High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions

Abstract
:1. Introduction
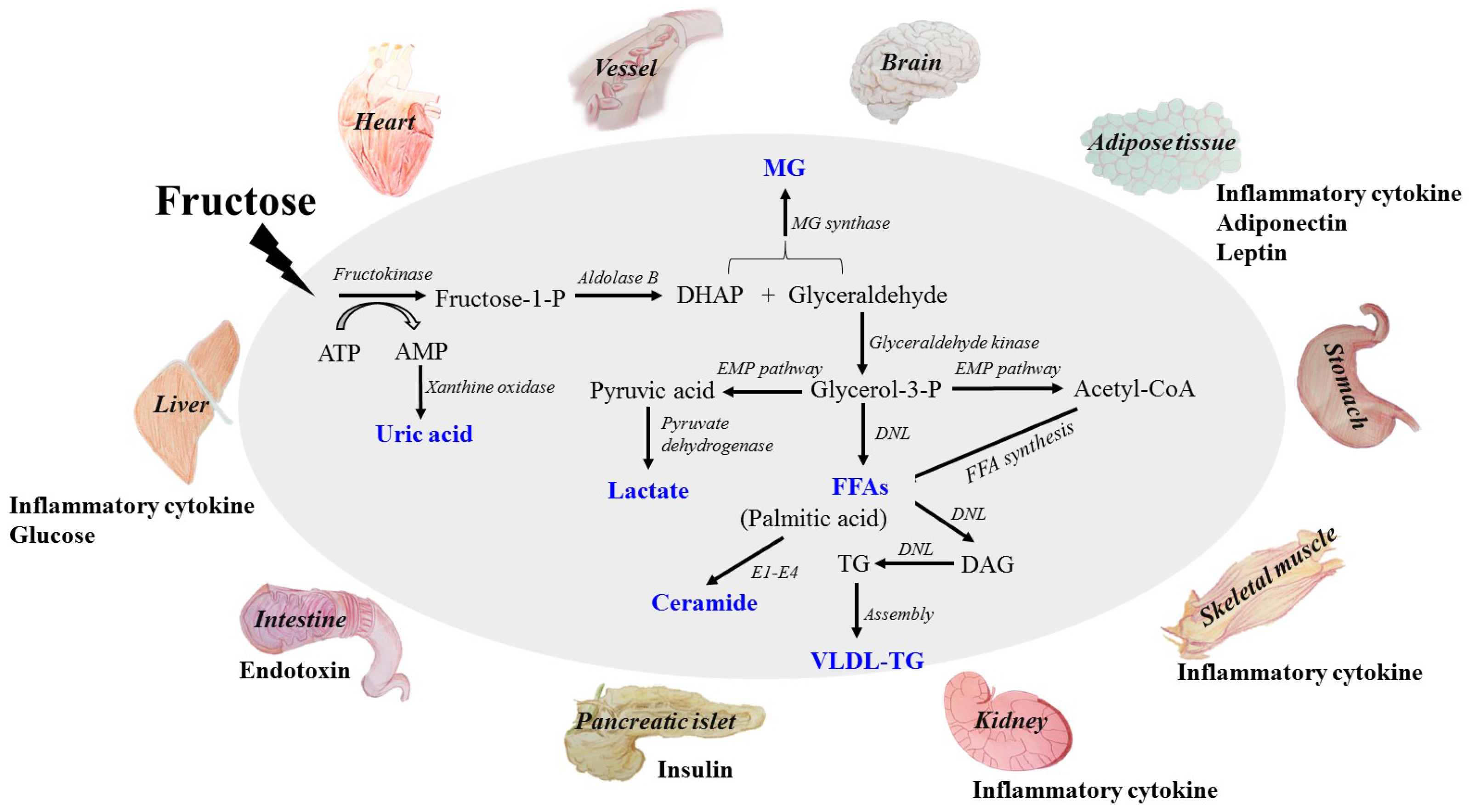
2. Absorption and Metabolism of Fructose
3. Direct Dangerous Factors under High Fructose Consumption
3.1. Glucose
3.2. Lactate
3.3. Free Fatty Acids (FFAs)
3.4. Uric Acid (UA)
3.5. Methylglyoxal (MG)
4. Indirect Dangerous Factors in Tissue and Organ Dysfunctions under High Fructose Consumption
4.1. Inflammatory Cytokines
4.2. Adiponectin
4.3. Leptin
4.4. Endotoxin
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACC | acetyl CoA carboxylase |
| AdipoR | adiponectin receptor |
| AGEs | advanced glycation endproducts |
| AS160 | Akt substrate-160 kDa |
| AMPK | AMP-activated protein kinase |
| ApoB100 | apolipoprotein B100 |
| ATG7 | autophagy-related gene 7 |
| Bax | Bcl-2-associated X protein |
| ChREBP | carbohydrate response element binding protein |
| Cideb | cell death-inducing DFF45-like effector b |
| CVD | cardiovascular disease |
| CPT1 | carnitine-palmitoyl-CoA transferase-1 |
| CXCL8 | chemokine interleukin 8 |
| CKD | chronic kidney diseases |
| COX-2 | cyclooxygenase-2 |
| DNL | de novo lipogenesis |
| DR5 | death receptor 5 |
| DAG | diacylglycerol |
| DHAP | dihydroxyacetone phosphate |
| ER | endoplasmic reticulum |
| FGF-21 | fibroblast growth factor-21 |
| FKN | fractalkine |
| FFA | free fatty acid |
| FFAR1 | free fatty acid receptor 1 |
| KHK | fructokinase |
| FATPs | FA transport proteins |
| FoxO1 | Forkhead box protein O1 |
| RAGE | glycation end products |
| GS | glycogen synthase |
| G6P | glucose-6-phosphatase |
| GLUT5 | glucose transporter 5 |
| HK | hexokinase |
| HDL | high-density lipoprotein |
| HFCS | high-fructose corn syrup |
| HUVECs | human umbilical endothelial cells |
| NPY | hunger peptide neuropeptide Y |
| HIF-1 | hypoxia-inducible factor-1 |
| iNOS | inducible NOS |
| IR | insulin receptor |
| CAM-1 | intercellular adhesion molecule-1 |
| IL | interleukin |
| IIRAK4/1 | IL-1R-associated kinase 4/1 |
| IKK-β | IκB kinase-β |
| JNK | c-jun N-terminal kinase |
| LEPR | leptin receptor |
| LCN-2 | lipocalin-2 |
| LBP | lipopolysaccharide-binding protein |
| LDL | low-density lipoprotein |
| LDLR | LDL receptor |
| LAMP2 | lysosomal-associated membrane protein 2 |
| MDA | malondialdehyde |
| MetS | metabolic syndrome |
| MG | methylglyoxal |
| MAP1LC3β | microtubule-associated protein 1 light chain 3 beta |
| MCP-1 | monocyte chemotactic protein-1 |
| NOX4 | NADPH oxidase 4 |
| NLRP3 | NOD-like receptor superfamily, pyrin domain containing 3 |
| NAFLD | non-alcoholic fatty liver |
| NF-κB | nuclear factor kappa B |
| OAT1 | organic anion transporter 1 |
| PYY | peptide YY3-36 |
| PPAR-δ | peroxisome proliferator-activated receptor-δ |
| PI3K | phosphatidylinositol 3-kinase |
| PFK | phosphofructokinase |
| PAI-1 | plasminogen activator inhibitor-1 |
| PEPCK | phosphoenolpyruvate carboxykinase |
| PUFA | polyunsaturated fatty acid |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| PGE2 | prostaglandin E2 |
| PKB/Akt | protein kinase B |
| PKC | protein kinase C |
| PP1c | protein phosphatase 1c |
| PTP-1B | protein tyrosine phosphatase-1B |
| POMC | satiety peptide pro-opiomelanocortin |
| ROS | reactive oxygen species |
| RST | renal specific transporter |
| RAAS | renin-angiotensin-aldosterone system |
| CD36 | scavenger receptor 36 |
| SR-BI | scavenger receptor class B type I |
| LKB1 | serine-threonine protein kinase 1 |
| SGBS | Simpson-Golabi-Behmel syndrome |
| α-SMA | α-smooth muscle actin |
| SphK1 | sphingosine kinase SphK1 |
| S1P | sphingosine 1-phosphate |
| SCD-1 | stearoyl-coenzyme A desaturase-1 |
| SREBP-1c | sterol regulatory element-binding protein 1c |
| TR | taste receptor |
| TXNIP | thioredoxin-interacting protein |
| TF | tissue factor |
| TGF-β1 | transforming growth factor-β1 |
| TRIB3 | tribbles homolog 3 |
| TCA | tricarboxylic acid cycle |
| TG | triglyceride |
| TLR4 | toll-like receptor 4 |
| TNF-α | tumor necrosis factor-α |
| T2DM | type 2 diabetes |
| UAT | urate transporter |
| UA | uric acid |
| VLDL | very low-density lipoprotein |
| WAT | white adipose tissue |
| XO | xanthine oxidase |
References
- Marchesini, G.; Forlani, G.; Cerrelli, F.; Manini, R.; Natale, S.; Baraldi, L.; Ermini, G.; Savorani, G.; Zocchi, D.; Melchionda, N. WHO and ATPIII proposals for the definition of the metabolic syndrome in patients with type 2 diabetes. Diabet. Med. 2004, 21, 383–387. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E. Insulin resistance. A multifaceted syndrome responsible for niddm, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991, 14, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.F.; Butler, R.N.; Brooks, D.A. Intestinal fructose transport and malabsorption in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G202–G206. [Google Scholar] [CrossRef] [PubMed]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, S754–S765. [Google Scholar]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Tounian, P.; Schneiter, P.; Henry, S.; Jéquier, E.; Tappy, L. Effects of infused fructose on endogenous glucose production, gluconeogenesis, and glycogen metabolism. Am. J. Physiol. 1994, 13, 710–717. [Google Scholar] [CrossRef]
- Froesch, E.R. Fructose metabolism in adipose tissue. Acta Med. Scand. Suppl. 1972, 542, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Dirlewanger, M.; Schneiter, P.; Jequier, E.; Tappy, L. Effects of fructose on hepatic glucose metabolism in humans. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E907–E911. [Google Scholar] [PubMed]
- Baena, M.; Sangüesa, G.; Dávalos, A.; Latasa, M.-J.; Sala-Vila, A.; Sánchez, R.M.; Roglans, N.; Laguna, J.C.; Alegret, M. Fructose, but not glucose, impairs insulin signaling in the three major insulin-sensitive tissues. Sci. Rep. 2016, 6, 26149. [Google Scholar] [CrossRef] [PubMed]
