1. Introduction
Mycotoxins are a diverse, large group of fungal secondary metabolites that exert severe toxic effects on human and animals worldwide. A recent study revealed that, among the nearly 20,000 feed and feed raw material samples which were collected from Asia, Europe, Americas, Africa, and Middle East, 72% contained at least one of the five major mycotoxins (aflatoxin B
1, AFB
1; zearalenone, ZEN; deoxynivalenol, DON; fumonisin B
1, FB
1; and ocharatoxin A, OTA). See
Figure 1 for structures [
1]. In the United States, an annual loss of
$52.1 million to
$1.68 billion is estimated to be caused by merely aflatoxin contamination in the corn industry [
2].
To minimize mycotoxin contamination, agronomic means have proven to be effective in reducing the risk but are still inadequate [
1]. Physical and chemical treatments of food and feed do not remove mycotoxins effectively because these secondary metabolites are often insensitive. In feed industry, inert adsorbents such as montmorillonite are widely used to bind mycotoxins in the animal’s gastrointestinal tract. However, binding of mycotoxins to adsorbents is effective for only limited kinds of mycotoxins such as AFB
1 because of the inherent selective nature of adsorbents [
3]. Therefore, microbial and, in essence, enzymatic detoxification are increasingly regarded as an attractive method to control mycotoxin contamination. However, mycotoxins are structurally distinct to each other and, more importantly, different types of mycotoxins usually co-exist in the food, feed, and feed raw materials. For example, more than 38% of the samples were detected with co-contamination of two or even more mycotoxins [
1]. To deal with the complexity of mycotoxin contamination, a feasible route toward economic detoxification would ideally be to use one or a few enzymes. This requires that enzymes must have a wide substrate specificity on multiple mycotoxins.
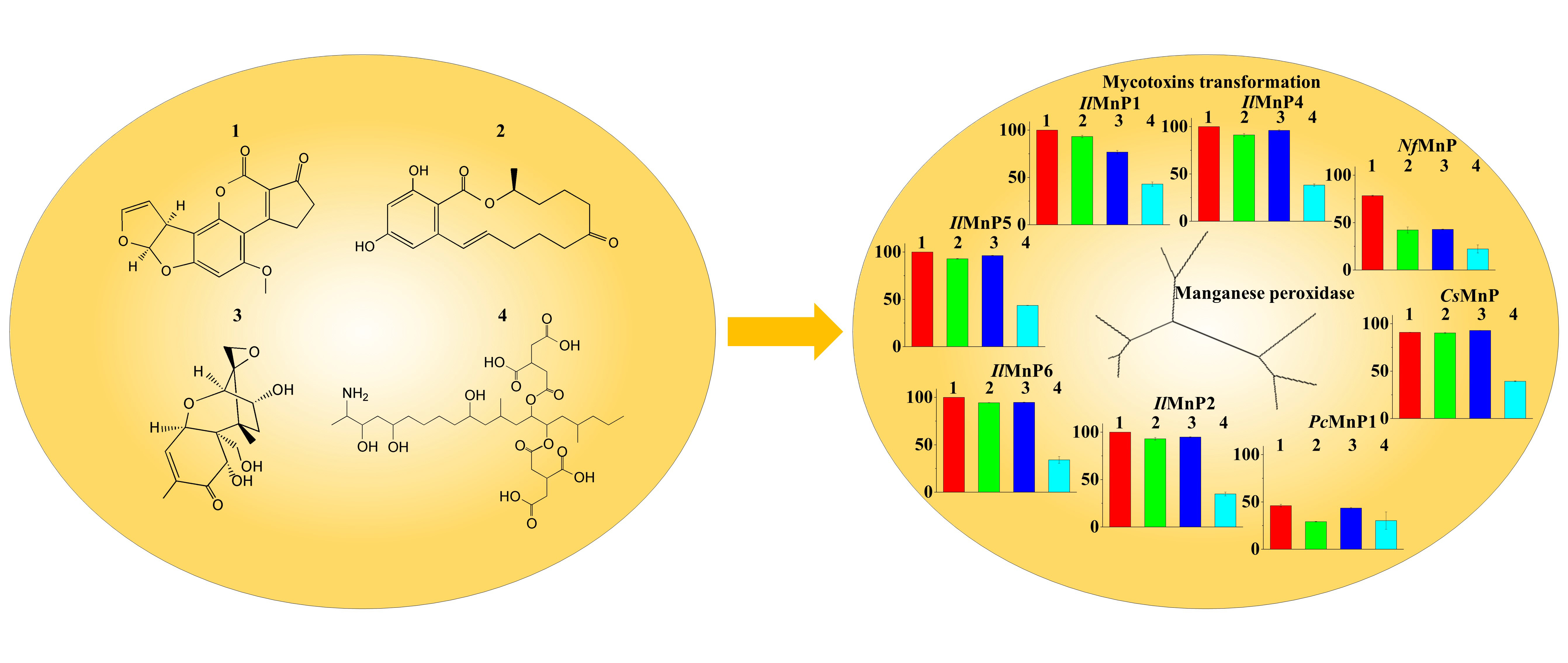
Most of the identified enzymes that are able to detoxify mycotoxins can be grouped into hydrolase, transferase, epimerase, and oxidoreductase. Although most discovered enzymes, if not all, are reported to detoxify only a certain type of mycotoxins, among the oxidoreductases, a special group of enzymes targeting lignin (i.e., laccase and lignin-modifying peroxidases) have intriguing promiscuous substrate specificity. It is thus noted that the lignin-degrading white rot fungi have been successfully used in mycotoxin detoxification [
4,
5]. In addition, part of mycotoxins may be structurally similar to a lignin monomer or its derivatives. For example, AFB
1 has a coumarin structure (
Figure 1), which is a derivative of the lignin monomer
p-coumaryl alcohol [
6]. Indeed, laccase (a multicopper oxidase catalyzing lignin degradation or polymerization) [
7] and manganese peroxidase (MnP, a Mn
2+-dependent lignin-degrading peroxidase) [
8] are occasionally found to be able to degrade AFB
1 [
9,
10,
11]. MnP has a higher redox potential than laccase, suggestive of its higher ability to react with more recalcitrant substrates. MnP is widely distributed in lignin-degrading white-rot filamentous fungi. However, despite the previous finding of MnP to degrade AFB
1, it remains unknown if MnP can serve as a candidate enzyme to degrade multiple mycotoxins. In this study, we determined the ability of MnP to act on other mycotoxins and if the ability to degrade multiple mycotoxins is a feature shared by MnPs. The recombinantly produced MnPs used in this study were from
Irpex lacteus CD2,
Phanerochaete chrysosporium, and
Ceriporiopsis subvermispora. All these fungi have not been reported to have mycotoxin-degrading ability. However, they all express MnPs involved in lignin degradation.
