Inborn Errors of Fructose Metabolism. What Can We Learn from Them?


Abstract
:1. Introduction
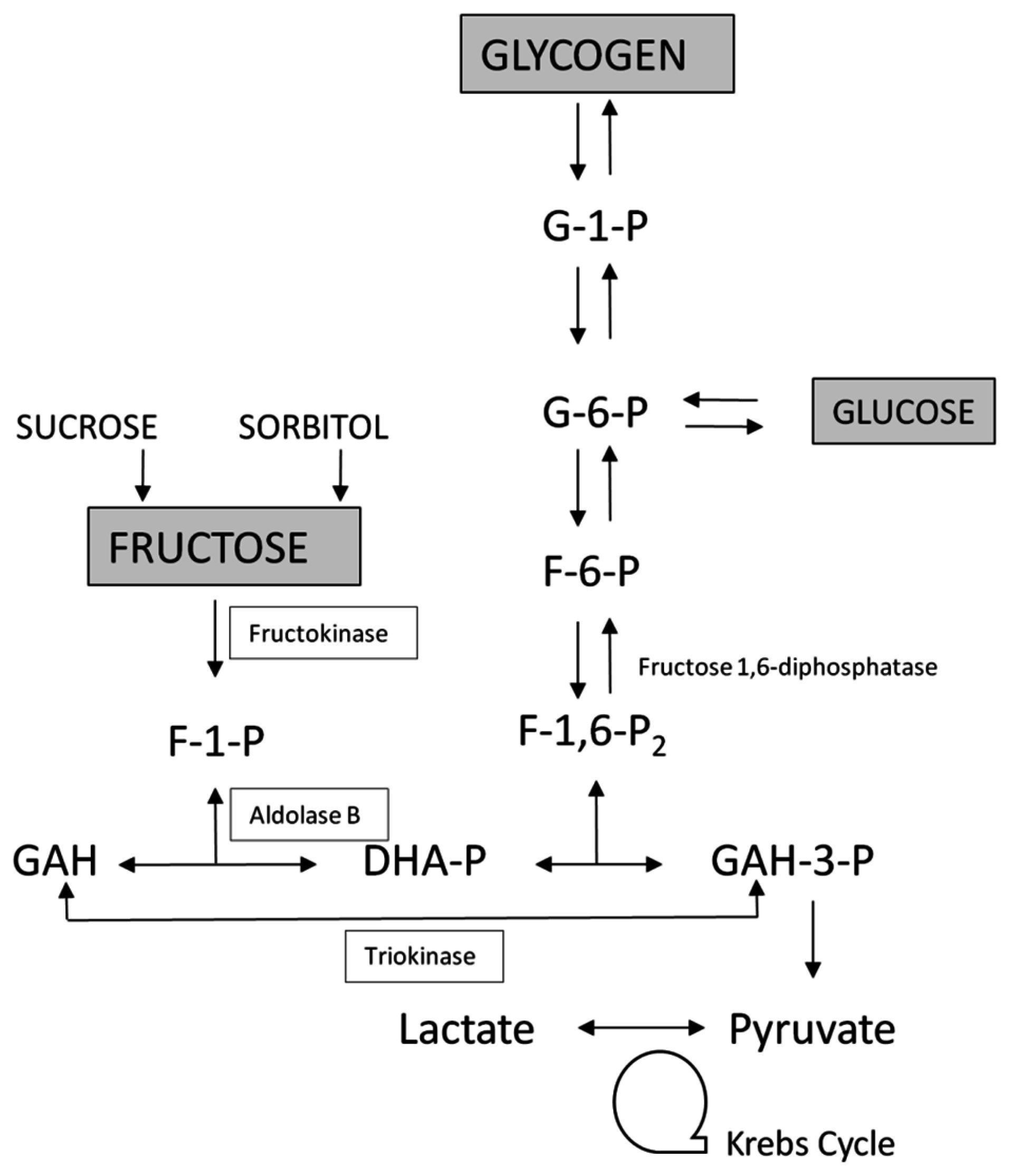
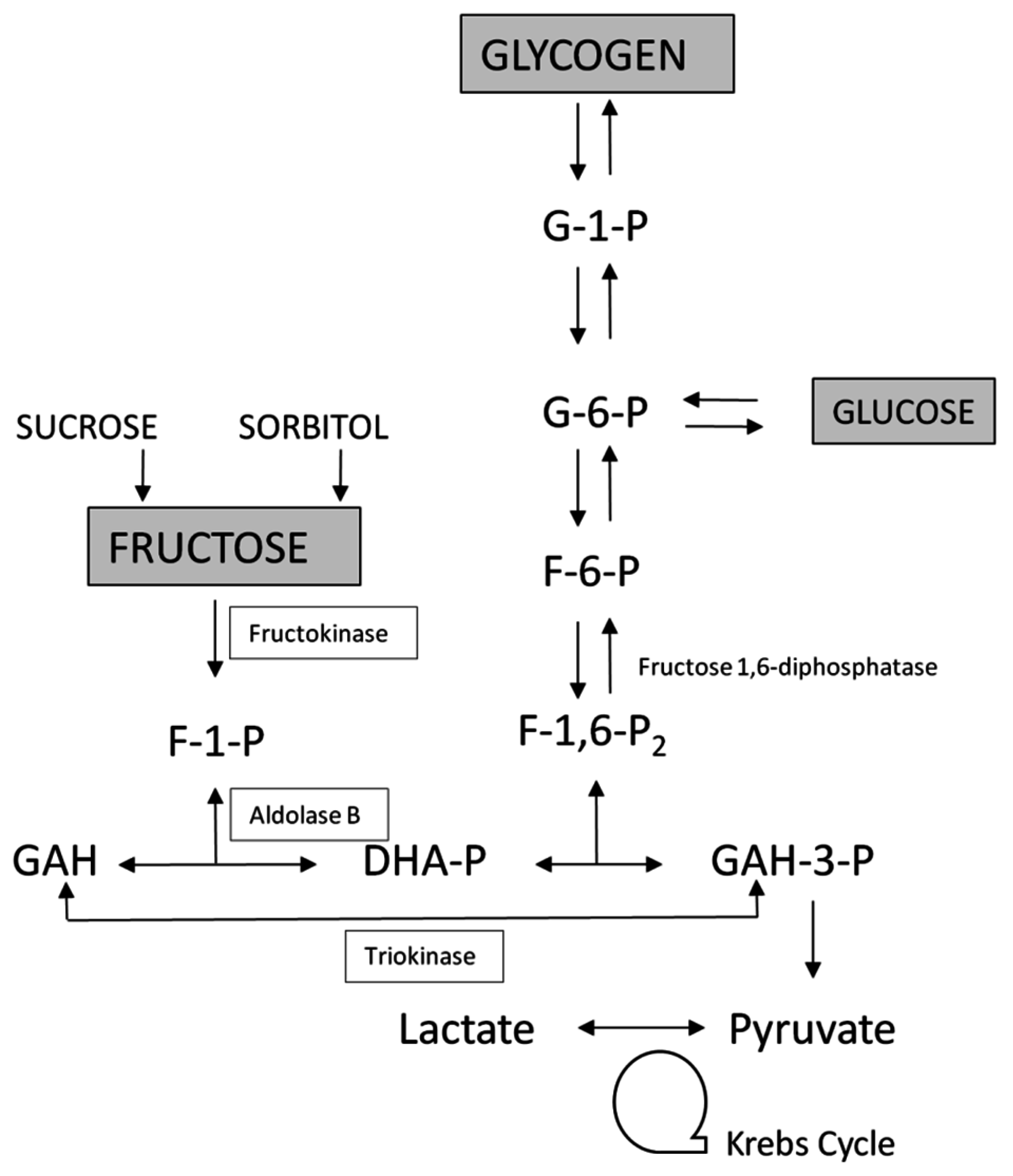
2. An Overview of Fructose Metabolism
2.1. Enzyme of Fructose Metabolism
2.2. Fructose Toxicity
3. Inborn Errors of Fructose Metabolism: A Model for Fructose Toxicity
3.1. Essential Fructosuria-Hepatic Fructokinase Deficiency (OMIM 229800)
3.2. Hereditary Fructose Intolerance and Accumulation of F-1-P (OMIM 229600)
3.3. Fructose-1,6-Bisphosphatase Deficiency (OMIM 229700)
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Havel, P.J. Dietary fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 2005, 63, 133–157. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among us children and adults: The third national health and nutrition examination survey. Medscape J. Med. 2008, 10, 160. [Google Scholar] [PubMed]
- Hwang, J.J.; Jiang, L.; Hamza, M.; Dai, F.; Belfort-DeAguiar, R.; Cline, G.; Rothman, D.L.; Mason, G.; Sherwin, R.S. The human brain produces fructose from glucose. JCI Insight 2017, 2, e90508. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G. Inborn errors of fructose metabolism. Annu. Rev. Nutr. 1994, 14, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G. Fructose: Metabolism and short-term effects on carbohydrate and purine metabolic pathways. Prog. Biochem. Pharmacol. 1986, 21, 1–32. [Google Scholar] [PubMed]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter glut5 in health and disease. Am. J. Physiology. Endocrinol Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, B.; Gitzelmann, R.; Van Den Berghe, D. Disorders of fructose metabolism. In The Metabolic and Molecular Basis of Inherited Disease; Scriver, C., Ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 1489–1520. [Google Scholar]
- Tappy, L.; Egli, L.; Tran, C. Fructose, high fructose corn syrup, sucrose and health. In Metabolism of Nutritive Sweeteners in Humans; Ripper, J.M., Ed.; Springer: New York, NY, USA, 2014; pp. 35–50. [Google Scholar]
- Salway, J.G. Fructose metabolism. In Metabolism at a Glance; Blackwell Publishing: Nashville, TN, USA, 2004; p. 104. [Google Scholar]
- Hollak, C.E.M.; Lachmann, R.H. Disorders of fructose metabolism. In Inherited Metabolic Disease in Adults; Oxford: New York, NY, USA, 2016; p. 25. [Google Scholar]