- Aeberli, I.; Gerber, P.A.; Hochuli, M.; Kohler, S.; Haile, S.R.; Gouni-Berthold, I.; Berthold, H.K.; Spinas, G.A.; Berneis, K. Low to moderate sugar-sweetened beverage consumption impairs glucose and lipid metabolism and promotes inflammation in healthy young men: A randomized controlled trial. Am. J. Clin. Nutr. 2011, 94, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, M.; Raji, L.; Prabhu, D.; Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. High-fructose diet is as detrimental as high-fat diet in the induction of insulin resistance and diabetes mediated by hepatic/pancreatic endoplasmic reticulum (ER) stress. Mol. Cell. Biochem. 2016, 423, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Kyriazis, G.A.; Soundarapandian, M.M.; Tyrberg, B. Sweet taste receptor signaling in beta cells mediates fructose-induced potentiation of glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. USA 2012, 109, E524–E532. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Wang, W.; Fan, C.Y.; Wang, M.X.; Zhang, X.; Hu, Q.H.; Kong, L.D. Quercetin preserves β-cell mass and function in fructose-induced hyperinsulinemia through modulating pancreatic Akt/FoxO1 activation. Evid.-Based Complement. Altern. Med. eCAM 2013, 2013, 303902. [Google Scholar]
- Heden, T.D.; Liu, Y.; Park, Y.M.; Nyhoff, L.M.; Winn, N.C.; Kanaley, J.A. Moderate amounts of fructose- or glucose-sweetened beverages do not differentially alter metabolic health in male and female adolescents. Am. J. Clin. Nutr. 2014, 100, 796–805. [Google Scholar] [CrossRef] [PubMed]
- DiGirolamo, M.; Newby, F.D.; Lovejoy, J. Lactate production in adipose tissue: A regulated function with extra-adipose implications. FASEB J. 1992, 6, 2405–2412. [Google Scholar] [PubMed]
- Kristensen, M.; Albertsen, J.; Rentsch, M.; Juel, C. Lactate and force production in skeletal muscle. J. Physiol. 2005, 562, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.; Boros, L.G.; Nolen, G.T.; Chang, C.W.; Wabitsch, M.; Beger, R.D.; Kaput, J. Fructose alters intermediary metabolism of glucose in human adipocytes and diverts glucose to serine oxidation in the one-carbon cycle energy producing pathway. Metabolites 2015, 5, 364–385. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, J.; Newby, F.D.; Gebhart, S.S.; Digirolamo, M. Insulin resistance in obesity is associated with elevated basal lactate levels and diminished lactate appearance following intravenous glucose and insulin. Metab. Clin. Exp. 1992, 41, 22. [Google Scholar] [CrossRef]
- Choi, C.S.; Kim, Y.B.; Lee, F.N.; Zabolotny, J.M.; Kahn, B.B.; Youn, J.H. Lactate induces insulin resistance in skeletal muscle by suppressing glycolysis and impairing insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E233–E240. [Google Scholar] [CrossRef] [PubMed]
- Leite, T.C.; Coelho, R.G.; Da Silva, D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Sola-Penna, M. Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Lett. 2011, 585, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.M.; Fabris, R.; Bassetto, F.; Serra, R.; Leturque, A.; Federspil, G.; Girard, J.; Vettor, R. Hyperlactatemia reduces muscle glucose uptake and glut-4 mrna while increasing (e1alpha)pdh gene expression in rat. Am. J. Physiol. 1999, 276, E922. [Google Scholar] [PubMed]
- Leturque, A.; Loizeau, M.; Vaulont, S.; Salminen, M.; Girard, J. Improvement of insulin action in diabetic transgenic mice selectively overexpressing GLUT4 in skeletal muscle. Diabetes 1996, 45, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D.; Brechtel, G.; Wallace, P.; Edelman, S.V. Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am. J. Physiol. 1988, 255, E769–E774. [Google Scholar] [PubMed]
- Regazzetti, C.; Peraldi, P.; Gremeaux, T.; Najem-Lendom, R.; Ben-Sahra, I.; Cormont, M.; Bost, F.; Le Marchand-Brustel, Y.; Tanti, J.F.; Giorgetti-Peraldi, S. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes 2009, 58, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, W.; Chuang, G.C.; Hill, H.S.; Tian, L.; Fu, Y.; Moellering, D.R.; Garvey, W.T. Role of TRIB3 in regulation of insulin sensitivity and nutrient metabolism during short-term fasting and nutrient excess. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E908–E916. [Google Scholar] [CrossRef] [PubMed]
- Marette, A. Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 377. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- De Stefanis, D.; Mastrocola, R.; Nigro, D.; Costelli, P.; Aragno, M. Effects of chronic sugar consumption on lipid accumulation and autophagy in the skeletal muscle. Eur. J. Nutr. 2017, 56, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, S.; Toufektsian, M.C.; Grauzam, S.; de Leiris, J.; Ghezzi, C.; Boucher, F.; Sulpice, T. Cardiac dysfunction in rats with dietary-induced insulin resistance associated with pharmacologically-induced dyslipidemia. Curr. Pharm. Des. 2013, 19, 6906–6911. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, S.; Nestorov, J.; Matic, G.; Elakovic, I. Fructose-enriched diet induces inflammation and reduces antioxidative defense in visceral adipose tissue of young female rats. Eur. J. Nutr. 2017, 56, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, P.; Susztak, K. Sweet debate: Fructose versus glucose in diabetic kidney disease. J. Am. Soc. Nephrol. 2014, 25, 2386–2388. [Google Scholar] [CrossRef] [PubMed]
- Pokrywczynska, M.; Flisinski, M.; Jundzill, A.; Krzyzanowska, S.; Brymora, A.; Deptula, A.; Bodnar, M.; Kloskowski, T.; Stefanska, A.; Marszalek, A.; et al. Impact of fructose diet and renal failure on the function of pancreatic islets. Pancreas 2014, 43, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Vilela, T.C.; Taschetto, L.; Vuolo, F.; Petronilho, F.; Dal-Pizzol, F.; Streck, E.L.; Ferreira, G.C.; Schuck, P.F. Evaluation of the effects of fructose on oxidative stress and inflammatory parameters in rat brain. Mol. Neurobiol. 2014, 50, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. Chrebp regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P.; Cheng, M.L.; Hung, C.Y.; Wang, C.H.; Hsieh, P.S.; Shiao, M.S.; Chen, J.K.; Li, D.E.; Hung, L.M. Docosapentaenoic acid and docosahexaenoic acid are positively associated with insulin sensitivity in rats fed high-fat and high-fructose diets. J. Diabetes 2016. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating tnf-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Bergman, R.N.; Ader, M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol. Metab. 