Simultaneous Determination of Ergot Alkaloids in Swine and Dairy Feeds Using Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry


 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
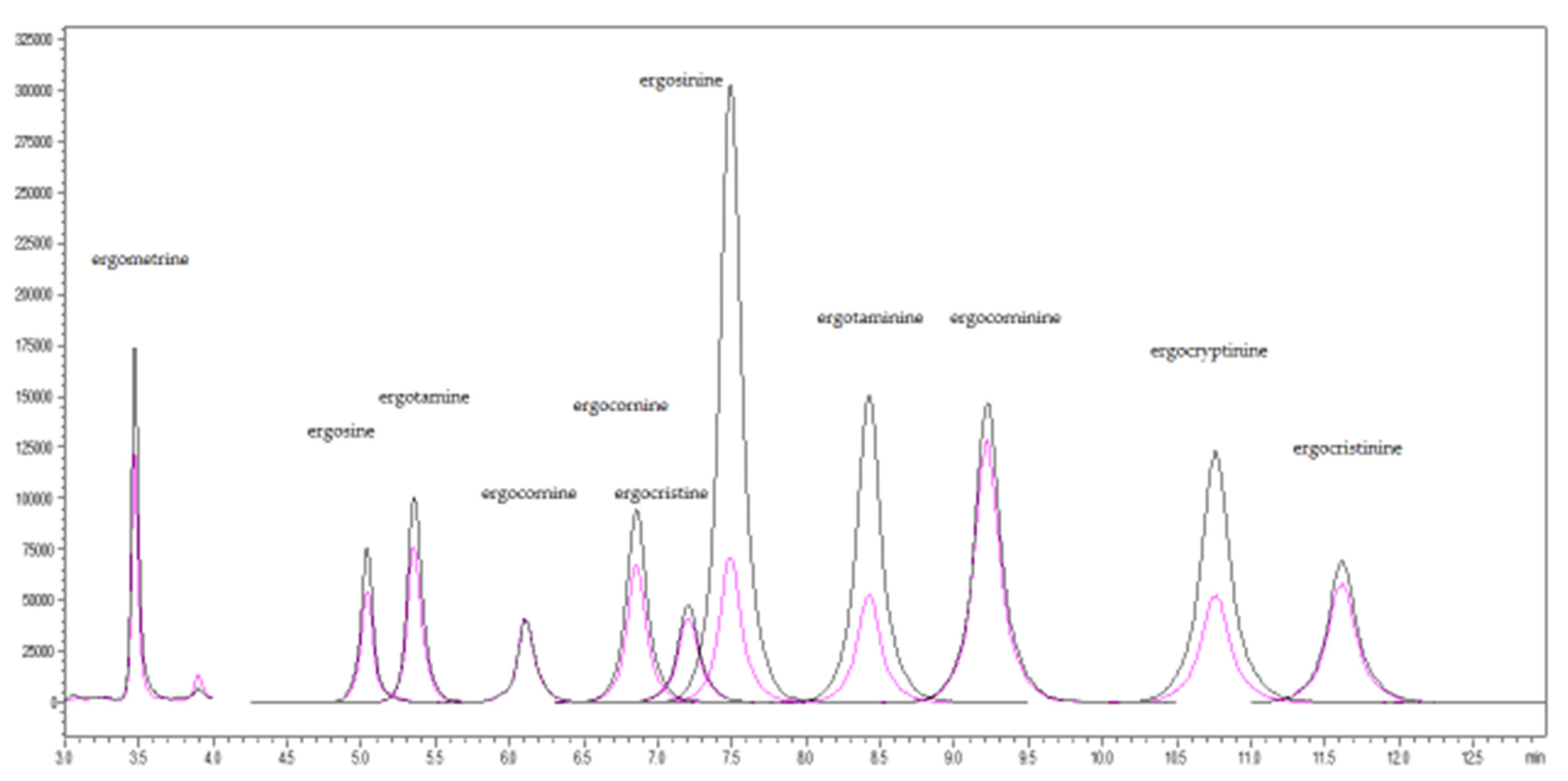
2.1. Method Validation
2.2. Matrix Effect Study
2.3. Occurrence of EAs in Swine and Dairy Feeds
3. Conclusions
4. Materials and Methods
4.1. Reagents and Materials
4.2. Analytical Standards
4.3. Preparation of Standards Solution
4.4. Sample Collection
4.5. Sample Preparation
4.6. UHPLC-MS/MS Analysis
4.7. Method Validation Procedure
4.8. Matrix Effects Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Miró-Abella, E.; Herrero, P.; Canela, N.; Arola, L.; Borrull, F.; Ras, R.; Fontanals, N. Determination of mycotoxins in plant-based beverages using QuEChERS and liquid chromatography–tandem mass spectrometry. Food Chem. 2017, 229, 366–372. [Google Scholar] [CrossRef]
- Dzuman, Z.; Zachariasova, M.; Lacina, O.; Veprikova, Z.; Slavikova, P.; Hajslova, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef]
- Aupanun, S.; Poapolathep, S.; Giorgi, M.; Imsilp, K.; Poapolathep, A. An overview of toxicology and toxicokinetics of fusarenon-X, a type B trichothecene mycotoxin. J. Vet. Med. Sci. 2017, 79, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Worldwide Regulation for Mycotoxins in Food and Feed in 2003. Available online: https//www.fao.org/docrep/007/y5499e/y5499e00.htm (accessed on 8 July 2021).