I. lacteus CD2 encodes seven MnPs [
12], with two of them having the ability to oxidize the recalcitrant non-phenolic lignin model compound veratryl alcohol in the presence of malonate [
13].
P. chrysosporium and
C. subvermispora are the representative simultaneous and selective white rot lignocellulose degraders, respectively [
14], both of which also encode MnP enzymes [
15,
16]. Therefore, five MnPs from
I. lacteus CD2 (which can be recombinantly produced) and one each representative MnP from
P. chrysosporium and
C. subvermispora were tested for their ability to degrade multiple mycotoxins.
3. Discussion
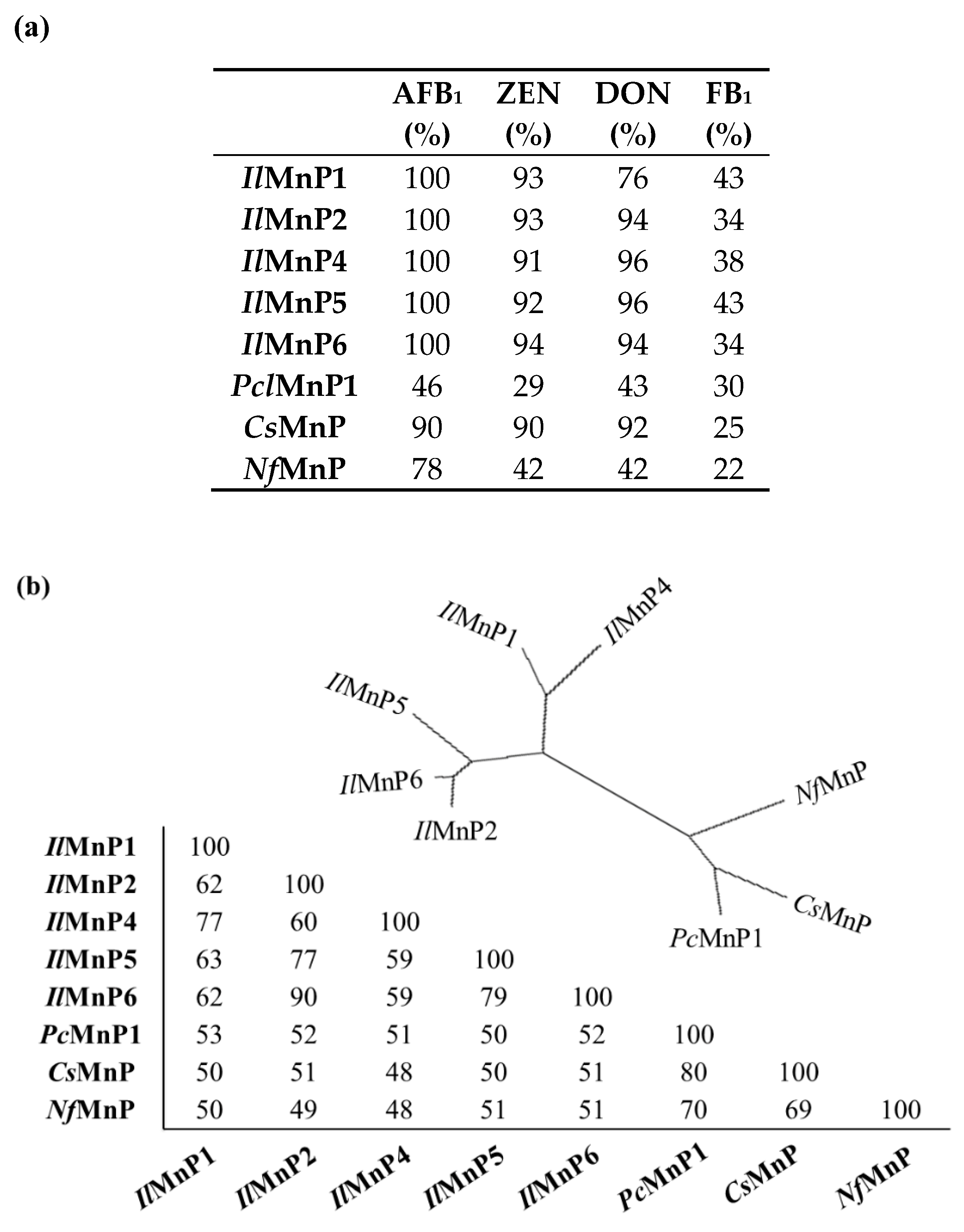
Currently, there are approximately 500 known mycotoxins [
28]. The structural diversity, in combination with co-contamination of multiple mycotoxins in food or feed, dwarfs the endeavors to use one or a limited number of enzymes for detoxification. However, in this study, we demonstrated that, MnPs can degrade at least four major mycotoxins instead of only one (i.e., AFB
1, as have been reported previously) [
11]. Although OTA has a phenolic hydroxyl group which is commonly the well-accepted substrate of MnPs, the tested MnPs were ineffective in its degradation. The underlying reason remains unknown.