2000, 11, 351–356. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Befroy, D.E.; Codella, R.; Kim, S.; Reznick, R.M.; Hwang, Y.J.; Liu, Z.X.; Lee, H.Y.; Distefano, A.; Samuel, V.T.; et al. Paradoxical effects of increased expression of pgc-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 19926–19931. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Li, Y.C.; Kong, L.D.; Hu, Q.H. Curcumin inhibits hepatic protein-tyrosine phosphatase 1b and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology 2010, 51, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Lupi, R.; Dotta, F.; Marselli, L.; Del, G.S.; Masini, M.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Shimano, H.; Yamamoto, T.; Ishikawa, M.; Kumadaki, S.; Matsuzaka, T.; Nakagawa, Y.; Yahagi, N.; Nakakuki, M.; Hasty, A.H.; et al. Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of SREBP-1c in pancreatic islets. Diabetes 2008, 57, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Amery, C.M.; Nattrass, M. Fatty acids and insulin secretion. Br. J. Nutr. 2000, 2, 213. [Google Scholar] [CrossRef]
- Hu, M.; Lin, H.; Yang, L.; Cheng, Y.; Zhang, H. Interleukin-22 restored mitochondrial damage and impaired glucose-stimulated insulin secretion through down-regulation of uncoupling protein-2 in INS-1 cells. J. Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, W.; Würdemann, W.; Plötz, T.; Jörns, A.; Lenzen, S.; Elsner, M. Antagonism between saturated and unsaturated fatty acids in ros mediated lipotoxicity in rat insulin-producing cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Song, Y.; Zhang, L.J.; Li, F.F.; Gu, Y.; Zhang, J.; Dong, W.P.; Xue, L.; Zhang, L.Y.; Liu, F. Cell death-inducing DFF45-like effector b (Cideb) is present in pancreatic beta-cells and involved in palmitate induced beta-cell apoptosis. Diabetes/Metab. Res. Rev. 2012, 28, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Yang, L.; Yan, S.; Wei, W.; Liang, L.; Zheng, H.; Cai, X. The effect of FFAR1 on pioglitazone-mediated attenuation of palmitic acid-induced oxidative stress and apoptosis in βTC6 cells. Metab. Clin. Exp. 2013, 63, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Habibi, J.; Ford, D.A.; Nistala, R.; Lastra, G.; Manrique, C.; Dunham, M.M.; Ford, K.D.; Thyfault, J.P.; Parks, E.J.; et al. Dipeptidyl peptidase-4 inhibition ameliorates western diet-induced hepatic steatosis and insulin resistance through hepatic lipid remodeling and modulation of hepatic mitochondrial function. Diabetes 2015, 64, 1988–2001. [Google Scholar] [CrossRef] [PubMed]
- Vila, L.; Roglans, N.; Alegret, M.; Sánchez, R.M.; Vázquez-Carrera, M.; Laguna, J.C. Suppressor of cytokine signaling-3 (SOCS-3) and a deficit of serine/threonine (Ser/Thr) phosphoproteins involved in leptin transduction mediate the effect of fructose on rat liver lipid metabolism. Hepatology 2008, 48, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.; Chang, H.; Azhar, S.; Reaven, G.M. Tissue-dependent activation of protein kinase c in fructose-induced insulin resistance. Endocrine 1995, 3, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Loffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Protein kinase c and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol activation of protein kinase cepsilon and hepatic insulin resistance. Cell Metab. 2012, 15, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Brozinick, J.T.; Hawkins, E.; Hoang Bui, H.; Kuo, M.S.; Tan, B.; Kievit, P.; Grove, K. Plasma sphingolipids are biomarkers of metabolic syndrome in non-human primates maintained on a western-style diet. Int. J. Obes. 2013, 37, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.J.; Baker, C.; Naples, M.; Samsoondar, J.; Zhang, R.; Qiu, W.; Sacco, J.; Adeli, K. Inhibition of sphingolipid synthesis improves dyslipidemia in the diet-induced hamster model of insulin resistance: Evidence for the role of sphingosine and sphinganine in hepatic VLDL-apoB100 overproduction. Atherosclerosis 2013, 228, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Cigliano, L.; Liverini, G.; Iossa, S. Increased skeletal muscle mitochondrial efficiency in rats with fructose-induced alteration in glucose tolerance. Br. J. Nutr. 2013, 110, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Kralik, S.F.; Liu, P.; Leffler, B.J.; Elmendorf, J.S. Ceramide and glucosamine antagonism of alternate signaling pathways regulating insulin- and osmotic shock-induced glucose transporter 4 translocation. Endocrinology 2002, 143, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, D.M.; Gu, T.T.; Ding, X.Q.; Fan, C.Y.; Zhu, Q.; Shi, Y.W.; Hong, Y.; Kong, L.D. Morin reduces hepatic inflammation-associated lipid accumulation in high fructose-fed rats via inhibiting sphingosine kinase 1/sphingosine 1-phosphate signaling pathway. Biochem. Pharmacol. 2013, 86, 1791. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Tussellino, M.; Carotenuto, R.; Liverini, G.; Iossa, S. Fructose supplementation worsens the deleterious effects of short-term high-fat feeding on hepatic steatosis and lipid metabolism in adult rats. Exp. Physiol. 2014, 99, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Liverini, G.; Iossa, S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur. J. Nutr. 2013, 52, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Maurya, C.K.; Arha, D.; Avisetti, D.R.; Prathapan, A.; Raj, P.S.; Raghu, K.G.; Kalivendi, S.V.; Tamrakar, A.K. Fructose induces mitochondrial dysfunction and triggers apoptosis in skeletal muscle cells by provoking oxidative stress. Apoptosis 2015, 20, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Cree-Green, M.; Gupta, A.; Coe, G.V.; Baumgartner, A.D.; Pyle, L.; Reusch, J.E.; Brown, M.S.; Newcomer, B.R.; Nadeau, K.J. Insulin resistance in type 2 diabetes youth relates to serum free fatty acids and muscle mitochondrial dysfunction. J. Diabetes Complicat. 2017, 31, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology 2015, 149, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.L.; Zhang, D.M.; Ma, C.H.; Zhang, J.H.; Jia, K.K.; Liu, J.H.; Wang, R.; Kong, L.D. Cinnamaldehyde and allopurinol reduce fructose-induced cardiac inflammation and fibrosis by attenuating CD36-mediated TLR4/6-IRAK4/1 signaling to suppress NLRP3 inflammasome activation. Sci. Rep. 2016, 6, 27460. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.C.; Liu, C.P.; Cheng, W.H.; Chen, B.R.; Lu, P.J.; Cheng, P.W.; Ho, W.Y.; Sun, G.C.; Liou, J.C.; Tseng, C.J. Caffeine intake improves fructose-induced hypertension and insulin resistance by enhancing central insulin signaling. Hypertension 2014, 63, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Coburn, C.T.; Hajri, T.; Ibrahimi, A.; Abumrad, N.A. Role of CD36 in membrane transport and utilization of long-chain fatty acids by different tissues. J. Mol. Neurosc. MN 2001, 16, 117–121. [Google Scholar] [CrossRef]
