsuji P. Sulforaphane and Brain Health: From Pathways of Action to Effects on Specific Disorders. Nutrients. 2025; 17(8):1353. https://doi.org/10.3390/nu17081353
Chicago/Turabian StyleFahey, Jed W., Hua Liu, Holly Batt, Anita A. Panjwani, and Petra Tsuji. 2025. "Sulforaphane and Brain Health: From Pathways of Action to Effects on Specific Disorders" Nutrients 17, no. 8: 1353. https://doi.org/10.3390/nu17081353
APA StyleFahey, J. W., Liu, H., Batt, H., Panjwani, A. A., & Tsuji, P. (2025). Sulforaphane and Brain Health: From Pathways of Action to Effects on Specific Disorders. Nutrients, 17(8), 1353. https://doi.org/10.3390/nu17081353