Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease



{kind=link}
{kind=link}
Abstract
:1. Dietary Sugars as a Risk for Health
1.1. Sugar Consumption in Modern Society
1.2. Fructose Consumption and Prevalence of Metabolic Diseases
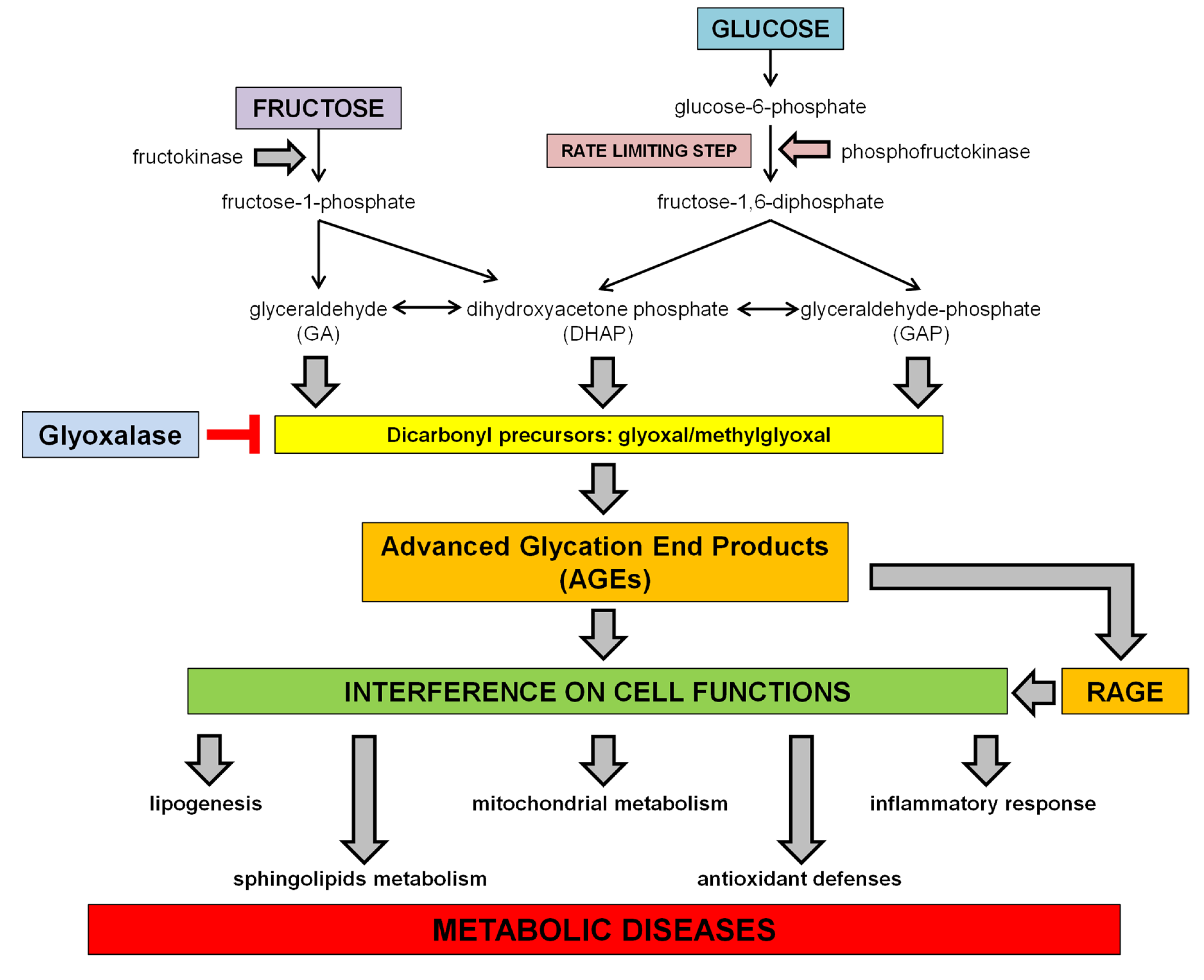
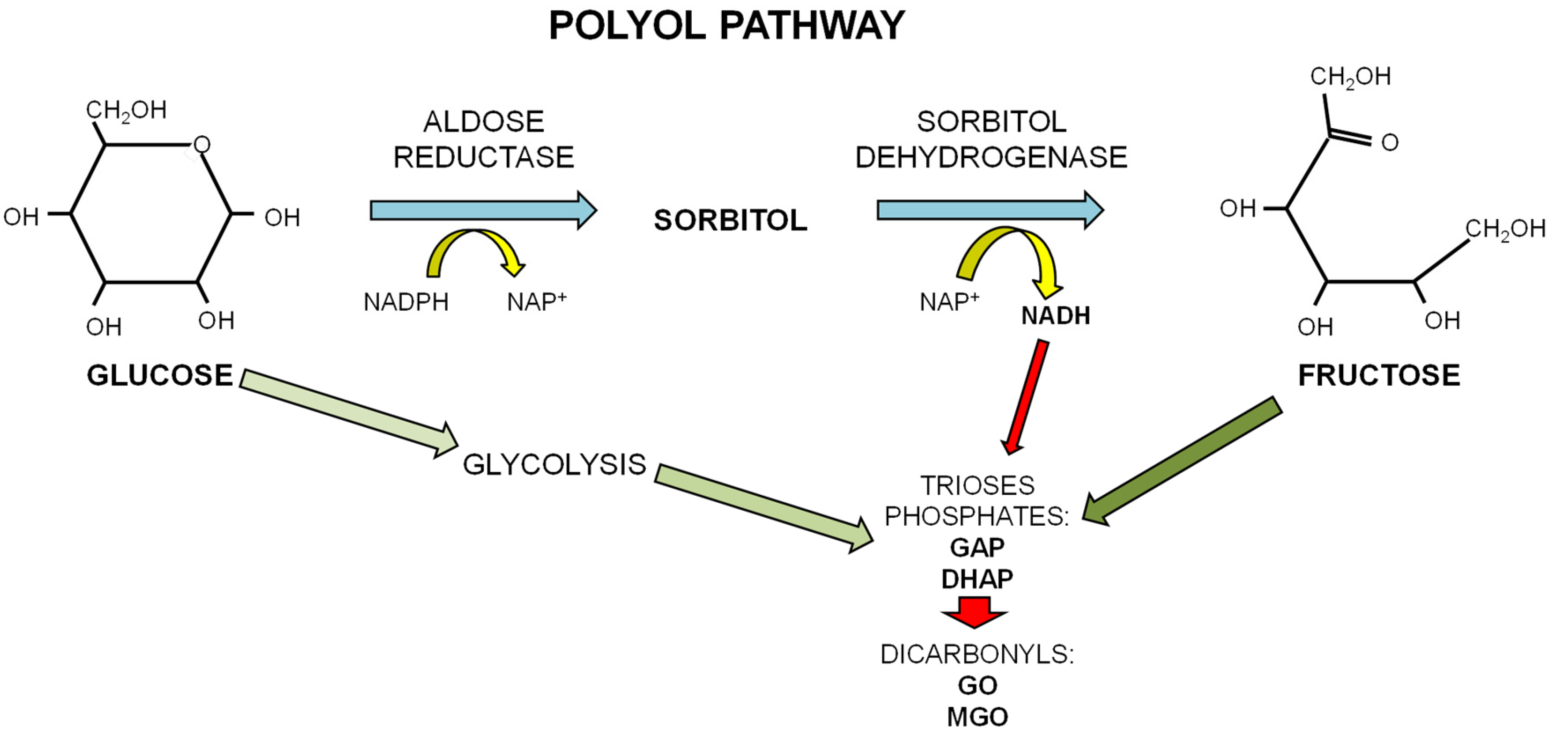
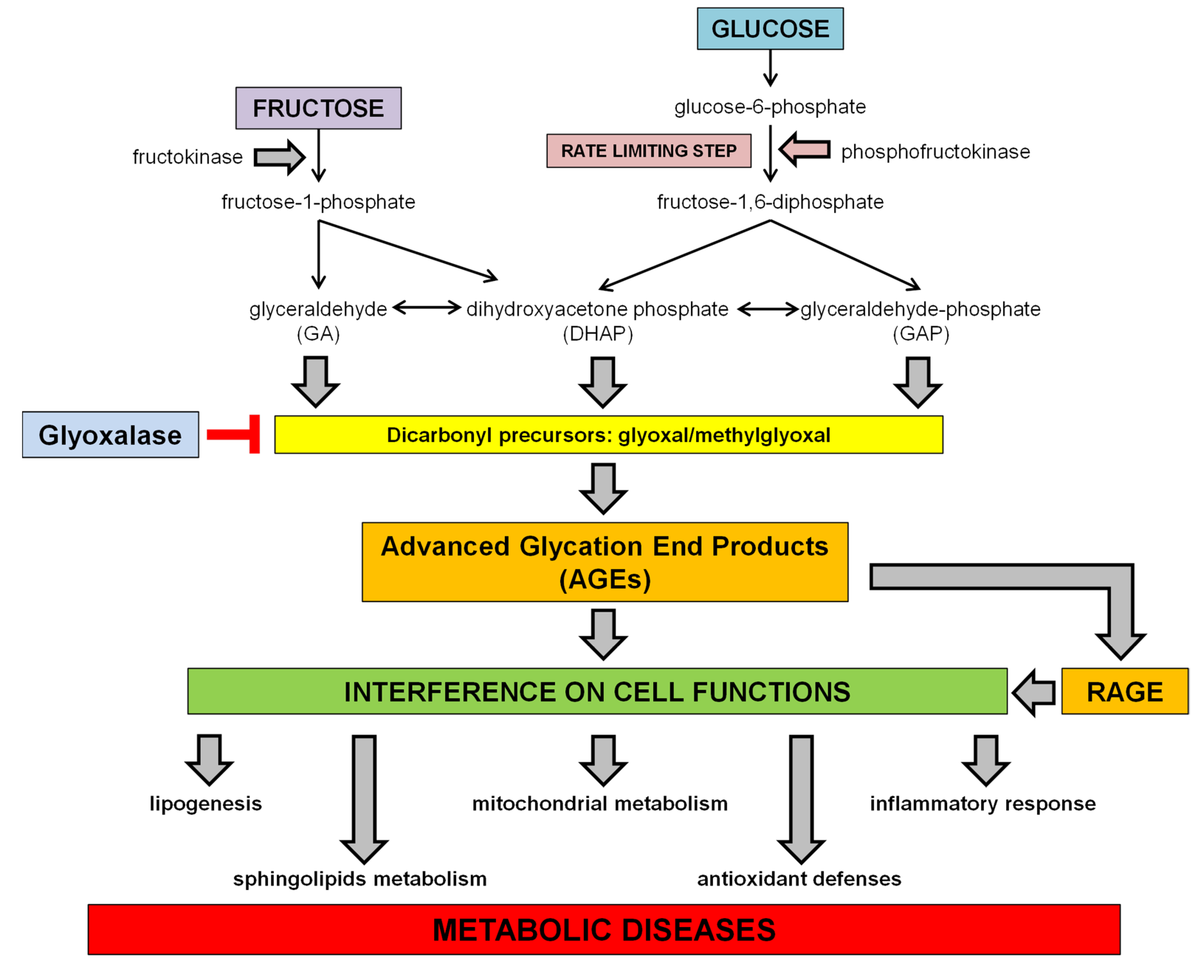
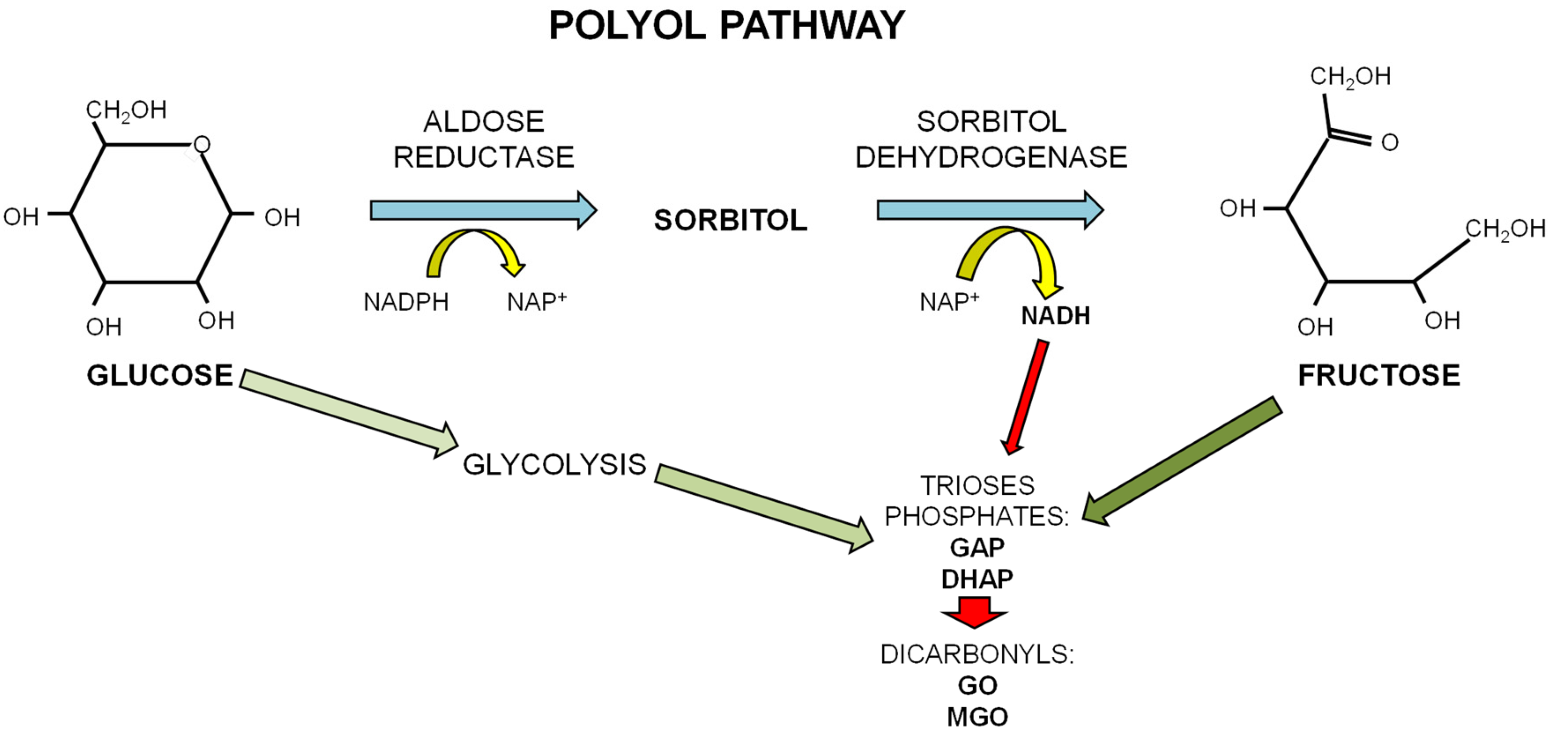
1.3. Dietary Fructose and Glucose Metabolism
2. Dietary Sugars and Glycation
2.1. Advanced Glycation Endproducts (AGEs)
2.2. In Vitro Protein Glycation of Different Sugars
2.3. Dietary Intake of Exogenous AGEs and of Simple Sugars as Potential Sources for Endogenous AGEs
3. Dietary Sugar-Induced Glycation: Interference on Cell Functions
3.1. Interference with the SCAP/SREBP Lipogenic Pathway
3.2. Interference on Sphingolipids Metabolism
3.3. Interference on Inflammatory Response
3.4. Interference with Mitochondrial Function and Oxidative Stress
4. Dietary Sugar-Induced Glycation and Pathogenic Role in Diseases
4.1. Data from Animal Studies
4.2. Data from Human Studies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCrory, M.A.; Shaw, A.C.; Lee, J.A. Energy and nutrient timing for weight control. Endocrinol. Metab. Clin. N. Am. 2016, 45, 689–718. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L. Q&A: “Toxic” effects of sugar. Should we be afraid of fructose? BMC Biol. 2012, 10, 42. [Google Scholar] [CrossRef]
- Marriot, B.P.; Olsho, L.; Haden, L.; Connor, P. Intake of Added sugars andselected nutrients in the United States, National Health and Nutrition Examination Survey (NHANES) 2003–2006. Crit. Rev. Food Sci. Nutr. 2010, 50, 228–258. [Google Scholar] [CrossRef] [PubMed]
- Avena, N.M.; Rada, P.; Hoebel, B.G. Evidence for sugar addiction: Behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci. Biobehav. Rev. 2008, 32, 20–39. [Google Scholar] [CrossRef] [PubMed]
- Kendig, M.D. Cognitive and behavioural effects of sugar consumption in rodents. A review. Appetite 2014, 80, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, B.; Flood, V.M.; Burlutksy, G.; Louie, J.C.; Mitchell, P. Association between carbohydrate nutrition and prevalence of depressive symptoms in older adults. Br. J. Nutr. 2016, 116, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.A. New unsweetened truths about sugar. JAMA 2014, 174, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Després, J.P.; Hu, F.B. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation 2010, 121, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, Z.; Gregg, E.W.; Flanders, W.D.; Merritt, R.; Hu, F.B. Added sugar intake and cardiovascular diseases mortality among US adults. JAMA Intern. Med. 2014, 174, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Adeli, K. Dietary fructose and the metabolic syndrome. Curr. Opin. Gastroenterol. 2008, 24, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Collino, M.; Rogazzo, M.; Medana, C.; Nigro, D.; Boccuzzi, G.; Aragno, M. Advanced glycation end products promote hepatosteatosis by interfering with SCAP-SREBP pathway in fructose-drinking mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G398–G407. [Google Scholar] [CrossRef] [PubMed]