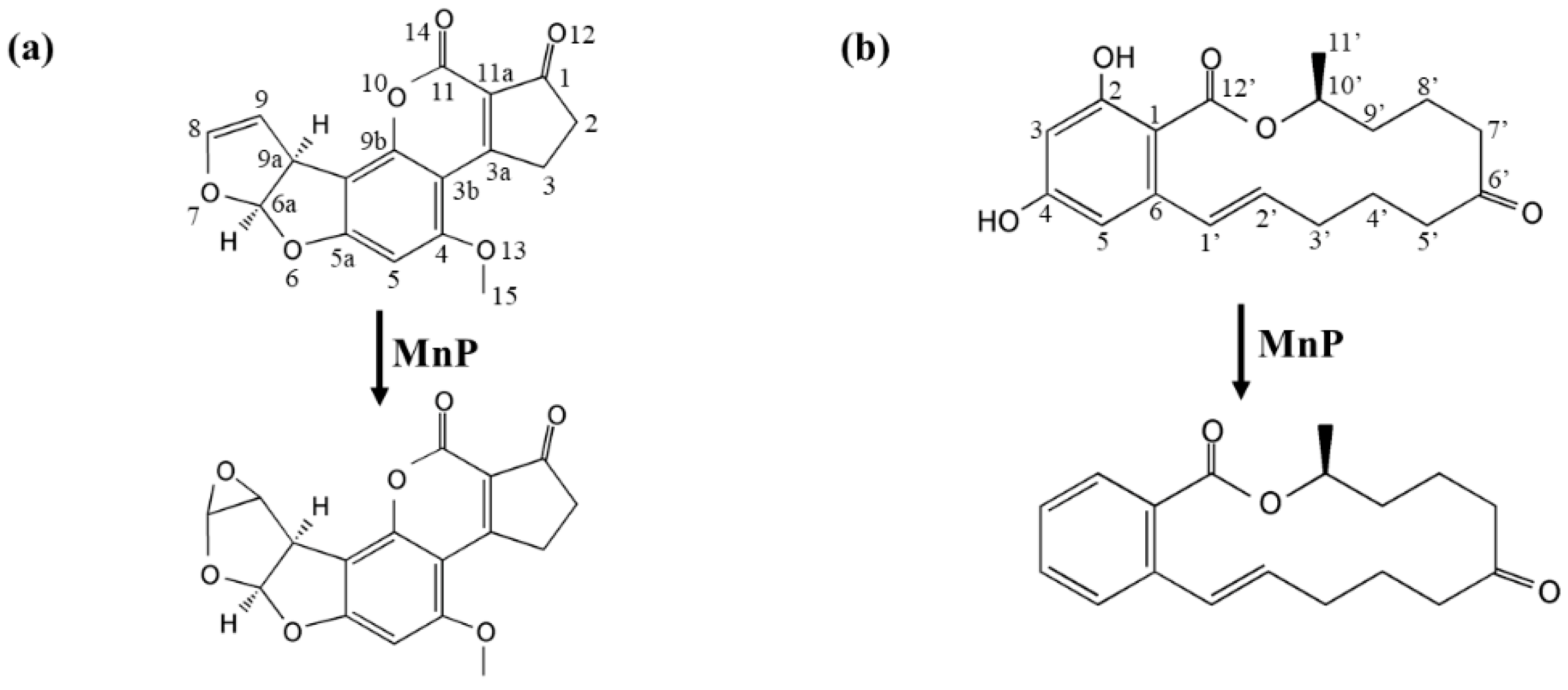
For AFB
1, degradation was mainly through the free radicals, which were generated by interaction of oxidized Mn
3+ with the dicarboxylic acid malonate. AFB
1-8,9-epoxide is labile in aqueous solution and its autonomous hydrolysis product can be monitored at 365 nm [
29], the same wavelength used to detect disappearance of AFB
1. Therefore, these together suggested that most chromophore groups (such as the benzene ring) in AFB
1 had been eliminated in MnP treatment and the observed AFB
1-8,9-epoxide (in UHPLC/MS-MS analysis) and new product peaks in HPLC analysis are likely minor parts of the products. The fact that AFB
1-8,9-epoxide only constituted a small fraction of the products in our experiment is of significance. This is because AFB