- Baena, M.; Sanguesa, G.; Hutter, N.; Beltran, J.M.; Sanchez, R.M.; Roglans, N.; Alegret, M.; Laguna, J.C. Liquid fructose in western-diet-fed mice impairs liver insulin signaling and causes cholesterol and triglyceride loading without changing calorie intake and body weight. J. Nutr. Biochem. 2017, 40, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Pan, Y.; Chonan, R.; Batey, R.; Rong, X.; Yamahara, J.; Wang, J.; Li, Y. Mitigation of insulin resistance by mangiferin in a rat model of fructose-induced metabolic syndrome is associated with modulation of CD36 redistribution in the skeletal muscle. J. Pharmacol. Exp. Ther. 2016, 356, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Oppelt, S.A.; Zhang, W.; Tolan, D.R. Specific regions of the brain are capable of fructose metabolism. Brain Res. 2017, 1657, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. New insights into the roles of proteins and lipids in membrane transport of fatty acids. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M. Influences of hepatic portal receptors on hypothalamic feeding and satiety centers. Am. J. Physiol. 1973, 225, 1089–1095. [Google Scholar] [PubMed]
- Bode, J.C.; Zelder, O.; Rumpelt, H.J.; Wittkamp, U. Depletion of liver adenosine phosphates and metabolic effects of intravenous infusion of fructose or sorbitol in man and in the rat. Eur J. Clin. Investig. 1973, 3, 436–441. [Google Scholar] [CrossRef]
- Kanuri, G.; Spruss, A.; Wagnerberger, S.; Bischoff, S.C.; Bergheim, I. Role of tumor necrosis factor alpha (TNFalpha) in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J. Nutr. Biochem. 2011, 22, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.H.; Wolfgang, M.; Tokutake, Y.; Chohnan, S.; Lane, M.D. Differential effects of central fructose and glucose on hypothalamic malonyl-coa and food intake. Proc. Natl. Acad. Sci. USA 2008, 105, 16871–16875. [Google Scholar] [CrossRef] [PubMed]
- Kinote, A.; Faria, J.A.; Roman, E.A.; Solon, C.; Razolli, D.S.; Ignacio-Souza, L.M.; Sollon, C.S.; Nascimento, L.F.; de Araujo, T.M.; Barbosa, A.P.; et al. Fructose-induced hypothalamic ampk activation stimulates hepatic pepck and gluconeogenesis due to increased corticosterone levels. Endocrinology 2012, 153, 3633–3645. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B. Resolved: There is sufficient scientific evidence that decreasing sugar-sweetened beverage consumption will reduce the prevalence of obesity and obesity-related diseases. Obes. Rev. Off. J. Int. Assoc. Stud. Obes. 2013, 14, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Miyashita, M.; Cho, B.H.; Nakamura, M.T. Replacing dietary glucose with fructose increases chrebp activity and SREBP-1 protein in rat liver nucleus. Biochem. Biophys. Res. Commun. 2009, 390, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Singh, A.B.; Azhar, S.; Seidah, N.G.; Liu, J. High-fructose feeding promotes accelerated degradation of hepatic LDL receptor and hypercholesterolemia in hamsters via elevated circulating PCSK9 levels. Atherosclerosis 2015, 239, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Kalra, J. Biochemical mechanism of hypercholesterolemia-induced atherosclerosis. Clin. Biochem. 1993, 26, 128. [Google Scholar] [CrossRef]
- Spady, D.K.; Dietschy, J.M. Interaction of dietary cholesterol and triglycerides in the regulation of hepatic low density lipoprotein transport in the hamster. J. Clin. Investig. 1988, 81, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Medici, V.; Bremer, A.A.; Lee, V.; Lam, H.D.; Nunez, M.V.; Chen, G.X.; Keim, N.L.; Havel, P.J. A dose-response study of consuming high-fructose corn syrup-sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am. J. Clin. Nutr. 2015, 101, 1144. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.S.; Tremblay, M.; Batal, R.; Jacques, H.; Rodriguez, C.; Steiner, G.; Mamer, O.; Davignon, J. Increased apoc-iii production is a characteristic feature of patients with hypertriglyceridemia. Atherosclerosis 2004, 177, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Pavlic, M.; Valero, R.; Duez, H.; Xiao, C.; Szeto, L.; Patterson, B.W.; Lewis, G.F. Triglyceride-rich lipoprotein-associated apolipoprotein c-iii production is stimulated by plasma free fatty acids in humans. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Arai, T.; Ji, Y.; Rinninger, F.; Tall, A.R. Liver-specific overexpression of scavenger receptor BI decreases levels of very low density lipoprotein ApoB, low density lipoprotein ApoB, and high density lipoprotein in transgenic mice. J. Biol. Chem. 1998, 273, 32920–32926. [Google Scholar] [CrossRef] [PubMed]
- Lino, M.; Farr, S.; Baker, C.; Fuller, M.; Trigatti, B.; Adeli, K. Intestinal scavenger receptor class b type i as a novel regulator of chylomicron production in healthy and diet-induced obese states. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G350–G359. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tuttle, K.R.; Short, R.A.; Johnson, R.J. Hypothesis: Fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat. Clin. Pract. Nephrol. 2005, 1, 80. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef] [PubMed]