- Soares, E.; Prediger, R.D.; Nunes, S.; Castro, A.A.; Viana, S.D.; Lemos, C.; De Souza, C.M.; Agostinho, P.; Cunha, R.A.; Carvalho, E.; et al. Spatial memory impairments in a prediabetic rat model. Neuroscience 2013, 250, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Newens, K.; Walton, J. A review of sugar consumption from nationally representative dietary surveys across the world. J. Hum. Nutr. Diet. 2016, 29, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.; Sadeghirad, B.; Lytvyn, L.; Slavin, J.; Johnston, B.C. The Scientific Basis of Guideline Recommendations on Sugar Intake: A Systematic Review. Ann. Intern. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Schillinger, D.; Kearns, C. Guidelines to Limit Added Sugar Intake: Junk Science or Junk Food? Ann. Intern. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kearns, C.E.; Schmidt, L.A.; Glantz, S.A. Sugar Industry and Coronary Heart Disease Research: A Historical Analysis of Internal Industry Documents. JAMA Intern. Med. 2016, 176, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- 1Mozaffarian, D.; Hao, T.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Changes in diet and lifestyle and long-term weight gain in women and men. N. Engl. J. Med. 2011, 364, 2392–2404. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.B.; Manson, J.E.; Ludwig, D.S.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 2004, 292, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Livesey, G.; Taylor, R. Fructose consumption and consequences for glycation, plasma triacylglycerol, and body weight: Meta-analyses and meta-regression models of intervention studies. Am. J. Clin. Nutr. 2008, 88, 1419–1437. [Google Scholar] [PubMed]
- Mock, K.; Lateef, S.; Benedito, V.A.; Tou, J.C. High-fructose corn syrup-55 consumption alters hepatic lipid metabolism and promotes triglyceride accumulation. J. Nutr. Biochem. 2017, 39, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Collino, M.; Benetti, E.; Rogazzo, M.; Mastrocola, R.; Yaqoob, M.M.; Aragno, M.; Thiemermann, C.; Fantozzi, R. Reversal of the deleterious effects of chronic dietary HFCS-55 intake by PPAR-δ agonism correlates with impaired NLRP3 inflammasome activation. Biochem. Pharmacol. 2013, 85, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Collino, M.; Mastrocola, R.; Nigro, D.; Chiazza, F.; Aragno, M.; D’Antona, G.; Minetto, M.A. Variability in myosteatosis and insulin resistance induced by high-fat diet in mouse skeletal muscles. BioMed Res. Int. 2014, 2014, 569623. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Mastrocola, R.; Rogazzo, M.; Chiazza, F.; Aragno, M.; Fantozzi, R.; Collino, M.; Minetto, M.A. High sugar intake and development of skeletal muscle insulin resistance and inflammation in mice: A protective role for PPAR-δ agonism. Mediat. Inflamm. 2013, 2013, 509502. [Google Scholar] [CrossRef] [PubMed]