1-8,9-epoxide is not a desirable product and it irreversibly attaches to guanine residues to generate highly mutagenic DNA adducts [
30].
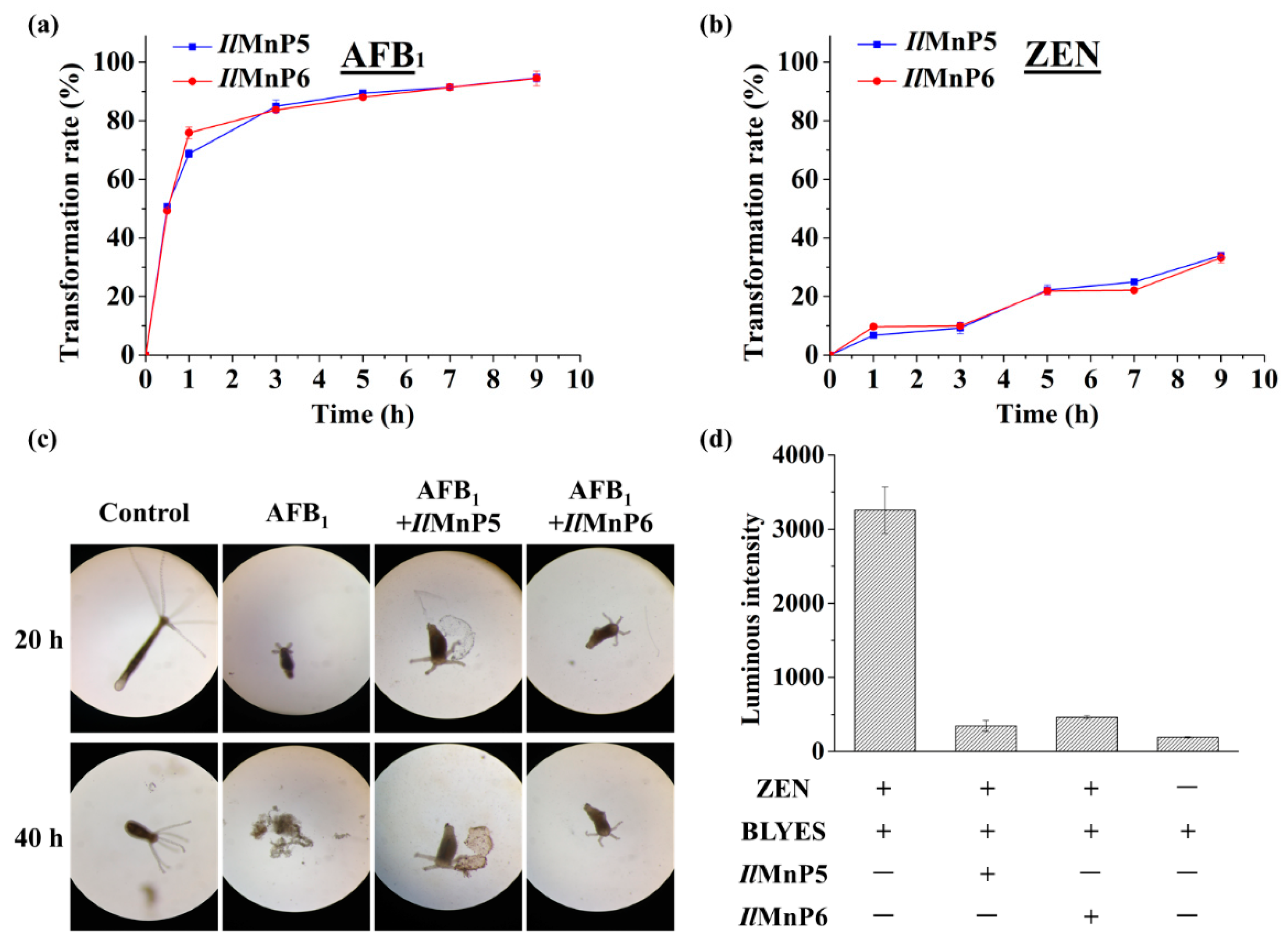
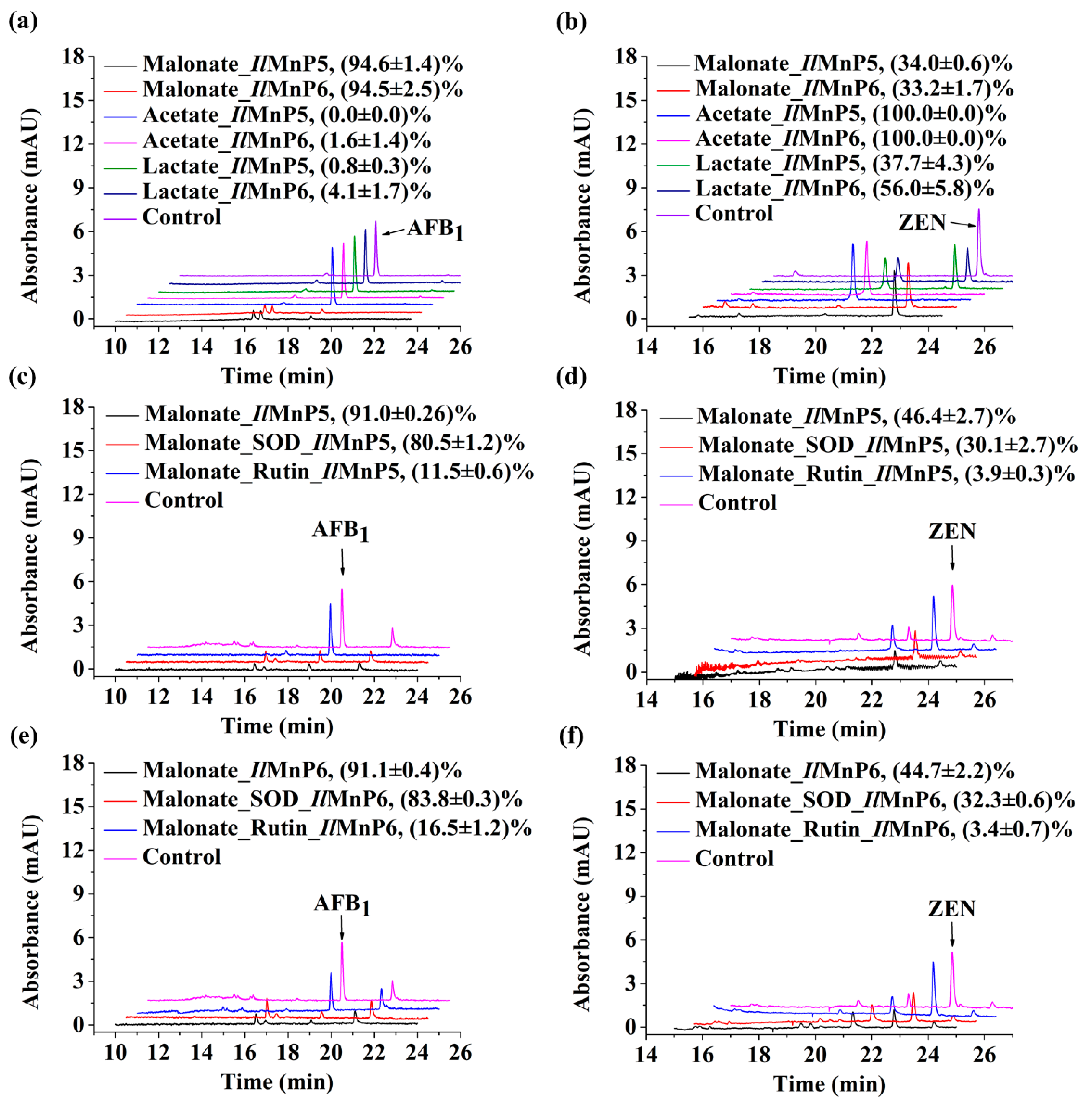
The nature of other and major complex degradation products remains to be unveiled. This finding appears to be applicable to MnP-catalyzed ZEN degradation, as well. However, since the 20.3 min product peak was present, it could be imagined that direct reaction of MnP with ZEN could also happen. With these observed, there appeared to be some competition between the two pathways leading to ZEN degradation, i.e., direct reaction of a MnP with ZEN and indirect reaction via free radicals, since the degradation rates of ZEN by IlMnP5 and IlMnP6 in lactate buffer were higher than those in malonate buffer but lower than those in acetate buffer.