- Perheentupa, J.; Raivio, K. Fructose-induced hyperuricaemia. Lancet 1967, 2, 528–531. [Google Scholar] [CrossRef]
- Bergstrom, J.; Hultman, E.; Roch-Norlund, A.E. Lactic acid accumulation in connection with fructose infusion. Acta Med. Scand. 1968, 184, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Hers, H.G. Misuses for fructose. Nature 1970, 227, 421. [Google Scholar] [CrossRef] [PubMed]
- Woods, H.F.; Alberti, K.G. Dangers of intravenous fructose. Lancet 1972, 2, 1354–1357. [Google Scholar] [CrossRef]
- Rey, M.; Behrens, R.; Zeilinger, G. Fatal consequences of fructose infusion in undiagnosed fructose intolerance. Dtsch. Med. Wochenschr. 1988, 113, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M. The genetic consequences of our sweet tooth. Nat. Rev. Genet. 2002, 3, 481–487. [Google Scholar] [PubMed]
- Johnston, R.D.; Stephenson, M.C.; Crossland, H.; Cordon, S.M.; Palcidi, E.; Cox, E.F.; Taylor, M.A.; Aithal, G.P.; Macdonald, I.A. No difference between high-fructose and high-glucose diets on liver triacylglycerol or biochemistry in healthy overweight men. Gastroenterology 2013, 145, 1016–1025.e2. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.M.; Berghe, G.V.D.; Walter, J.H. Disorders of fructose metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment, 5th ed.; Springer: London, UK, 2012; pp. 157–164. [Google Scholar]
- Laron, Z. Essential benign fructosuria. Arch. Dis. Child. 1961, 36, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Boesiger, P.; Buchli, R.; Meier, D.; Steinmann, B.; Gitzelmann, R. Changes of liver metabolite concentrations in adults with disorders of fructose metabolism after intravenous fructose by 31p magnetic resonance spectroscopy. Pediatric. Res. 1994, 36, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Leonidas, J.C. Essential fructosuria. N. Y. State J. Med. 1965, 65, 2257–2259. [Google Scholar] [PubMed]
- Petersen, A.; Steinmann, B.; Gitzelmann, R. Essential fructosuria: Increased levels of fructose 3-phosphate in erythrocytes. Enzyme 1992, 46, 319–323. [Google Scholar] [PubMed]