- Truswell, A.S.; Seach, J.M.; Thornburn, A.W. Incomplete absorption of pure fructose in healthy subjects and the facilitating effect of fructose. Am. J. Clin. Nutr. 1988, 48, 1424–1430. [Google Scholar] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.; van Hinsbergh, V.W. Fructose-mediated non-enzymatic glycation: Sweet coupling or bad modification. Diabetes Metab. Res. Rev. 2004, 20, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Levi, B.; Werman, M.J. Long-term fructose consumption accelerates glycation and several age-related variables in male rats. J. Nutr. 1998, 128, 1442–1449. [Google Scholar] [PubMed]
- Gillery, P.; Jaisson, S. Post-translational modification derived products (PTMDPs): Toxins in chronic diseases? Clin. Chem. Lab. Med. 2014, 52, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. 1), 3–27. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Ueda, S.; Okuda, S. Food-derived advanced glycation end products (AGEs): A novel therapeutic target for various disorders. Curr. Pharm. Des. 2007, 13, 2832–2836. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Pathologic role of dietary advanced glycation end products in cardiometabolic disorders, and therapeutic intervention. Nutrition 2016, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Bunn, H.F.; Higgins, P.J. Reaction of monosaccharides with proteins: Possible evolutionary significance. Science 1981, 213, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Kashiwagi, A.; Tanaka, Y.; Asahina, T.; Kikkawa, R.; Shigeta, Y. Significance of fructose-induced protein oxidation and formation of advanced glycation end product. J. Diabetes Complicat. 1995, 9, 87–91. [Google Scholar] [CrossRef]
- Suárez, G.; Rajaram, R.; Oronsky, A.L.; Gawinowicz, M.A. Nonenzymatic glycation of bovine serum albumin by fructose (fructation). Comparison with the Maillard reaction initiated by glucose. J. Biol. Chem. 1989, 264, 3674–3679. [Google Scholar] [PubMed]
- Luers, L.; Rysiewski, K.; Dumpitak, C.; Birkmann, E. Kinetics of advanced glycation end products formation on bovine serum albumin with various reducing sugars and dicarbonyl compounds in equimolar ratios. Rejuvenation Res. 2012, 15, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Galiniak, S.; Bartosz, G. Kinetics of glycoxidation of bovine serum albumin by methylglyoxal and glyoxal and its prevention by various compounds. Molecules 2014, 19, 4880–4896. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.J.; Lim, S.I. The effect of sugar, amino acid, metal ion, and NaCl on model Maillard reaction under pH control. Amino Acids 2004, 27, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Namiki, M. Role of sugar fragmentation in an early stage browning of amino-carbonyl reaction of sugar with amino acid. Agric. Biol. Chem. 1986, 50, 1965–1970. [Google Scholar]
- Kawasaki, T.; Akanuma, H.; Yamanouchi, T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care 2002, 25, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Araki, N.; Koh, N.; Nakamura, J.; Horiuchi, S.; Hotta, N. Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem. Biophys. Res. Commun. 