The huge number of mycotoxins prevents us from testing the efficacy of the eight MnPs in their degradation one by one. However, given the wide reactivity of free radicals generated by MnPs, amended by direct interaction of MnPs with substrates containing the phenolic hydroxy, phenylamine, and azino linkages [
8], it is very likely that other mycotoxins can also be degraded. More importantly, it appears that the ability to degrade multiple mycotoxins is not restricted to one specific MnP but tends to be a common feature shared by this kind of enzymes. In this study, although the toxicity of degradation products of DON and FB
1 was not evaluated, we observed much reduced toxicity (AFB
1) or estrogenicity (ZEN) for AFB
1 and ZEN degraded by
IlMnP5 and
IlMnP6 using the hydra and BLYES yeast assays, suggesting the application potential of these enzymes in mycotoxin detoxification. Nevertheless, it has to be admitted that these preliminary assays used much simplified model systems and cannot completely reflect the true scenarios where mycotoxins exert their roles in human and animals. For example, in addition to its toxicity, AFB
1 is well-known for its carcinogenicity and causes cancer in the liver, kidney, and colon [
31]. Therefore, a detailed study of the nature of the complex degradation products for all the tested mycotoxins, as well as their toxicity to cultured cells and animal models have to be systematically investigated. Two or more mycotoxins are commonly observed to co-exist but the types of mycotoxins vary among different foods and feeds [
32]. If the MnP-catalyzed degradation products of certain specific combinations of mycotoxins are, in future, proved to be of less or even no toxicity to animals and human, MnP can truly serve as a candidate enzyme for detoxification because of its capability to degrade multiple mycotoxins.
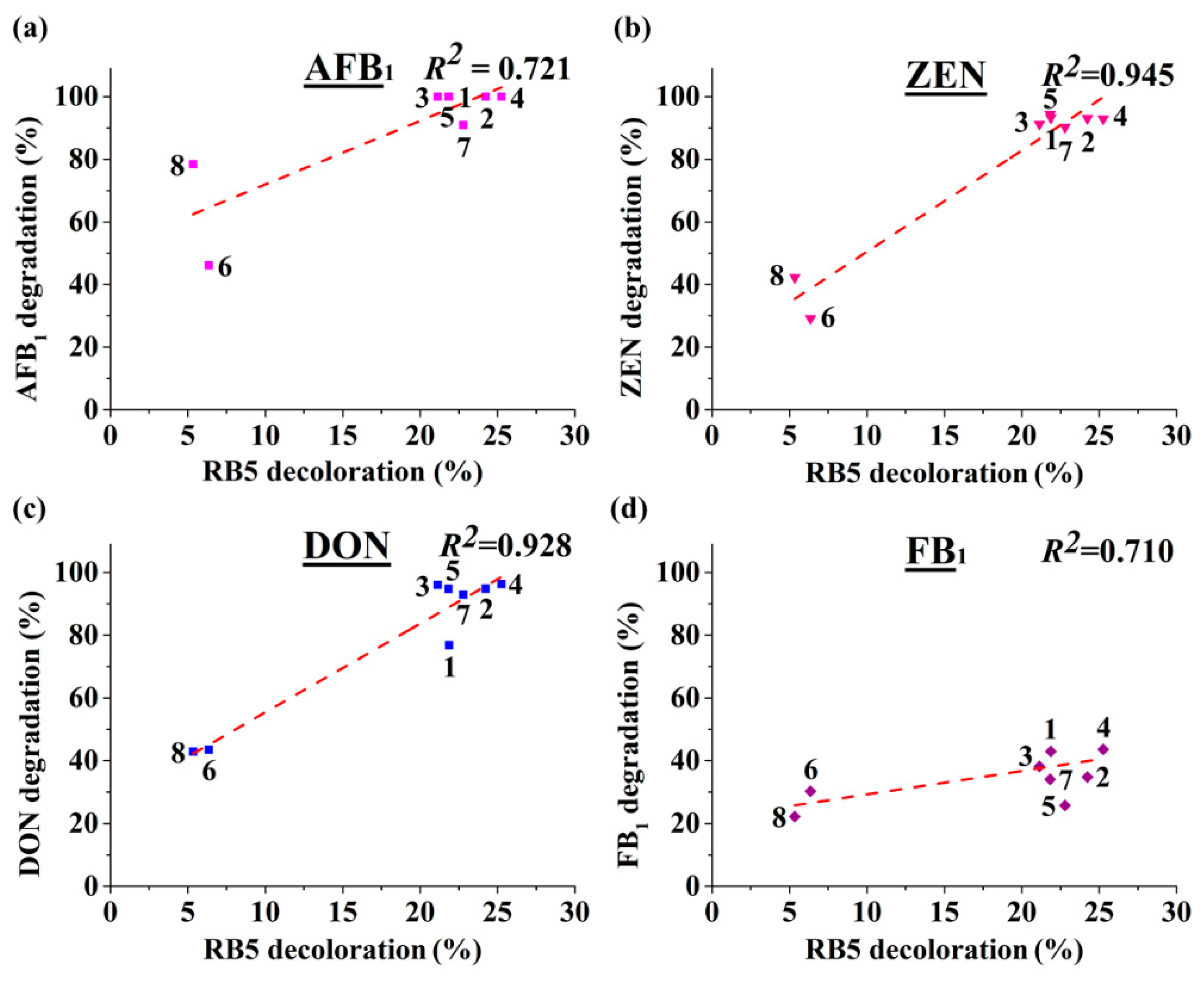
We also demonstrated that RB5 decolorization may be used as a starting point to dictate discovery of new MnPs, through which the risk of handling mycotoxin will be greatly reduced. As MnP is classified in a large although expanding CAZy (Carbohydrate Active Enzymes) family (
http://www.cazy.org/AA2.html) [
33], it is therefore expected that the current finding of broad-spectrum mycotoxin-degrading enzymes will be used as a guiding framework to facilitate future discovery of robust mycotoxin-degrading enzymes with desirable properties. Additionally, this method can also be used for high-throughput screening of MnP variants with improved properties such as higher velocity of transformation, which may be generated from (semi)rational design or directed evolution. This is important since the time of incubation with the four mycotoxins is as long as 72 h in this study (
Figure 5). From a practical perspective, a MnP with higher efficiency as well as higher velocity of transformation would be much more preferred.
