Lifetime Exposure to a Constant Environment Amplifies the Impact of a Fructose-Rich Diet on Glucose Homeostasis during Pregnancy

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Regular Monitoring of Body Weight and Plasma Metabolites
2.3. Oral Glucose Tolerance Tests (OGTT)
2.4. Determination of Body Composition and Tissue Collection
2.5. RNA Extraction and Determination of Hepatic Gene Expression
2.6. Statistical Analysis
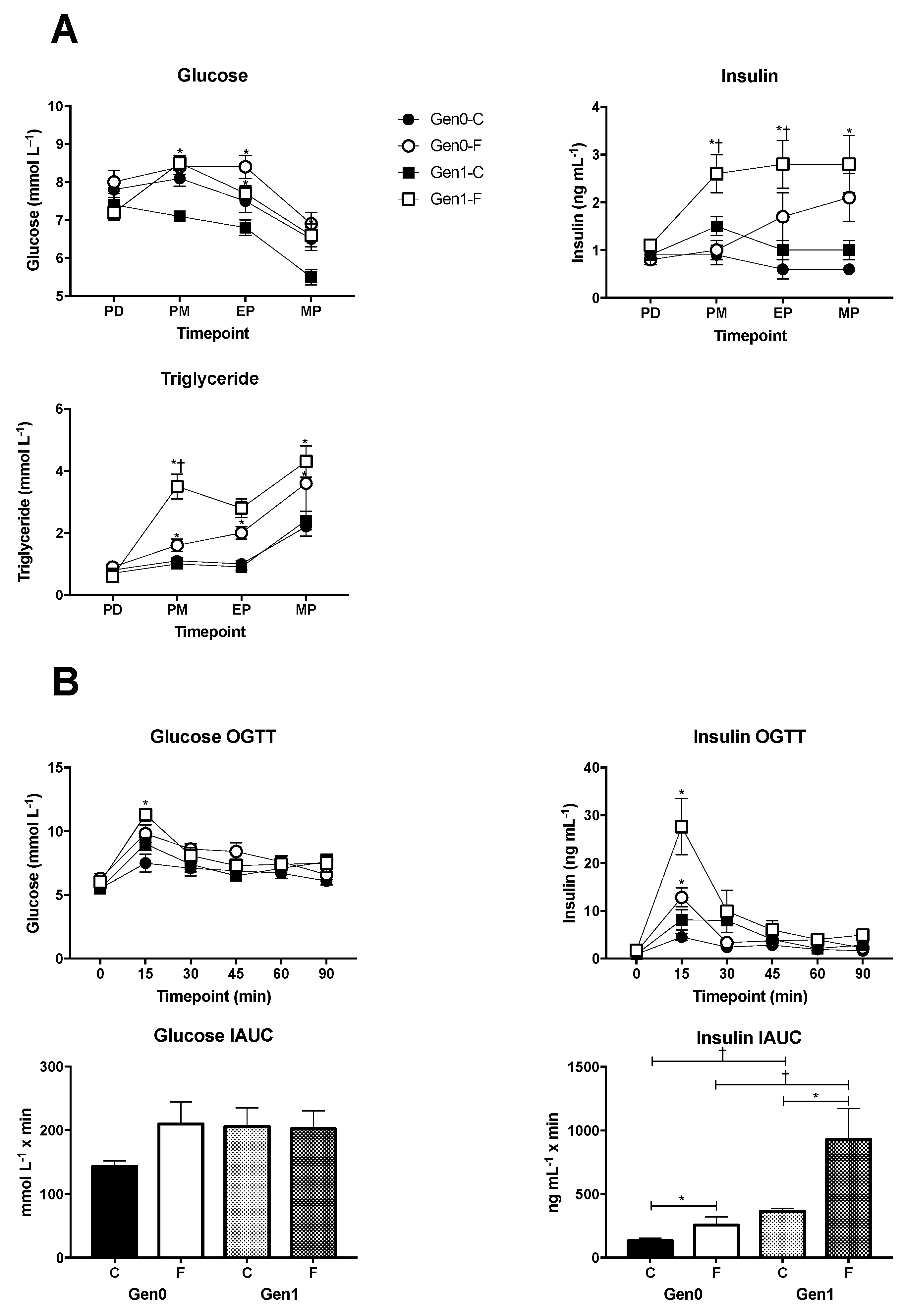
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roche, H.M.; Phillips, C.; Gibney, M.J. The metabolic syndrome: The crossroads of diet and genetics. Proc. Nutr. Soc. 2005, 64, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Ganda, O.P. Migration and its impact on adiposity and type 2 diabetes. Nutrition 2007, 23, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E.; Sebert, S.P.; Hyatt, M.A.; Budge, H. Nutritional programming of the metabolic syndrome. Nat. Rev. Endocrinol. 2009, 5, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, C.B. Dietary fructose effects on lipoprotein metabolism and risk for coronary artery disease. Am. J. Clin. Nutr. 1993, 58, 800S–809S. [Google Scholar] [PubMed]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. BMJ 2015, 351, h3576. [Google Scholar] [CrossRef] [PubMed]
- Sloboda, D.M.; Li, M.; Patel, R.; Clayton, Z.E.; Yap, C.; Vickers, M.H. Early Life Exposure to Fructose and Offspring Phenotype: Implications for Long Term Metabolic Homeostasis. J. Obes. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lineker, C.; Kerr, P.M.; Nguyen, P.; Bloor, I.; Astbury, S.; Patel, N.; Budge, H.; Hemmings, D.G.; Plane, F.; Symonds, M.E.; et al. High fructose consumption in pregnancy alters the perinatal environment without increasing metabolic disease in the offspring. Reprod. Fertil. Dev. 2016, 28, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Alzamendi, A.; Del Zotto, H.; Castrogiovanni, D.; Romero, J.; Giovambattista, A.; Spinedi, E. Oral Metformin Treatment Prevents Enhanced Insulin Demand and Placental Dysfunction in the Pregnant Rat Fed a Fructose-Rich Diet. Int. Sch. Res. Not. 2012, 2012, e757913. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, L.; Panadero, M.I.; Rodrigo, S.; Roglans, N.; Otero, P.; Álvarez-Millán, J.J.; Laguna, J.C.; Bocos, C. Liquid fructose in pregnancy exacerbates fructose-induced dyslipidemia in adult female offspring. J. Nutr. Biochem. 2016, 32, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Clayton, Z.E.; Yap, C.; Sloboda, D.M. Maternal fructose intake during pregnancy and lactation alters placental growth and leads to sex-specific changes in fetal and neonatal endocrine function. Endocrinology 2011, 152, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Kumazawa, M.; Sato, S. Fructose intake during pregnancy up-regulates the expression of maternal and fetal hepatic sterol regulatory element-binding protein-1c in rats. Endocrine 2012, 44, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Bayol, S.A.; Simbi, B.H.; Stickland, N.C. A maternal cafeteria diet during gestation and lactation promotes adiposity and impairs skeletal muscle development and metabolism in rat offspring at weaning. J. Physiol. 