1996, 228, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, F.K.; Alli, I.; Yaylayan, V.A. Reactivities of d-glucose and d-fructose during glycation of bovine serum albumin. J. Agric. Food Chem. 1999, 47, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Cai, W.; Tripp, E.; Pyzik, R.; Yee, K.; Goldberg, L.; Tansman, L.; Chen, X.; Mani, V.; Fayad, Z.A.; et al. Oral AGE restriction ameliorates insulin resistance in obese individuals with the metabolic syndrome: A randomised controlled trial. Diabetologia 2016, 59, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Savige, G.S. Dietary advanced glycation end-product restriction for the attenuation of insulin resistance, oxidative stress and endothelial dysfunction: A systematic review. Eur. J. Clin. Nutr. 2013, 67, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Cannizzaro, L.; Rossoni, G.; Savi, F.; Altomare, A.; Marinello, C.; Saethang, T.; Carini, M.; Payne, D.M.; Pisitkun, T.; Aldini, G.; et al. Regulatory landscape of AGE-RAGE-oxidative stress axis and its modulation by PPARγ activation in high fructose diet-induced metabolic syndrome. Nutr. Metab. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [PubMed]
- Nseir, W.; Nassar, F.; Assy, N. Soft drinks consumption and nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Abid, A.; Taha, O.; Nseir, W.; Farah, R.; Grosovski, M.; Assy, N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J. Hepatol. 2009, 51, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Liverini, G.; Iossa, S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur. J. Nutr. 2013, 52, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.; Bruce, W.R.; Dong, Q.; Bruce, J.; Mehta, R.; O’Brien, P.J. Fructose and carbonyl metabolites as endogenous toxins. Chem. Biol. Interact. 2009, 178, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Nigro, D.; Chiazza, F.; Medana, C.; Dal Bello, F.; Boccuzzi, G.; Collino, M.; Aragno, M. Fructose-derived advanced glycation end-products drive lipogenesis and skeletal muscle reprogramming via SREBP-1c dysregulation in mice. Free Radic. Biol. Med. 2016, 91, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Nigro, D.; Menotti, F.; Cento, A.; Serpe, L.; Chiazza, F.; Dal Bello, F.; Romaniello, F.; Medana, C.; Collino, M.; Aragno, M.; et al. Chronic administration of saturated fats and fructose differently affect SREBP activity resulting in different modulation of Nrf2 and Nlrp3 inflammasome pathways in mice liver. J. Nutr. Biochem. 2017, 42, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Lê, K.A.; Ith, M.; Kreis, R.; Faeh, D.; Bortolotti, M.; Tran, C.; Boesch, C.; Tappy, L. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am. J. Clin. Nutr. 2009, 89, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- De Stefanis, D.; Mastrocola, R.; Nigro, D.; Costelli, P.; Aragno, M. Effects of chronic sugar consumption on lipid accumulation and autophagy in the skeletal muscle. Eur. J. Nutr. 2017, 56, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Havel, P.J. Dietary fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 2005, 63, 133–157. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Wallig, M.A.; Chung, B.H.; Nara, T.Y.; Cho, B.H.; Nakamura, M.T. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim. Biophys. Acta 2008, 1782, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, L.C.; Hellerstein, M.K.; Seidman, C.E.; Neese, R.A.; Tremaroli, J.D.; Hirsch, J. Relationship between carbohydrate-induced hypertriglyceridemia and fatty acid synthesis in lean and obese subjects. J. Lipid Res. 2000, 41, 595–604. [Google Scholar] [PubMed]