One drawback of using MnPs for mycotoxins degradation (and potentially, detoxification) is the difficulty in obtaining large quantities of such enzymes. White-rot fungi, such as
P. chrysosporium,
C. subvermispora, and
I. lacteus, are a natural microbial source rich in MnP genes. However, the white rots are divided into simultaneous and selective lignocellulose degraders, which differ in their way of regulating expression of the lignin-degrading enzymes (including MnPs) [
14]. For instance, while
P. chrysosporium gradually increases expression of MnPs (and other lignin-degrading enzymes) during culture on lignocellulose [
15], the selective lignocellulose degraders
C. subvermispora and
I. lacteus only express MnPs at the early stage of culturing [
12,
34]. Therefore, care has to be taken in monitoring the expression of MnPs if such enzymes are to be produced from these microorganisms. MnPs can also be made in
Pichia pastoris [
35] or filamentous fungi [
36]; however, they are notoriously hard to express recombinantly. In addition, the prosthetic group hemin has to be supplemented to the culture as an expensive additive for the formation of functional proteins. Thus future efforts can be placed in genetic engineering of the commonly used platform industrial microbes to facilitate large scale, heterologous production of MnPs.
Although MnPs were the only enzymes investigated in this study, because of the important role of free radicals involved, it is proposed that other oxidoreductases may also have the ability to degrade mycotoxins. Specifically, another lignin-degrading enzyme, laccase [
37] (and probably other peroxidases as well), may also be used in mycotoxin degradation once a reaction condition is settled up favoring generation of mycotoxin-attacking radicals. Mediators (i.e., small molecules reacting with a laccase) play key roles in dictating the scope of a laccase and amplifying its catalyzing efficiency [
38]. Both artificial and natural mediators can be screened if laccase is to be employed in mycotoxin degradation. Indeed, the Ery4 laccase from
Pleurotus eryngii has been used to degrade AFB
1, ZEN, FB
1, and OTA in presence of a few structurally defined chemicals, such as acetosyringone and syringaldehyde, albeit with lower efficiency [
31]. Interesting, the laccase/mediator system can degrade OTA but cannot act on DON, while the MnP/malonate system can degrade DON but not OTA. All in all, the versatility of manganese peroxidase makes it a candidate enzyme to simultaneously degrade multiple and much differing mycotoxins in food and feed, although a systematic investigation of the toxicity of the degradation products to animals and human has to be implemented ahead of that.
5. Materials and Methods
5.1. Chemicals and Other Materials
AFB1, ZEN, DON, and the commercial Nematoloma frowardii manganese peroxidase (NfMnP) were purchased from Sigma-Aldrich (St. Louis, MO, USA). FB1 and OTA were purchased from Pribolab (Beijing, China). Hemin was purchased from TCI (Tokyo, Japan). DNA polymerase, T4 ligase, and chromatographic grade reagents (acetonitrile, methanol, methanoic acid, acetic acid, and trifluoroacetic acid) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). DNase I and TransScript One-Step gDNA Removal and cDNA Synthesis Supermix with oligo(dT) were purchased from TransGen (Beijing, China). Isopropyl-β-D-thiogalactoside (IPTG), SOD, DTT, and Rutin were purchased from Solarbio (Beijing, China). TRIZOL was from Invitrogen (Carlsbad, CA, USA). Lysozyme was purchased from Amresco (Solon, OH, USA). Ni-NTA agarose was purchased from QIAGEN (Duesseldorf, Germany). All other chemicals were of analytical grade or chromatographically pure, and were commercially available.
5.2. Plasmids, Bacterial Strains, and Cultural Conditions
The plasmids used in this study for expression of recombinant MnPs were pET-28a-
IlMnP1, pET28a-
IlMnP2, pET28a-
IlMnP4, pET28a-
IlMnP5, pET28a-
IlMnP6, pET28a-
PcMnP1, and pCold I-
CsMnP (
Figure S6).
Irpex lacteus CD2 was isolated from Shennong Nature Reserve (Hubei province, China) and maintained at 4 °C on potato-dextrose agar (PDA) plate. The
Escherichia coli Trans1-T1 was used for gene cloning and plasmid propagation. The
E. coli BL21 (DE3) strain was used for the expression of enzymes. These
E. coli strains were cultivated at 37 °C with constant shaking at 220 rpm in Luria-Bertani (LB) broth medium: tryptone (10 g), yeast extract (5 g), and NaCl (10 g) in water (1 L, pH 7.0) containing appropriate antibiotics.
5.3. Construction of Recombinant Plasmids
The primers used in this study are listed in
Table S1.
I. lacteus CD2 was grown for 5 d in the basal liquid medium. Total RNA was extracted using the TRIZOL reagent according to the manufacturer’s instructions and reverse transcribed to the first strand cDNA using the TransScript One-Step gDNA Removal and cDNA Synthesis Supermix with oligo (dT). The
Phanerochaete chrysosporium PcMnP1 (GenBank: J04980.1) [
25] and
Ceriporiopsis subvermispora CsMnP (GenBank: MG190336.1) [
26] genes devoid of the sequences encoding the signal peptide were synthesized by BGI (Beijing, China). The three-dimensional structures of two homologs of
PcMnP1 and
CsMnP have been solved, which can be found at the protein structure database (
http://www.rcsb.org/) with the entry numbers of 1MNP [
39] (or 1YYD [
40]) and 4CZN [
41], respectively.