- Moretti, A.; Pascale, M.; Logrieco, A.F. Mycotoxin risks under a climate change scenario in Europe. Trends Food Sci. Tech. 2019, 84, 38–40. [Google Scholar] [CrossRef]
- Komarova, E.L.; Tolkachev, O.N. The chemistry of peptide ergot alkaloids. Part I. Classification and chemistry of ergot peptides. Pharm. Chem. J. 2001, 35, 37–45. [Google Scholar]
- Malachova, A.; Sulyok, M.; Beltran, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 2014, 1362, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urga, K.; Debella, A.; Medihn, Y.W.; Agata, N.; Bayu, A.; Zewdie, W. Laboratory studies on the outbreak of gangrenous ergotism associated with consumption of contaminated barley in Arsi, Ethiopia. Ethiop. J. Health Dev. 2002, 16, 317–323. [Google Scholar] [CrossRef]
- Krska, R.; Crews, C. Significance, chemistry and determination of ergot alkaloids: A review. Food Addit. Contam. Part A 2008, 25, 722–731. [Google Scholar] [CrossRef]
- Scott, P.M. Ergot alkaloids: Extent of human and animal exposure. World Mycotoxin J. 2009, 2, 141–149. [Google Scholar] [CrossRef]
- Craig, A.M.; Klotz, J.L.; Duringer, J.M. Cases of ergotism in livestock and associated ergot alkaloid concentrations in feed. Front. Chem. 2015, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Malysheva, S.V.; Larionova, D.A.; Di Mavungu, J.D.; De Saeger, S. Pattern and distribution of ergot alkaloids in cereals and cereal products from European countries. World Mycotoxin J. 2014, 7, 217–230. [Google Scholar] [CrossRef]
- Maruo, V.M.; Bracarense, A.P.; Metayer, J.P.; Vilarino, M.; Oswald, I.P.; Pinton, P. Ergot alkaloids at doses close to EU regulatory limits induce alterations of the liver and intestine. Toxins 2018, 10, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tittlemier, S.A.; Drul, D.; Roscoe, M.; McKendry, T. Occurrence of ergot and ergot alkaloids in Western Canadian wheat and other cereals. J. Agric. Food Chem. 2015, 63, 6644–6650. [Google Scholar] [CrossRef] [PubMed]
- Wyka, S.A.; Mondo, S.J.; Liu, M.; Nalam, V.; Broders, K.D. A large accessory genome, high recombination rates, and selection of secondary metabolite genes help maintain global distribution and broad host range of the fungal plant pathogen Claviceps purpurea. bioRxiv 2020. [Google Scholar] [CrossRef]
- Guo, Q.; Shao, B.; Du, Z.; Zhang, J. Simultaneous determination of 25 ergot alkaloids in cereal samples by ultra-performance liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2016, 64, 7033–7039. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.; Tischler, J. Determination of ergot alkaloids in feed by HPLC. Mycotoxin Res. 2008, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.P.; Pereboom-de Fauw, D.P.; Hoogenboom, R.L.; de Stoppelaar, J.; de Nijs, M. Tropane and ergot alkaloids in grain-based products for infants and young children in the Netherlands in 2011–2014. Food Addit. Contam. Part B 2015, 8, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Kodisch, A.; Oberforster, M.; Raditschnig, A.; Rodermann, B.; Tratwal, A.; Danielewicz, J.; Kobas, M.; Schmiedchen, B.; Eifler, J.; Gordillo, A.; et al. Covariation of ergot severity and alkaloid content measured by HPLC and one Elisa method in inoculated winter rye across three isolates and three European countries. Toxins 2020, 12, 676. [Google Scholar] [CrossRef]
- Schummer, C.; Zandonella, I.; van Nieuwenhuyse, A.; Moris, G. Epimerization of ergot alkaloids in feed. Heliyon 2020, 6, e04336. [Google Scholar] [CrossRef]
- Arroyo-Manzanares, N.; Rodriguez-Estevez, V.; Garcia-Campana, A.M.; Castellon-Rendon, E.; Gamiz-Gracia, L. Determination of principal ergot alkaloids in swine feeding. J. Sci. Food Agric. 2021, 101, 5214–5224. [Google Scholar] [CrossRef] [PubMed]
- Di Mavungu, J.D.; Malysheva, S.A.; Sanders, M.; Larionova, D.; Robbens, J.; Dubruel, P.; van Peteghem, C.; de Saeger, S. Development and validation of a new LC-MS/MS method for the simultaneous determination of six major ergot alkaloids and their corresponding epimers. Application to some food and feed commodities. Food Chem. 2012, 135, 292–303. [Google Scholar] [CrossRef]
- Versilovskis, A.; Mulder, P.J.; Pereboom-de Fauw, P.K.H.; de Stoppelaar, J.; de Nijs, M. Simultaneous quantification of ergot and tropane alkaloids in bread in the Netherlands by LC-MS/MS. Food Addit. Contam. Part B 2020, 13, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Malysheva, S.V.; di Mavungu, J.D.; Goryacheva, I.Y.; de Saeger, S. A systematic assessment of the variability of matrix effects in LC-MS/MS analysis of ergot alkaloids in cereals and evaluation of method robustness. Anal. Bioanal. Chem. 2013, 405, 5595–5604. [Google Scholar] [CrossRef] [PubMed]
- Babic, J.; Tavcar-Kalcher, G.; Celar, F.A.; Kos, K.; Cervek, M.; Jackovac-Strajn, B. Ergot and ergot alkaloids in cereal grains intended for animal feeding collected in Slovenia: Occurrence, pattern and correlations. Toxins 2020, 12, 730. [Google Scholar] [CrossRef] [PubMed]
- SANTE, J. European Commission Document No. SANTE/11813/. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed. 2017, pp. 3357–3367. Available online: https//ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf (accessed on 9 July 2021).