- Janevski, M.; Ratnayake, S.; Siljanovski, S.; McGlynn, M.A.; Cameron-Smith, D.; Lewandowski, P. Fructose containing sugars modulate mRNA of lipogenic genes ACC and FAS and protein levels of transcription factors ChREBP and SREBP1c with no effect on body weight or liver fat. Food Funct. 2012, 3, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhao, L.; Chen, Y.; Moorhead, J.F.; Varghese, Z.; Powis, S.H.; Minogue, S.; Sun, Z.; Ruan, X.Z. Advanced glycation end products (AGEs) increase human mesangial foam cell formation by increasing Golgi SCAP glycosylation in vitro. Am. J. Physiol. Ren. Physiol. 2011, 301, F236–F243. [Google Scholar] [CrossRef] [PubMed]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Iwaki, S.; Koike, K.; Yuda, Y.; Nagasaki, A.; Ohkawa, R.; Yatomi, Y.; Furumoto, T.; Tsutsui, H.; Sobel, B.E.; et al. Increased plasma sphingosine-1-phosphate in obese individuals and its capacity to increase the expression of plasminogen activator inhibitor-1 in adipocytes. Coron. Artery Dis. 2013, 24, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Cigliano, L.; Liverini, G.; Iossa, S. The effect of high-fat—High-fructose diet on skeletal muscle mitochondrial energetics in adult rats. Eur. J. Nutr. 2015, 54, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Brozinick, J.T.; Hawkins, E.; Hoang Bui, H.; Kuo, M.S.; Tan, B.; Kievit, P.; Grove, K. Plasma sphingolipids are biomarkers of metabolic syndrome in non-human primates maintained on a Western-style diet. Int. J. Obes. 2013, 37, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, K.; Wiernsperger, N.; Lagarde, M.; El Bawab, S. Bimodal effect of advanced glycation end products on mesangial cell proliferation is mediated by neutral ceramidase regulation and endogenous sphingolipids. J. Biol. Chem. 2004, 279, 34343–34352. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, K.; Hao, J.; Huang, J.; Yang, Z.; Xiong, F.; Liu, P.; Huang, H. Polydatin attenuates AGEs-induced upregulation of fibronectin and ICAM-1 in rat glomerular mesangial cells and db/db diabetic mice kidneys by inhibiting the activation of the SphK1-S1P signaling pathway. Mol. Cell. Endocrinol. 2016, 427, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Koves, T.R.; Cadete, V.J.; Zhang, L.; Jaswal, J.S.; Swyrd, S.J.; Lopaschuk, D.G.; Proctor, S.D.; Keung, W.; Muoio, D.M.; et al. Inhibition of de novo ceramide synthesis reverses diet induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 2010, 59, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, D.M.; Gu, T.T.; Ding, X.Q.; Fan, C.Y.; Zhu, Q.; Shi, Y.W.; Hong, Y.; Kong, L.D. Morin reduces hepatic inflammation-associated lipid accumulation in high fructose-fed rats via inhibiting sphingosine kinase 1/sphingosine 1-phosphate signaling pathway. Biochem Pharmacol. 2013, 86, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Collino, M.; Penna, C.; Nigro, D.; Chiazza, F.; Fracasso, V.; Tullio, F.; Alloatti, G.; Pagliaro, P.; Aragno, M. Maladaptive Modulations of NLRP3 Inflammasome and Cardioprotective Pathways Are Involved in Diet-Induced Exacerbation of Myocardial Ischemia/Reperfusion Injury in Mice. Oxid. Med. Cell. Longev. 2016, 2016, 3480637. [Google Scholar] [CrossRef] [PubMed]