PcMnP1 has 79% amino acid sequence identity with 1MNP (or 1YYD) while
CsMnP has 84% identity with 4CZN. The
I. lacteus,
P. chrysosporium, and
C. subvermispora MnP genes were amplified with gene specific primers using the following conditions: 95 °C for 2 min; then 30 cycles of 95 °C for 30 s, 54 °C for 30 s, and 72 °C for 1 min. The amplified genes of
IlMnP enzymes and
PcMnP1 were restriction digested with
EcoRI/
NotI, gel purified, and ligated into the pET-28a(+) plasmid pre-digested with the same enzymes to obtain pET-28a-
IlMnP1, pET28a-
IlMnP2, pET28a-
IlMnP4, pET28a-
IlMnP5, pET28a-
IlMnP6, and pET28a-
PcMnP1 (
Figure S6). The PCR product of
CsMnP was digested with
NdeI/
BamHI, gel purified, and ligated into the pre-digested pCold I (digested with
NdeI/
BamHI) to obtain pCold I-
CsMnP. The seven recombinant plasmids were transformed into
E. coli BL21(DE3) competent cells for gene expression.
5.4. Expression of Manganese Peroxidases
The
E. coli BL21(DE3) strains harboring pET-28a-
IlMnP1, pET-28a-
IlMnP2, pET-28a-
IlMnP4, pET-28a-
IlMnP5, pET-28a-
IlMnP6, or pET-28a-
PcMnP1 were cultured in LB medium supplemented with 50 μg/mL of kanamycin at 37 °C overnight with shaking at 220 rpm. These pre-cultures were individually inoculated into 200 mL LB medium. The culture was continued at 37 °C for approximately 2 h. When the optical density at 600 nm (OD
600) reached 0.6–0.8, IPTG was added to a final concentration of 1 mM for induction of MnPs expression [
13]. For the
E. coli BL21(DE3) strain containing pCold I-
CsMnP, when OD
600 reached 0.5, the bacterium was rapidly chilled to 10 °C by soaking the culture flask in a water-ice bath, followed by shaking at 220 rpm at 10 °C for 30 min. IPTG was added to a final concentration of 0.2 mM and the cells were grown at 10 °C with shaking at 220 rpm for 24 h. Six hour after IPTG was added, 1 M CaCl
2 and 10 g/L hemin were supplied continuously for another 9 h at rates of 22 μL/h and 220 μL/h to the 200 mL LB culture medium, respectively. After induction, the cells were harvested by centrifugation at 12,000
g for 2 min [
26].
5.5. Refolding and Purification of the Recombinant MnPs
For
E. coli expressing recombinant MnPs from
I. lacteus and
P. chrysosporium MnPs, the harvested cell pellets were re-suspended in 50 mM Tris-HCl, 10 mM EDTA, and 5 mM DTT (pH 8.0). Lysozyme was added to a final concentration of 2 mg/mL and the cells were incubated on ice for 1 h. Then, 20 μL of DNase I was added and the incubation was continued on ice for 30 min. Subsequently, the cells were centrifuged at 12,000
g for 30 min at 4 °C. The cell debris was washed twice with 20 mM Tris-HCl, 1 mM EDTA, and 5 mM DTT (pH 8.0), followed by incubation in 50 mM Tris-HCl, 8 M urea, 1 mM EDTA, and 1 mM DTT (pH 8.0) on ice for 1 h. To optimize the parameters for recovery of active enzyme from the inclusion bodies, the refolding was carried out in different conditions. The parameters including concentrations of urea, GSSG, and hemin and pH were investigated, while the concentrations of enzyme, EDTA, and DTT were kept constant during the refolding. The efficiency of refolding was indicated by the MnP activity. Refolding of the MnPs were conducted under the optimized conditions (pH 9.5, 50 mM Tris-HCl buffer, 0.6 M urea, 0.5 mM GSSG, 0.1 mM DTT, 10 μM hemin, 5 mM CaCl
2, 0.1 mg/mL protein for
IlMnP1; pH 9.5 50 mM Tris-HCl buffer, 0.5 M urea, 0.7 mM GSSG, 0.1 mM DTT, 10 μM hemin, 5 mM CaCl
2, 0.1 mg/mL protein for
IlMnP2,
IlMnP5 and
IlMnP6; pH 9.5 50 mM Tris-HCl buffer, 0.5 M urea, 0.7 mM GSSG, 0.1 mM DTT, 10 μM hemin, 5 mM CaCl
2, 0.1 mg/mL protein for
IlMnP4; pH 9.5 50 mM Tris-HCl buffer, 1 M urea, 0.4 mM GSSG, 0.1 mM DTT, 10 μM hemin, 5 mM CaCl
2, 0.1 mg/mL protein for
PcMnP1) for 10 h at 15 °C. After refolding, the crude enzymes were centrifuged at 12,000
g for 10 min at 4 °C and the insoluble fractions were discarded. The supernatants containing the refolded MnP were concentrated through a 10 kDa cut-of centrifuge filter, followed by dialysis against buffers of different pHs (pH 6.0, 20 mM Na
2HPO
4-citric acid buffer for
IlMnP1; pH 5.0 20 mM HAc-NaAc buffer for
IlMnP2,
IlMnP5,
IlMnP6; pH 6.5 20 mM Na
2HPO
4-citric acid buffer for
IlMnP4 and
PcMnP1). The crude enzymes were further purified by a HiTrap Q HP anion exchange column (GE Health, Fairfeld, CT) pre-equilibrated with the same buffer. The proteins were eluted with a linear gradient of 0–1.0 M NaCl, and fractions containing pure and active enzymes were pooled [
13].
The
E. coli cell pellet expressing recombinant
CsMnP was harvested from 100 mL of culture medium, re-suspended in 10 mL of 50 mM Tris-HCl buffer (pH 8.0) containing 1 mM CaCl
2, and homogenized by sonication. After centrifugation at 12,000
g for 30 min, the cell debris was discarded and the supernatant was collected, which was passed through a Ni affinity column resin. The resin was washed with 50 mM Na
2HPO
4-NaH
2PO
4 buffer (pH 7.5) containing 500 mM NaCl, 1 mM CaCl
2, and 20 mM imidazole to remove the nonspecifically bound proteins. The bound MnPs were eluted with 50 mM Tris-HCl (pH 7.5) containing 500 mM NaCl, 1 mM CaCl
2, and 40/60/80/100/200/500 mM imidazole, respectively. The eluents were analyzed for purity by SDS-PAGE. The purified MnPs were stored in 50 mM Tris-HCl (pH 7.5) at 4 °C containing 1 mM CaCl
2 until used [
26]. The purified recombinant manganese peroxidases were analyzed on SDS-PAGE (
Figure S3).