- Szwergold, B.S.; Kappler, F.; Brown, T.R. Identification of fructose 3-phosphate in the lens of diabetic rats. Science 1990, 247, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Romano, S.; Mention, K.; Vassault, A.; Rabier, D.; Polak, M.; Robert, J.J.; de Keyzer, Y.; de Lonlay, P. What’s new in metabolic and genetic hypoglycaemias: Diagnosis and management. Eur. J. Pediatr. 2008, 167, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, B.; Gitzelmann, R. The diagnosis of hereditary fructose intolerance. Helv. Paediatr. Acta 1981, 36, 297–316. [Google Scholar] [PubMed]
- Santer, R.; Rischewski, J.; von Weihe, M.; Niederhaus, M.; Schneppenheim, S.; Baerlocher, K.; Kohlschutter, A.; Muntau, A.; Posselt, H.G.; Steinmann, B.; et al. The spectrum of aldolase b (aldob) mutations and the prevalence of hereditary fructose intolerance in central europe. Hum. Mutat. 2005, 25, 594. [Google Scholar] [CrossRef] [PubMed]
- Coffee, E.M.; Yerkes, L.; Ewen, E.P.; Zee, T.; Tolan, D.R. Increased prevalence of mutant null alleles that cause hereditary fructose intolerance in the american population. J. Inherit. Metab. Dis. 2010, 33, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Oberhaensli, R.D.; Rajagopalan, B.; Taylor, D.J.; Radda, G.K.; Collins, J.E.; Leonard, J.V.; Schwarz, H.; Herschkowitz, N. Study of hereditary fructose intolerance by use of 31p magnetic resonance spectroscopy. Lancet 1987, 2, 931–934. [Google Scholar] [CrossRef]
- Stirpe, F.; Della Corte, E.; Bonetti, E.; Abbondanza, A.; Abbati, A.; De Stefano, F. Fructose-induced hyperuricaemia. Lancet 1970, 2, 1310–1311. [Google Scholar] [CrossRef]
- Jamnik, J.; Rehman, S.; Blanco Mejia, S.; de Souza, R.J.; Khan, T.A.; Leiter, L.A.; Wolever, T.M.; Kendall, C.W.; Jenkins, D.J.; Sievenpiper, J.L. Fructose intake and risk of gout and hyperuricemia: A systematic review and meta-analysis of prospective cohort studies. BMJ Open 2016, 6, e013191. [Google Scholar] [CrossRef] [PubMed]
- Rosset, R.; Surowska, A.; Tappy, L. Pathogenesis of cardiovascular and metabolic diseases: Are fructose-containing sugars more involved than other dietary calories? Curr. Hypertens. Rep. 2016, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase c and a isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Oppelt, S.A.; Sennott, E.M.; Tolan, D.R. Aldolase-b knockout in mice phenocopies hereditary fructose intolerance in humans. Mol. Genet. Metab. 