- Bakker, P.J.; Butter, L.M.; Kors, L.; Teske, G.J.; Aten, J.; Sutterwala, F.S.; Florquin, S.; Leemans, J.C. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int. 2014, 85, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.H.; Zhang, X.; Pan, Y.; Li, Y.C.; Kong, L.D. Allopurinol, quercetin and rutin ameliorate renal NLRP3 inflammasome activation and lipid accumulation in fructose-fed rats. Biochem. Pharmacol. 2012, 84, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Pan, Y.; Wang, R.; Kang, L.L.; Xue, Q.C.; Wang, X.N.; Kong, L.D. Quercetin inhibits AMPK/TXNIP activation and reduces inflammatory lesions to improve insulin signaling defect in the hypothalamus of high fructose-fed rats. J. Nutr. Biochem. 2014, 25, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ding, X.Q.; Gu, T.T.; Song, L.; Li, J.M.; Xue, Q.C.; Kong, L.D. Pterostilbene and allopurinol reduce fructose-induced podocyte oxidative stress and inflammation via microRNA-377. Free Radic. Biol. Med. 2015, 83, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Chiazza, F.; Couturier-Maillard, A.; Benetti, E.; Mastrocola, R.; Nigro, D.; Cutrin, J.C.; Serpe, L.; Aragno, M.; Fantozzi, R.; Ryffel, B.; et al. Targeting the NLRP3 inflammasome to reduce diet-induced metabolic abnormalities in mice. Mol. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.L.; Zhang, D.M.; Ma, C.H.; Zhang, J.H.; Jia, K.K.; Liu, J.H.; Wang, R.; Kong, L.D. Cinnamaldehyde and allopurinol reduce fructose-induced cardiac inflammation and fibrosis by attenuating CD36-mediated TLR4/6-IRAK4/1 signaling to suppress NLRP3 inflammasome activation. Sci. Rep. 2016, 6, 27460. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; You, H.; Li, X.M.; Liu, T.H.; Wang, P.; Wang, B.E. HMGB1 promotes the synthesis of pro-IL-1β and pro-IL-18 by activation of p38 MAPK and NF-κB through receptors for advanced glycation end-products in macrophages. Asian Pac. J. Cancer Prev. 2012, 13, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.J.; Yang, H.Y.; Pai, M.H.; Wu, C.H.; Chen, J.R. Long-term administration of advanced glycation end-product stimulates the activation of NLRP3 inflammasome and sparking the development of renal injury. J. Nutr. Biochem. 2017, 39, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yang, G.; Chen, H.Y.; Hsu, D.K.; Tomilov, A.; Olson, K.A.; Dehnad, A.; Fish, S.R.; Cortopassi, G.; Zhao, B.; et al. Galectin-3 regulates inflammasome activation in cholestatic liver injury. FASEB J. 2016, 30, 4202–4213. [Google Scholar] [CrossRef] [PubMed]