- Madlala, H.P.; Maarman, G.J.; Ojuka, E. Uric acid and transforming growth factor in fructose-induced production of reactive oxygen species in skeletal muscle. Nutr. Rev. 2016, 74, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Lanaspa, M.A.; Cristobal-Garcia, M.; Garcia-Arroyo, F.; Soto, V.; Cruz-Robles, D.; Nakagawa, T.; Yu, M.A.; Kang, D.H.; Johnson, R.J. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp. Nephrol. 2012, 121, e71–e78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Meng, Y.H.; Chang, S.; Zhang, R.Y.; Shi, C. High fructose causes cardiac hypertrophy via mitochondrial signaling pathway. Am. J. Transl. Res. 2016, 8, 4869–4880. [Google Scholar] [PubMed]
- Wang, W.; Ding, X.Q.; Gu, T.T.; Song, L.; Li, J.M.; Xue, Q.C.; Kong, L.D. Pterostilbene and allopurinol reduce fructose-induced podocyte oxidative stress and inflammation via microrna-377. Free Radic. Biol. Med. 2015, 83, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lan, Z.; Lin, Q.; Mi, X.; He, Y.; Wei, L.; Lin, Y.; Zhang, Y.; Deng, X. Polydatin ameliorates renal injury by attenuating oxidative stress-related inflammatory responses in fructose-induced urate nephropathic mice. Food Chem. Toxicol. 2013, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, N.; Kannappan, S.; Anuradha, C.V. Genistein modulates nf-kappab-associated renal inflammation, fibrosis and podocyte abnormalities in fructose-fed rats. Eur. J. Pharmacol. 2011, 667, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lee, S.H.; Kim, Y.G.; Kim, S.Y.; Seo, J.W.; Choi, Y.W.; Kim, D.J.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 308, F993–F1003. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.H.; Zhang, X.; Pan, Y.; Li, Y.C.; Kong, L.D. Allopurinol, quercetin and rutin ameliorate renal NLRP3 inflammasome activation and lipid accumulation in fructose-fed rats. Biochem. Pharmacol. 2012, 84, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.X.; Yu, R.; Xu, M.X.; Li, P.Q.; Fan, C.Y.; Li, J.M.; Kong, L.D. Betaine prevented fructose-induced NAFLD by regulating LXRα/PPARα pathway and alleviating ER stress in rats. Eur. J. Pharmacol. 2015, 770, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Pan, Y.; Wang, R.; Kang, L.L.; Xue, Q.C.; Wang, X.N.; Kong, L.D. Quercetin inhibits AMPK/TXNIP activation and reduces inflammatory lesions to improve insulin signaling defect in the hypothalamus of high fructose-fed rats. J. Nutr. Biochem. 2014, 25, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.H.; Kang, L.L.; Ren, H.M.; Zhang, D.M.; Kong, L.D. Simiao pill ameliorates renal glomerular injury via increasing Sirt1 expression and suppressing NF-κb/NLRP3 inflammasome activation in high fructose-fed rats. J. Ethnopharmacol. 2015, 172, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, D.M.; Liu, J.H.; Hu, L.S.; Xue, Q.C.; Ding, X.Q.; Kong, L.D. Wuling san protects kidney dysfunction by inhibiting renal TLR4/MyD88 signaling and NLRP3 inflammasome activation in high fructose-induced hyperuricemic mice. J. Ethnopharmacol. 2015, 169, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; Lanaspa, M.A.; Rivard, C.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Jalal, D.; Andres-Hernando, A.; Tanabe, K.; Madero, M.; Li, N.; et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metab. Clin. Exp. 2011, 60, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.F.; He, C.T.; Chen, Y.T.; Hsieh, P.S. Lipoic acid suppresses portal endotoxemia-induced steatohepatitis and pancreatic inflammation in rats. World J. Gastroenterol. WJG 2013, 19, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Segal, M.; Afsar, B.; Kang, D.H.; Rodriguez-Iturbe, B.; Johnson, R.J. The role of uric acid in the pathogenesis of human cardiovascular disease. Heart 2013, 99, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hu, X.; Shi, M.; Knepper, M.A.; Ecelbarger, C.A. Effects of dietary fat, nacl, and fructose on renal sodium and water transporter abundances and systemic blood pressure. Am. J. Physiol. Ren. Physiol. 2004, 287, F1204–F1212. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Johnson, R.J. Uric acid predicts clinical outcomes in heart failure insights regarding the role of xanthine oxidase and uric acid in disease pathophysiology. Circulation 2003, 107, 1951. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Yoon, Y.; Lee, K.Y.; Hien, T.T.; Kang, K.W.; Kim, K.C.; Lee, J.; Lee, M.Y.; Lee, S.M.; Kang, D.H.; et al. Uric acid induces endothelial dysfunction by vascular insulin resistance associated with the impairment of nitric oxide synthesis. FASEB J. 2014, 28, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, P.; Pellegrino, G.; Conte, S.; Maresca, F.; Pacifico, F.; Leonardi, A.; Trimarco, B. Fructose induces prothrombotic phenotype in human endothelial cells: A new role for “added sugar” in cardio-metabolic risk. J. Thromb. Thrombolysis 2015, 40, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Toschi, V.; Gallo, R.; Lettino, M.; Fallon, J.T.; Gertz, S.D.; Fernandez-Ortiz, A.; Chesebro, J.H.; Badimon, L.; Nemerson, Y.; Fuster, V.; et al. Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation 1997, 95, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.H.; Wang, C.; Li, J.M.; Zhang, D.M.; Kong, L.D. Allopurinol, rutin, and quercetin attenuate hyperuricemia and renal dysfunction in rats induced by fructose intake: Renal organic ion transporter involvement. Am. J. Physiol. Ren. Physiol. 2009, 297, F1080. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Demarco, V.G.; Jia, G.; Sun, Z.; Nistala, R.; Meininger, G.A.; Sowers, J.R. The role of tissue renin-angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front. Endocrinol. 2013, 4, 161. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, O.; Kosugi, T.; Roncal, C.; Mu, W.; Heinig, M.; Cirillo, P.; Sanchez-Lozada, L.G.; Johnson, R.J.; Nakagawa, T. Fructose induces the inflammatory molecule icam-1 in endothelial cells. J. Am. Soc. Nephrol. 2008, 19, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.S.; Mu, W.; Cirillo, P.; Reungjui, S.; Zhang, L.; Roncal, C.; Sautin, Y.Y.; Johnson, R.J.; Nakagawa, T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2007, 293, F1256–F1261. [Google Scholar] [CrossRef] [PubMed]