2005, 567, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Nivoit, P.; Morens, C.; Assche, F.A.V.; Jansen, E.; Poston, L.; Remacle, C.; Reusens, B. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 2009, 52, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Investig. 2009, 119, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T.J.; Moyce, B.L.; Kereliuk, S.M.; Dolinsky, V.W. Influence of maternal overnutrition and gestational diabetes on the programming of metabolic health outcomes in the offspring: Experimental evidence. Biochem. Cell Biol. 2014, 93, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, F.C.; Meschia, G. Principal substrates of fetal metabolism. Physiol. Rev. 1978, 58, 499–527. [Google Scholar] [PubMed]
- Rawana, S.; Clark, K.; Zhong, S.; Buison, A.; Chackunkal, S.; Jen, K.L. Low dose fructose ingestion during gestation and lactation affects carbohydrate metabolism in rat dams and their offspring. J. Nutr. 1993, 123, 2158–2165. [Google Scholar] [PubMed]
- Bowe, J.E.; Franklin, Z.J.; Hauge-Evans, A.C.; King, A.J.; Persaud, S.J.; Jones, P.M. Metabolic phenotyping guidelines: Assessing glucose homeostasis in rodent models. J. Endocrinol. 2014, 222, G13–G25. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, S.; Blair, A.R.; Deluca, N.; Fam, B.C.; Proietto, J. Evaluating the glucose tolerance test in mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1323–E1332. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Proietto, J. The feto-placental glucose steal phenomenon is a major cause of maternal metabolic adaptation during late pregnancy in the rat. Diabetologia 1994, 37, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115–e115. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bocarsly, M.E.; Powell, E.S.; Avena, N.M.; Hoebel, B.G. High-fructose corn syrup causes characteristics of obesity in rats: Increased body weight, body fat and triglyceride levels. Pharmacol. Biochem. Behav. 2010, 97, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Teff, K.L.; Elliott, S.S.; Tschöp, M.; Kieffer, T.J.; Rader, D.; Heiman, M.; Townsend, R.R.; Keim, N.L.; D’Alessio, D.; Havel, P.J. Dietary Fructose Reduces Circulating Insulin and Leptin, Attenuates Postprandial Suppression of Ghrelin, and Increases Triglycerides in Women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Wood, I.S.; Trayhurn, P. Glucose transporters (GLUT and SGLT): Expanded families of sugar transport proteins. Br. J. Nutr. 2003, 89, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Giacchetti, G.; Novello, M.; Colussi, G.; Cavarape, A.; Sechi, L.A. Cellular mechanisms of insulin resistance in rats with fructose-induced hypertension. Am. J. Hypertens. 2003, 16, 973–978. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Chehade, J.; Hurd, R.; Haas, M.J. Monosaccharide-enriched diets cause hyperleptinemia without hypophagia. Nutrition 2000, 16, 439–441. [Google Scholar] [CrossRef]