- Pejnovic, N.N.; Pantic, J.M.; Jovanovic, I.P.; Radosavljevic, G.D.; Djukic, A.L.; Arsenijevic, N.N.; Lukic, M.L. Galectin-3 is a regulator of metaflammation in adipose tissue and pancreatic islets. Adipocyte 2013, 2, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Jeftic, I.; Jovicic, N.; Pantic, J.; Arsenijevic, N.; Lukic, M.L.; Pejnovic, N. Galectin-3 Ablation Enhances Liver Steatosis, but Attenuates Inflammation and IL-33-Dependent Fibrosis in Obesogenic Mouse Model of Nonalcoholic Steatohepatitis. Mol. Med. 2015, 21, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Pejnovic, N.N.; Pantic, J.M.; Jovanovic, I.P.; Radosavljevic, G.D.; Milovanovic, M.Z.; Nikolic, I.G.; Zdravkovic, N.S.; Djukic, A.L.; Arsenijevic, N.N.; Lukic, M.L. Galectin-3 deficiency accelerates high-fat diet-induced obesity and amplifies inflammation in adipose tissue and pancreatic islets. Diabetes 2013, 62, 1932–1944. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Keim, N.L.; et al. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr. Metab. 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Chess, D.J.; Stanley, W.C. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc. Res. 2008, 79, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Valentim, I.B.; de Araújo, O.R.; Ataide, T.D.R.; Goulart, M.O. Development of nonalcoholic hepatopathy: Contributions of oxidative stress and advanced glycation end products. Int. J. Mol. Sci. 2013, 14, 19846–19866. [Google Scholar] [CrossRef] [PubMed]
- Hipkiss, A.R. Aging risk factors and Parkinson’s disease: Contrasting roles of common dietary constituents. Neurobiol. Aging 2014, 35, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Wainwright, G.; Mascitelli, L. Nutrition and Alzheimer’s disease: The detrimental role of a high carbohydrate diet. Eur. J. Intern. Med. 2011, 22, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Nigro, D.; Cento, A.S.; Chiazza, F.; Collino, M.; Aragno, M. High-fructose intake as risk factor for neurodegeneration: Key role for carboxy methyllysine accumulation in mice hippocampal neurons. Neurobiol. Dis. 2016, 89, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y. Toll-like receptor-6 (TLR6) deficient mice are protected from myocardial fibrosis induced by high fructose feeding through anti-oxidant and inflammatory signaling pathway. Biochem. Biophys. Res. Commun. 2016, 473, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Zheng, F.; Striker, G.E.; Vlassara, H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am. J. Pathol. 2008, 173, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Tan, C.; Wang, Y.; Yang, S.; Tan, D. Betanin reduces the accumulation and cross-links of collagen in high-fructose-fed rat heart through inhibiting non-enzymatic glycation. Chem. Biol. Interact. 2015, 227, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Brouwers, O.; Gaens, K.H.; Wouters, K.; Cleutjens, J.P.; Janssen, B.J.; Miyata, T.; Stehouwer, C.D.; Schalkwijk, C.G. Delayed Intervention With Pyridoxamine Improves Metabolic Function and Prevents Adipose Tissue Inflammation and Insulin Resistance in High-Fat Diet-Induced Obese Mice. Diabetes 2016, 65, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Blaak, E.E.; et al. Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Beck-Nielsen, H.; Pedersen, O.; Lindskov, H.O. Impaired cellular insulin binding and insulin sensitivity induced by high-fructose feeding in normal subjects. Am. J. Clin. Nutr. 1980, 33, 273–278. [Google Scholar] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [PubMed]