- Prince, P.D.; Lanzi, C.R.; Toblli, J.E.; Elesgaray, R.; Oteiza, P.I.; Fraga, C.G.; Galleano, M. Dietary (−)-epicatechin mitigates oxidative stress, no metabolism alterations, and inflammation in renal cortex from fructose-fed rats. Free Radic. Biol. Med. 2015, 90, 35. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jia, X.; Chang, T.; Desai, K.; Wu, L. Attenuation of hypertension development by scavenging methylglyoxal in fructose-treated rats. J. Hypertens. 2008, 26, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Seefeld, K.; Witters, L.A.; Coleman, R.A. Amp-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: Evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 1999, 338 Pt 3, 783. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. Fructose surges damage hepatic adenosyl-monophosphate-dependent kinase and lead to increased lipogenesis and hepatic insulin resistance. Med. Hypotheses 2016, 93, 87. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. “Blinding” of amp-dependent kinase by methylglyoxal: A mechanism that allows perpetuation of hepatic insulin resistance? Med. Hypotheses 2009, 73, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Desai, K.; Kazachmov, M.; Yu, P.; Wu, L. Methylglyoxal production in vascular smooth muscle cells from different metabolic precursors. Metab. Clin. Exp. 2008, 57, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, R.; Desai, K.; Wu, L. Upregulation of aldolase b and overproduction of methylglyoxal in vascular tissues from rats with metabolic syndrome. Cardiovasc. Res. 2011, 92, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mak, T.C.; Banigesh, A.; Desai, K.; Wang, R.; Wu, L. Aldolase b knockdown prevents high glucose-induced methylglyoxal overproduction and cellular dysfunction in endothelial cells. PLoS ONE 2012, 7, e41495. [Google Scholar] [CrossRef] [PubMed]
- Dhar, I.; Dhar, A.; Wu, L.; Desai, K.M. Increased methylglyoxal formation with upregulation of renin angiotensin system in fructose fed Sprague dawley rats. PLoS ONE 2013, 8, e74212. [Google Scholar] [CrossRef] [PubMed]
- Porto, M.L.; Lirio, L.M.; Dias, A.T.; Batista, A.T.; Campagnaro, B.P.; Mill, J.G.; Meyrelles, S.S.; Baldo, M.P. Increased oxidative stress and apoptosis in peripheral blood mononuclear cells of fructose-fed rats. Toxicol. In Vitro 2015, 29, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Adeli, K. Dietary fructose and the metabolic syndrome. Curr. Opin. Gastroenterol. 2008, 24, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Pektas, M.B.; Koca, H.B.; Sadi, G.; Akar, F. Dietary fructose activates insulin signaling and inflammation in adipose tissue: Modulatory role of resveratrol. BioMed Res. Int. 2016, 2016, 8014252. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Park, Y.J.; Ham, M.; Kim, J.B. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol. Cells 2014, 37, 365–371. [Google Scholar]
- Jia, G.; Aroor, A.R.; Whaley-Connell, A.T.; Sowers, J.R. Fructose and uric acid: Is there a role in endothelial function? Curr. Hypertens. Rep. 2014, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kimura, I.; Inoue, D.; Ichimura, A.; Hirasawa, A. Free fatty acid receptors and their role in regulation of energy metabolism. Rev. Physiol. Biochem. Pharmacol. 2013, 164, 77–116. [Google Scholar] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. Tlr4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; Van, E.P.; Koenen, T.; Joosten, L.A.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [PubMed]
- Äijälä, M.; Malo, E.; Ukkola, O.; Bloigu, R.; Lehenkari, P.; Autioharmainen, H.; Santaniemi, M.; Kesäniemi, Y.A. Long-term fructose feeding changes the expression of leptin receptors and autophagy genes in the adipose tissue and liver of male rats: A possible link to elevated triglycerides. Genes Nutr. 2013, 8, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Teruel, T.; Hernandez, R.; Lorenzo, M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001, 50, 2563–2571. [Google Scholar] [CrossRef] [PubMed]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking tnf-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [PubMed]
- Arner, P. The adipocyte in insulin resistance: Key molecules and the impact of the thiazolidinediones. Trends Endocrinol. Metab. 2003, 14, 137–145. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. Irs-1-mediated inhibition of insulin receptor tyrosine kinase activity in tnf-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Higashimori, T.; Park, S.Y.; Choi, H.; Dong, J.; Kim, Y.J.; Noh, H.L.; Cho, Y.R.; Cline, G.; Kim, Y.B. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 2004, 53, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Mastrocola, R.; Rogazzo, M.; Chiazza, F.; Aragno, M.; Fantozzi, R.; Collino, M.; Minetto, M.A. High sugar intake and development of skeletal muscle insulin resistance and inflammation in mice: A protective role for ppar-δ agonism. Mediat. Inflamm. 2013, 2013, 509502. [Google Scholar] [CrossRef] [PubMed]
- Ropelle, E.R.; Pauli, J.R.; Cintra, D.E.; Silva, A.S.D.; Souza, C.T.D.; Guadagnini, D.; Carvalho, B.M.; Caricilli, A.M.; Katashima, C.K.; Carvalho-Filho, M.A. Targeted disruption of inducible nitric oxide synthase protects against aging, s-nitrosation, and insulin resistance in muscle of male mice. Diabetes 2013, 62, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, C.; Zhang, J.; Liu, B.; Du, Q. Resveratrol inhibits inflammation and ameliorates insulin resistant endothelial dysfunction via regulation of AMPK and sirt1 activities. J. Diabetes 2015, 184, 98–105. [Google Scholar]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Rodrigues, M.E.; Bekhbat, M.; Houser, M.C.; Chang, J.; Walker, D.I.; Jones, D.P.; Oller do Nascimento, C.M.; Barnum, C.J.; Tansey, M.G. Chronic psychological stress and high-fat high-fructose diet disrupt metabolic and inflammatory gene networks in the brain, liver, and gut and promote behavioral deficits in mice. Brain Behav. Immun. 2017, 59, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.X.; Yu, R.; Shao, L.F.; Zhang, Y.X.; Ge, C.X.; Liu, X.M.; Wu, W.Y.; Li, J.M.; Kong, L.D. Up-regulated fractalkine (FKN) and its receptor CX3CR1 are involved in fructose-induced neuroinflammation: Suppression by curcumin. Brain Behav. Immun. 2016, 58, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Bursać, B.N.; Vasiljević, A.D.; Nestorović, N.M.; Veličković, N.A.; Matić, G.M.; Djordjevic, A.D. High-fructose diet leads to visceral adiposity and hypothalamic leptin resistance in male rats—Do glucocorticoids play a role? J. Nutr. Biochem. 2014, 25, 446. [Google Scholar] [CrossRef] [PubMed]