5.6. Measurement of MnP Activity
The MnP activity was measured by monitoring the oxidation of 2,2′-Azino-
bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS, ε420 = 36,000 M
−1·cm
−1) at 420 nm, in a buffer containing 50 mM malonate, 1 mM ABTS, 1 mM MnSO
4, and 0.1 mM H
2O
2 (pH 5.0 and 25 °C) [
13]. One unit (1 U) of MnP activity was defined as the amount of enzyme that produced 1 μmol of product per minute under the standard assay condition.
5.7. Mycotoxin Degradation
The activities of the enzymes were first determined using ABTS as the substrate. Each of the calibrated enzymes (0.5 U/mL each) was then incubated with mycotoxins (AFB1 and ZEN 5 μg/mL; DON, and FB1, 10 μg/mL; OTA, 50 μg/mL) in 70 mM malonate buffer supplemented with 1 mM MnSO4 and 0.1 mM H2O2. The reaction was carried out at 30 °C. Periodically, samples were taken out and three volumes of methanol were added to terminate the reaction. Each reaction was repeated three times.
5.8. RB5 Decolorization
The reactions were carried out at 30 °C in a total volume of 200 μL containing 50 mM malonate buffer (pH 5.0), 0.1 mM H
2O
2, 1 mM Mn
2+, 0.25 U/mL each of the MnPs, and 50 μg/mL of dye. During the incubation, the color change was detected by measuring the OD
556 (RB5). The rate of decolorization was calculated using the following formula: decolorization (%) = [(Ai−At)/Ai] × 100, where Ai and At are the absorbance at the initial and a given time [
13].
5.9. BLYES Assay
The
Saccharomyces cerevisiae BLYES strain was inoculated into 30 mL YMM ((NH
4)
2SO
4) 1.7 g/L, CuSO
4 12 mg/L, FeSO
4 684 µg/L, KH
2PO
4 11.6 g/L, KOH 3.6 g/L, MgSO
4 171 µg/L, D-(+)-glucose 20 g/L, biotin 20 µg/L, pantothenic acid 400 µg/L, inositol 1 mg/L, pyridoxine 400 µg/L, thiamine 400 µg/L, adenine 42.7 mg/L, arginine HCl 17.1 mg/L, aspartic acid 100 mg/L, glutamic acid 85.5 mg/L, histidine 42.73 mg/L, isoleucine 25.64 mg/L, lysine HCl 25.64 mg/L, methionine 17.1 mg/L, phenylalanine 21.4 mg/L, serine 320.4 mg/L, threonine 192 mg/L, tyrosine 25.7 mg/L) in a baked 250 mL glass flask. The cells were cultured at 28 °C with constant shaking at 200 rpm to an OD
600 of 0.6. ZEN was dissolved in 70 mM malonate buffer to 5 μg/mL supplemented with 1 mM MnSO
4 and 0.1 mM H
2O
2. Twenty microliter of appropriately diluted ZEN pre-treated or non-treated with
IlMnP5 or
IlMnP6 were mixed with 200 µL BLYES and the estrogenicity was checked by measuring the bioluminescence of the cells collected 6-h post treatment for analysis [
20,
21].
5.10. Hydra Assay
The hydra was maintained clean and free from bacteria and fungi contamination by treating with diluted iodine solution (2.7 ppm) periodically. The assay was performed by exposing the hydra to AFB
1 treated or nontreated with an MnP. Fifty μg/mL of AFB
1 were incubated with
IlMnP5 or
IlMnP6 (0.5 U/mL each) in 70 mM malonate buffer supplemented with 1 mM MnSO
4 and 0.1 mM H
2O
2. The reaction was carried out at 30 °C for 10 h. Each test dish contained 1 mL of test solution and three normal healthy hydra. The hydra were examined for signs of toxicity at 20 h and 40 h, respectively. The toxic endpoint was determined by the “tulip” or “disintegration” stage of the hydra. In each test, experimental treatments were compared with untreated and solvent controls [
19].
5.11. HPLC and LC-MS/MS Analyses
HPLC analysis of AFB
1, ZEN, DON, and OTA was performed using a SHIMADZU 20A series instrument (Kyoto, Japan) with an Agilent ZORBAX SB-C18 column (5 µm, 4.6 mm × 250 mm) (Santa Clara, CA, USA). The elution condition for AFB
1 and ZEN was set as: no acetonitrile (ACN), 4 min; 0–100% ACN, 25 min; 100% ACN, 6 min, at a flow rate of 0.8 mL/min. AFB
1 and ZEN were monitored at 365 nm or 316 nm [
42], respectively. The elution condition for DON was set as: 20% methanol, 20 min; 20–100% methanol, 1 min; 100% methanol, 6 min, at a flow rate of 0.8 mL/min. DON was monitored at 220 nm. The mobile phase for OTA was mixed CAN:H
2O:HAc (99:99:2) and the flow rate was 1.0 mL/min. OTA was monitored for its absorbance at 333 nm.
AFB1, ZEN transformation products were analyzed by using LC-MS/MS, which was carried out by coupling a SHIMADZU Nexera UHPLC system (Kyoto, Japan) to an AB-SCIEX 5600+ Triple TOF mass spectrometer (Waltham, Massachusetts, USA). The solvent A for LC is mixed ACN:methanol (1:1) and solvent B is 0.1% fomic acid. The program was set as: 30–70% solvent A, 10 min; 70% solvent A, 8 min; 100% solvent A, 2 min; 30% solvent A, 5 min. The parameters for MS analysis were: positive and high-sensitivity mode; GS1, 50 psi; GS2, 50 psi; curtain gas, 25 psi; temperature, 500 °C; ion spray voltage floating, 5, 500 V; CE energy, 35 V ± 15 V. Degradation of FB1 was analyzed by using UHPLC-MS/MS, which was carried out by the same method as above.
5.12. Phylogenetic Analysis
The alignment of the amino acid sequences of 8 MnPs were conducted using the clustalW algorithm of MEGA-X. Phylogenetic tree constructions were performed using the neighbor-joining method of MEGA-X. The reliability of the trees was tested by bootstrap analysis and the parameter was set to 1000. The other parameters used default values.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