2015, 114, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Douillard, C.; Mention, K.; Dobbelaere, D.; Wemeau, J.L.; Saudubray, J.M.; Vantyghem, M.C. Hypoglycaemia related to inherited metabolic diseases in adults. Orphanet J. Rare Dis. 2012, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Haymond, M.W. Hypoglycemia in infants and children. Endocrinol. Metab. Clin. North. Am. 1989, 18, 211–252. [Google Scholar] [PubMed]
- Saudubray, J.M.; Baumgartner, M.R.; Walter, J.H. Disorders of fructose metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment; Springer: Berlin, Germany, 2016; Volume 6, pp. 165–167. [Google Scholar]
- Huidekoper, H.H.; Visser, G.; Ackermans, M.T.; Sauerwein, H.P.; Wijburg, F.A. A potential role for muscle in glucose homeostasis: In vivo kinetic studies in glycogen storage disease type 1a and fructose-1,6-bisphosphatase deficiency. J. Inherit. Metab. Dis. 2010, 33, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Kikawa, Y.; Miyamaoto, J.; Sugimoto, S.; Adachi, M.; Ohura, T.; Mayumi, M. Intravenous glycerol therapy should not be used in patients with unrecognized fructose-1,6-bisphosphatase deficiency. Pediatr. Int. 2003, 45, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Gitzelmann, R.; Baerlocher, K.; Prader, A. Hereditary defects of fructose and galactose metabolism. Monatsschrift Kinderheilkd. 1973, 121, 174–180. [Google Scholar]
- Kikawa, Y.; Shin, Y.S.; Inuzuka, M.; Zammarchi, E.; Mayumi, M. Diagnosis of fructose-1,6-bisphosphatase deficiency using cultured lymphocyte fraction: A secure and noninvasive alternative to liver biopsy. J. Inherit. Metab. Dis. 2002, 25, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Sugita, G.; Tsuyoshi, H.; Nishijima, K.; Yoshida, Y. Fructose-1,6-bisphosphatase deficiency: A case of a successful pregnancy by closely monitoring metabolic control. JIMD Rep. 2014, 14, 115–118. [Google Scholar] [PubMed]
- Krishnamurthy, V.; Eschrich, K.; Boney, A.; Sullivan, J.; McDonald, M.; Kishnani, P.S.; Koeberl, D.D. Three successful pregnancies through dietary management of fructose-1,6-bisphosphatase deficiency. J. Inherit. Metab. Dis. 2007, 30, 819. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Tahrani, A.A.; Barnett, A.H. Future glucose-lowering drugs for type 2 diabetes. Lancet Diabetes Endocrinol. 2016, 4, 350–359. [Google Scholar] [CrossRef]