- Khodabandehloo, H.; Gorgani-Firuzjaee, S.; Panahi, G.; Meshkani, R. Molecular and cellular mechanisms linking inflammation to insulin resistance and β-cell dysfunction. Transl. Res. J. Lab. Clin. Med. 2015, 167. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, R.; Cianflone, K.; Mcgahan, J.P.; Berglund, L.; Bremer, A.A.; Keim, N.L.; Griffen, S.C.; Havel, P.J.; Stanhope, K.L. Effects of sugar-sweetened beverages on plasma acylation stimulating protein, leptin and adiponectin: Relationships with metabolic outcomes. Obesity 2013, 21, 2471–2480. [Google Scholar] [CrossRef] [PubMed]
- Teff, K.L.; Elliott, S.S.; Tschop, M.; Kieffer, T.J.; Rader, D.; Heiman, M.; Townsend, R.R.; Keim, N.L.; D’Alessio, D.; Havel, P.J. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Zou, C.; van der Westhuyzen, D.R.; Shao, J. Adiponectin reduces plasma triglyceride by increasing VLDL triglyceride catabolism. Diabetes 2008, 57, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, M.M.; Havel, P.J. Physiological, pharmacological, and nutritional regulation of circulating adiponectin concentrations in humans. Metab. Syndr. Relat. Disord. 2008, 6, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Hella, J.; Wiltrud, H.; Castañeda, T.R.; Annette, S.; Corinna, K.; Frank, D.; Bärbel, O.; Nawrocki, A.R.; Scherer, P.E.; Jochen, S. Consuming fructose-sweetened beverages increases body adiposity in mice. Obes. Res. 2005, 13, 1146. [Google Scholar]
- Rodrigues, D.F.; Henriques, M.C.; Oliveira, M.C.; Menezes-Garcia, Z.; Marques, P.E.; Souza Dda, G.; Menezes, G.B.; Teixeira, M.M.; Ferreira, A.V. Acute intake of a high-fructose diet alters the balance of adipokine concentrations and induces neutrophil influx in the liver. J. Nutr. Biochem. 2014, 25, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Marek, G.; Pannu, V.; Shanmugham, P.; Pancione, B.; Mascia, D.; Crosson, S.; Ishimoto, T.; Sautin, Y.Y. Adiponectin resistance and proinflammatory changes in the visceral adipose tissue induced by fructose consumption via ketohexokinase-dependent pathway. Diabetes 2015, 64, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Ykijärvinen, H.; Aminoff, A.; Bergholm, R.; Pietiläinen, K.H.; Westerbacka, J.; Talmud, P.J.; Humphries, S.E.; Hamsten, A.; Isomaa, B. Genetic variation in the adipor2 gene is associated with liver fat content and its surrogate markers in three independent cohorts. Eur. J. Endocrinol. 2009, 160, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Oike, Y.; Teratani, T.; Taguchi, T.; Noguchi, M.; Suzuki, T.; Mizutani, A.; Yokoyama, H.; Irie, R.; Sumimoto, H.; et al. Hepatic adipor2 signaling plays a protective role against progression of nonalcoholic steatohepatitis in mice. Hepatology 2008, 48, 458–473. [Google Scholar] [CrossRef] [PubMed]
- Tsatsanis, C.; Zacharioudaki, V.; Androulidaki, A.; Dermitzaki, E.; Charalampopoulos, I.; Minas, V.; Gravanis, A.; Margioris, A.N. Adiponectin induces tnf-alpha and il-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochem. Biophys. Res. Commun. 2005, 335, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Wanninger, J.; Neumeier, M.; Weigert, J.; Bauer, S.; Weiss, T.S.; Schäffler, A.; Krempl, C.; Bleyl, C.; Aslanidis, C.; Schölmerich, J. Adiponectin-stimulated CXCL8 release in primary human hepatocytes is regulated by ERK1/ERK2, p38 MAPK, NF-kappaB, and STAT3 signaling pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G611–G618. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, I.; Bechmann, L.P.; Odenthal, M.; Jochum, C.; Marquitan, G.; Drebber, U.; Gerken, G.; Gieseler, R.K.; Dienes, H.P.; Canbay, A. Adiponectin inhibits steatotic CD95/Fas up-regulation by hepatocytes: Therapeutic implications for hepatitis C. J. Hepatol. 2009, 50, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Lee, Y.J.; Lee, M.W.; Kim, S.M.; Jung, T.W. Full-length adiponectin protects hepatocytes from palmitate-induced apoptosis via inhibition of c-jun NH2 terminal kinase. FEBS J. 2009, 276, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.A.; Mariño, G.; Benyounès, A.; Shen, S.; Harper, F.; Maiuri, M.C.; Kroemer, G. Neuroendocrine regulation of autophagy by leptin. Cell Cycle 2011, 10, 2917. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Miyazaki, M.; Boucher, J.; Ntambi, J.M.; Kahn, C.R. Leptin suppresses stearoyl-coa desaturase 1 by mechanisms independent of insulin and sterol regulatory element-binding protein-1c. Diabetes 2006, 55, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Leclercq, I.; Brymora, J.M.; Xu, N.; Ramezani-Moghadam, M.; London, R.M.; Brigstock, D.; George, J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 2009, 137, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Baelemans, A.; Erlanson-Albertsson, C. Effects of sucrose, glucose and fructose on peripheral and central appetite signals. Regul. Pept. 2008, 150, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Priebs, J.; Landmann, M.; Degen, C.; Engstler, A.J.; Jin, C.J.; Gärttner, S.; Spruss, A.; Huber, O.; Bergheim, I. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem. 2015, 26, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Di Luccia, B.; Crescenzo, R.; Mazzoli, A.; Cigliano, L.; Venditti, P.; Walser, J.C.; Widmer, A.; Baccigalupi, L.; Ricca, E.; Iossa, S. Rescue of fructose-induced metabolic syndrome by antibiotics or faecal transplantation in a rat model of obesity. PLoS ONE 2015, 10, e0134893. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.; Celikbilek, M.; Guven, K. High fructose consumption can induce endotoxemia. Gastroenterology 2012, 143, e29. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Rivard, C.; Lanaspa, M.A.; Otabachian-Smith, S.; Ishimoto, T.; Cicerchi, C.; Cheeke, P.R.; Macintosh, B.; Hess, T. Fructokinase, fructans, intestinal permeability, and metabolic syndrome: An equine connection? J. Equine Vet. Sci. 2013, 33, 120. [Google Scholar] [CrossRef] [PubMed]