- Wehmeyer, M.H.; Zyriax, B.C.; Jagemann, B.; Roth, E.; Windler, E.; Zur Wiesch, J.S.; Lohse, A.W.; Kluwe, J. Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine 2016, 95, e3887. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Sugars, obesity, and cardiovascular disease: Results from recent randomized control trials. Eur. J. Nutr. 2016, 55 (Suppl. 2), 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A.; Lustig, R.H.; Caccavello, R.; Erkin-Cakmak, A.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Mulligan, K.; Schwarz, J.M. Short-term isocaloric fructose restriction lowers apoC-III levels and yields less atherogenic lipoprotein profiles in children with obesity and metabolic syndrome. Atherosclerosis 2016, 253, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Li, J.; Shi, J.; Yang, B.; Tang, J.; Truby, H.; Li, D. Acute metabolic and endocrine responses induced by glucose and fructose in healthy young subjects: A double-blinded, randomized, crossover trial. Clin. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [PubMed]
- Beeri, M.S.; Moshier, E.; Schmeidler, J.; Godbold, J.; Uribarri, J.; Reddy, S.; Sano, M.; Grossman, H.T.; Cai, W.; Vlassara, H.; et al. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech. Ageing Dev. 2011, 132, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Uribarri, J.; Zhu, L.; Chen, X.; Swamy, S.; Zhao, Z.; Grosjean, F.; Simonaro, C.; Kuchel, G.A.; Schnaider-Beeri, M.; et al. Oral glycotoxins are a modifiable cause of dementia and the metabolic syndrome in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 4940–4945. [Google Scholar] [CrossRef] [PubMed]
- DeChristopher, L.R.; Uribarri, J.; Tucker, K.L. Intake of high fructose corn syrup sweetened soft drinks is associated with prevalent chronic bronchitis in U.S. Adults, ages 20–55 years. Nutr. J. 2015, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- DeChristopher, L.R.; Uribarri, J.; Tucker, K.L. Intakes of apple juice, fruit drinks and soda are associated with prevalent asthma in US children aged 2–9 years. Public Health Nutr. 2016, 19, 123–130. [Google Scholar] [CrossRef] [PubMed]
- DeChristopher, L.R.; Uribarri, J.; Tucker, K.L. Intake of high-fructose corn syrup sweetened soft drinks, fruit drinks and apple juice is associated with prevalent arthritis in US adults, aged 20–30 years. Nutr. Diabetes 2016, 6, e199. [Google Scholar] [CrossRef] [PubMed]
- Bains, Y.; Gugliucci, A. Ilex paraguariensis and its main component chlorogenic acid inhibit fructose formation of advanced glycation endproducts with amino acids at conditions compatible with those in the digestive system. Fitoterapia 2016, 117, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 9, 385. https://doi.org/10.3390/nu9040385
Aragno M, Mastrocola R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients. 2017; 9(4):385. https://doi.org/10.3390/nu9040385
Chicago/Turabian StyleAragno, Manuela, and Raffaella Mastrocola. 2017. "Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease" Nutrients 9, no. 4: 385. https://doi.org/10.3390/nu9040385
APA StyleAragno, M., & Mastrocola, R. (2017). Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients, 9(4), 385. https://doi.org/10.3390/nu9040385