- van Poelje, P.D.; Potter, S.C.; Chandramouli, V.C.; Landau, B.R.; Dang, Q.; Erion, M.D. Inhibition of fructose 1,6-bisphosphatase reduces excessive endogenous glucose production and attenuates hyperglycemia in zucker diabetic fatty rats. Diabetes 2006, 55, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.; Van Poelje, P.D.; Bullough, D.; Watling, S.; Milad, M.; Stern, T.; Foyt, H.; Erion, M. Pronounced glucose (g) reduction in poorly controlled t2dm with mb07803, a novel fructose-1,6-bisphosphatase inhibitor (FBPase1) with reduced potential for acid-base disturbances vs the 1st generation FBPaseICS-917 [abstract]. Diabetes 2009, 58 (Suppl. 1A), LB4. [Google Scholar]
- Van Poelje, P.D.; Potter, S.C.; Erion, M.D. Fructose-1,6-bisphosphatase inhibitors for reducing excessive endogenous glucose production in type 2 diabetes. Handb. Exp. Pharmacol. 2011, 203, 279–301. [Google Scholar]

{kind=link}
| Name of the Disease | Enzyme Defect | OMIM Number | Main Clinical Symptoms | Gene Mutations/Inheritance | Treatment |
|---|---|---|---|---|---|
| Essential fructosuria | Fructokinase | 229800 | Asymptomatic | KHK/AR |
|
| Hereditary fructose intolerance | Aldolase B | 229600 | Abdominal pain, nausea, hypoglycemia symptoms, shock-like syndrome after fructose ingestion | ALDOB/AR |
|
| FBPase deficiency | FBPase | 229700 | Life-threatening episodes of hypoglycemia, coma triggered by a febrile episode, fasting or large amount of fructose ingestion (~1 g/kg BW) | FBP1/AR |
|
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, C. Inborn Errors of Fructose Metabolism. What Can We Learn from Them? Nutrients 2017, 9, 356. https://doi.org/10.3390/nu9040356
Tran C. Inborn Errors of Fructose Metabolism. What Can We Learn from Them? Nutrients. 2017; 9(4):356. https://doi.org/10.3390/nu9040356
Chicago/Turabian StyleTran, Christel. 2017. "Inborn Errors of Fructose Metabolism. What Can We Learn from Them?" Nutrients 9, no. 4: 356. https://doi.org/10.3390/nu9040356
APA StyleTran, C. (2017). Inborn Errors of Fructose Metabolism. What Can We Learn from Them? Nutrients, 9(4), 356. https://doi.org/10.3390/nu9040356