- . Spruss, A.; Kanuri, G.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Metformin protects against the development of fructose-induced steatosis in mice: role of the intestinal barrier function. Lab. Investig. 2012, 92, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Castagliuolo, I.; Di, L.V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518. [Google Scholar] [PubMed]
- Bruewer, M.; Luegering, A.; Kucharzik, T.; Parkos, C.A.; Madara, J.L.; Hopkins, A.M.; Nusrat, A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol. 2003, 171, 6164. [Google Scholar] [CrossRef] [PubMed]
- Frazier, T.H.; Dibaise, J.K.; Mcclain, C.J. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. J. Parenter. Enter. Nutr. 2011, 35, S14–S20. [Google Scholar] [CrossRef] [PubMed]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Königsrainer, A.; Maier, K.P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr. 2008, 138, 1452. [Google Scholar] [PubMed]
- Liu, J.; Zhuang, Z.J.; Bian, D.X.; Ma, X.J.; Xun, Y.H.; Yang, W.J.; Luo, Y.; Liu, Y.L.; Jia, L.; Wang, Y.; et al. Toll-like receptor-4 signalling in the progression of non-alcoholic fatty liver disease induced by high-fat and high-fructose diet in mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.J.; Engstler, A.J.; Ziegenhardt, D.; Bischoff, S.C.; Trautwein, C.; Bergheim, I. Loss of lipopolysaccharide-binding protein attenuates the development of diet-induced non-alcoholic fatty liver disease (NAFLD) in mice. J. Gastroenterol. Hepatol. 2017, 32, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Li, F.Y.L.; Lam, K.S.L.; Li, H.; Jia, W.; Yu, W.; Man, K.; Lo, C.M.; Li, X.; Xu, A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of x-box binding protein-1 in mice. Gut 2012, 61, 1058. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Xu, M.; Seyhan, H.A.; Ahmad, S.; Mihm, S.; Ramadori, G.; Schultze, F.C. Diet high in fructose leads to an overexpression of lipocalin-2 in rat fatty liver. World J. Gastroenterol. 2014, 20, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Carranza, A.; Litterio, M.C.; Prince, P.D.; Mayer, M.A.; Ingaramo, P.I.; Ronco, M.T.; Peredo, H.A.; Puyó, A.M.; Galleano, M. Lipopolysaccharide (LPS) induction of nitric oxide synthase-2 and cyclooxygenase-2 is impaired in fructose overloaded rats. Life Sci. 2011, 88, 307–313. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Organs Histopathological Changes | Dangerous Factors | Pathological Indexes | Molecular Mechanisms | |
|---|---|---|---|---|
| ↑ | ↓ | |||
| Adipose tissue Inflammation response Endothelial dysfunction | FFA UA | ROS production Inflammatory cytokine flux FFA uptake Adiponectin secretion Lipid accumulation Autophagy | Insulin sensitivity Leptin sensitivity Glucose uptake Oxygen availability | PKCθ/IKK-β/c-JNK [39,40,41] IRS/Akt/GLUT4 [60] FATPs/CD36 [70] RAAS [115] LEPR/Stearoyl-CoA desaturase [135,165,166] ATG7/LAMP2/MAP1LC3β [135] |
| Brain Appetite increase Psychological stress | FFA UA MG | ROS production Inflammation cytokine flux Food intake | Insulin sensitivity Leptin sensitivity | TNF-α/AMPK/malonyl-CoA [76,77] NLRP3/NF-κB [95] TLR4/NF-κB, FKN/CX3CR1 [148] PYY, NPY [149] |
| Heart/vessel Hypertrophy Endothelial dysfunction Plaque formation Vascular stiffness | FFA UA | ROS production FFA uptake Vascular tone RAGE production Blood pressure | Insulin sensitivity Glucose consumption Vascular vasodilation | HK/PFK [22] FATPs/CD36 [61] CD36/TLR4/6/IRAK4/1/NLRP3 [67] AMPK/malonyl-CoA [77] XO/eNOS [110,111] PI3K/Akt/eNOS [143] |
| Intestine Increased intestinal permeability | UA | Endotoxin translocation Bacterial composition disturbance Dysregulation of tight junction protein | Insulin sensitivity | SR-BI/ERK/ApoB [80] KHK/Occludin and ZO-1 [173,174] |
| Kidney CKD Endothelial dysfunction | UA MG | ROS production Inflammatory cytokine flux Dysregulation of renal organic ion transporters NO production Urine sodium retention | Insulin sensitivity UA clearance | HK/PFK [22] XO/eNOS [110,111] NLRP3/NF-κB [92,93,94,96] PGE2/Organic ion transporters [114] MAPK/TXNIP/NLRP3 [97,100,101,102,103,104,105] TLR4/MyD88/NF-κB [105] |
| Liver Steatosis NAFLD Fibrogenesis Endothelial dysfunction | Lactate FFA DAG Ceramide UA MG | Gluconeogenesis Glucose export ROS production DNL Inflammatory cytokine flux Lipid accumulation Mitochondrial dysfunction VLDL-secretion | Insulin sensitivity Glucose consumption Glucose uptake Oxygen availability | IRS/PI3K/Akt, ChREBP/SCD-1 [11] HK/PFK [22,91] ChREBP/G6Pase [36] Bax/cathepsin B/NF-κB/TNF-α [38] PTP1B/IRS/PI3K/Akt [42] PKC/Akt2/GS/G6Pase/PEPCK [56] SphK1/S1P/NF-κB [61] NOX4/PTP1c [66] SREBP-1c [80] PCSK9/LDLR [82] SR-BI/ERK [88] AMPK/ACC [120] LEPR/ATG7/LAMP2/MAP1LC3β [135] AdipoR1,2/NF-κB/CXCL8 [158,161] CD95, c-JNK [161,162] TLR-4/MyD88 [170] LCN-2 [182] |
| Pancreatic islet Glucose intolerance Increased β-cell mass Irregular insulin secretion | Glucose FFA UA | Inflammatory cytokines flux ER stress Apoptosis | Insulin sensitivity Leptin sensitivity | TR [14] Akt/FoxO1 [15] SREBP-1c/IRS-2/Akt [44] Cideb [48] FFAR1 [49] NF-κB [106] |
| Skeletal muscle Inflammation response Endothelial dysfunction | Lactate FFA Ceramide UA | ROS production FFA uptake Autophagy Inflammatory cytokine flux Lipid accumulation | Insulin sensitivity Glucose uptake Oxygen availability | PI3K/Akt [21] HK/PFK [22] GLUT4 [23,24] FATPs/CD36 [23,61,62] PKCθ/IKK-β/c-JNK [40] LKB1/AMPK/AS160/IRS [140] PPAR-δ/FGF-21 [141] NF-κB/IL-6/iNOS, ICAM-1 [142] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.-M.; Jiao, R.-Q.; Kong, L.-D. High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients 2017, 9, 335. https://doi.org/10.3390/nu9040335
Zhang D-M, Jiao R-Q, Kong L-D. High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients. 2017; 9(4):335. https://doi.org/10.3390/nu9040335
Chicago/Turabian StyleZhang, Dong-Mei, Rui-Qing Jiao, and Ling-Dong Kong. 2017. "High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions" Nutrients 9, no. 4: 335. https://doi.org/10.3390/nu9040335