- Gray, C.; Long, S.; Green, C.; Gardiner, S.M.; Craigon, J.; Gardner, D.S. Maternal Fructose and/or Salt Intake and Reproductive Outcome in the Rat: Effects on Growth, Fertility, Sex Ratio, and Birth Order. Biol. Reprod. 2013, 89, 51. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Lê, K.-A. Metabolic Effects of Fructose and the Worldwide Increase in Obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Age/Pregnancy Stage | Gen0-C | Gen0-F | Gen1-C | Gen1-F |
|---|---|---|---|---|
| Body Weight (g) | ||||
| Week 8/Pre-diet | 200.6 ± 3.3 | 204.1 ± 3.1 | 225.1 ± 6.3 † | 226.4 ± 5.7 † |
| Week 9 | 227.0 ± 2.0 | 238.3 ± 2.9 | 254.5 ± 6.6 † | 252.3 ± 6.5 † |
| Week 10 | 247.3 ± 2.3 | 257.2 ± 3.1 | 280.7 ± 6.8 † | 285.9 ± 7.7 † |
| Week 11/Pre-mating | 261.5 ± 3.0 | 277.7 ± 4.7 | 305.5 ± 7.4 † | 316.3 ± 9.3 † |
| Week 12/ Early pregnancy | 290.5 ± 2.3 | 304.9 ± 5.3 | 332.2 ± 6.8 † | 352.3 ± 12.9 † |
| Week 13/ Mid pregnancy | 329.4 ± 5.1 | 337.6 ± 5.9 | 383.8 ± 7.6 † | 393.3 ± 14.3 † |
| Week 14/ Late pregnancy | 386.5 ± 5.4 | 403.1 ± 6.7 | 428.7 ± 12.3 † | 444.4 ± 15.6 † |
| Body Composition (%) | ||||
| Fat mass at GD21 | 11.2 ± 0.7 | 15.2 ± 1.0 * | 11.4 ± 0.5 | 16.7 ± 1.3 * |
| Feto-placental unit | ||||
| Number of pups | 15.5 ± 1.7 | 16.0 ± 3.2 | 16.3 ± 2.3 | 17.0 ± 3.3 |
| Placental weight (g) | 0.53 ± 0.02 | 0.48 ± 0.02 | 0.57 ± 0.02 | 0.47 ± 0.01 * |
| Fetal weight (g) | 3.88 ± 0.19 | 3.36 ± 0.14 * | 4.07 ± 0.05 | 3.57 ± 0.09 * |
| The ratio of placental:fetal weight | 0.14 ± 0.01 | 0.15 ± 0.01 | 0.14 ± 0.00 | 0.13 ± 0.01 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, A.; Astbury, S.; Hoedl, A.; Nielsen, B.; Symonds, M.E.; Bell, R.C. Lifetime Exposure to a Constant Environment Amplifies the Impact of a Fructose-Rich Diet on Glucose Homeostasis during Pregnancy. Nutrients 2017, 9, 327. https://doi.org/10.3390/nu9040327
Song A, Astbury S, Hoedl A, Nielsen B, Symonds ME, Bell RC. Lifetime Exposure to a Constant Environment Amplifies the Impact of a Fructose-Rich Diet on Glucose Homeostasis during Pregnancy. Nutrients. 2017; 9(4):327. https://doi.org/10.3390/nu9040327
Chicago/Turabian StyleSong, Aleida, Stuart Astbury, Abha Hoedl, Brent Nielsen, Michael E. Symonds, and Rhonda C. Bell. 2017. "Lifetime Exposure to a Constant Environment Amplifies the Impact of a Fructose-Rich Diet on Glucose Homeostasis during Pregnancy" Nutrients 9, no. 4: 327. https://doi.org/10.3390/nu9040327
APA StyleSong, A., Astbury, S., Hoedl, A., Nielsen, B., Symonds, M. E., & Bell, R. C. (2017). Lifetime Exposure to a Constant Environment Amplifies the Impact of a Fructose-Rich Diet on Glucose Homeostasis during Pregnancy. Nutrients, 9(4), 327. https://doi.org/10.3390/nu9040327