- European Commission. Commission Regulation (EC) 472/2002 of March 12th 2002 amending Regulation (EC) 466/2001 setting maximum levels for certain contaminants if foodstuffs. Off. J. Eur. Communities 2002, L75, 18–20. [Google Scholar]
- European Commission. Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, L364, 5–24. [Google Scholar]
- European Commission. Directive 2002/32/EC of the European Parliament and of the Council of 7 May 2002 on undesirable substances in animal feed. Off. J. Eur. Communities 2002, L140, 10–21. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Ergot Alkaloid | LOD (ng/g) | LOQ (ng/g) | Calibration Range (ng/g) |
|---|---|---|---|
| Ergometrine | 0.25 | 0.5 | 0.5–100 |
| Ergosine | 0.25 | 0.5 | 0.5–100 |
| Ergocornine | 0.25 | 0.5 | 0.5–100 |
| Ergocryptine | 0.25 | 0.5 | 0.5–100 |
| Ergocristine | 0.25 | 0.5 | 0.5–100 |
| Ergotamine | 0.25 | 0.5 | 0.5–100 |
| Ergosinine | 0.25 | 0.5 | 0.5–100 |
| Ergocorninine | 0.25 | 0.5 | 0.5–100 |
| Ergocryptinine | 0.25 | 0.5 | 0.5–100 |
| Ergocristinine | 0.25 | 0.5 | 0.5–100 |
| Ergotaminine | 0.25 | 0.5 | 0.5–100 |
| Ergot Alkaloids | Spike Level, (ng/g) | Swine Feed | ||
|---|---|---|---|---|
| %Recovery, (%) | Intra-Day Precision, (%RSD) | Inter-Day Precision, (%RSD) | ||
| Ergometrine | 0.5 | 113.1 | 2.60 | 8.8 |
| 10.0 | 94.2 | 1.33 | 7.1 | |
| 100.0 | 96.2 | 2.95 | 4.7 | |
| Ergosine | 0.5 | 111.7 | 5.60 | 8.14 |
| 10.0 | 115.8 | 2.87 | 11.59 | |
| 100.0 | 109.2 | 2.88 | 13.38 | |
| Ergocornine | 0.5 | 105.3 | 3.55 | 4.80 |
| 10.0 | 115.1 | 9.39 | 15.69 | |
| 100.0 | 116.6 | 3.60 | 8.45 | |
| Ergocryptine | 0.5 | 118.9 | 4.21 | 7.22 |
| 10.0 | 109.0 | 11.44 | 11.62 | |
| 100.0 | 114.4 | 2.73 | 6.87 | |
| Ergocristine | 0.5 | 107.1 | 8.77 | 9.34 |
| 10.0 | 119.6 | 14.07 | 16.63 | |
| 100.0 | 120.0 | 7.45 | 8.95 | |
| Ergotamine | 0.5 | 116.9 | 2.70 | 9.76 |
| 10.0 | 117.3 | 8.68 | 15.67 | |
| 100.0 | 117.1 | 5.52 | 9.90 | |
| Ergosinine | 0.5 | 99.1 | 2.25 | 6.51 |
| 10.0 | 98.1 | 2.29 | 5.99 | |
| 100.0 | 97.0 | 1.96 | 5.09 | |
| Ergocorninine | 0.5 | 101.9 | 4.08 | 8.48 |
| 10.0 | 100.7 | 5.74 | 5.52 | |
| 100.0 | 100.0 | 3.07 | 5.84 | |
| Ergocryptinine | 0.5 | 110.9 | 2.84 | 5.86 |
| 10.0 | 106.7 | 4.52 | 10.13 | |
| 100.0 | 100.2 | 3.52 | 6.11 | |
| Ergocristinine | 0.5 | 111.3 | 3.96 | 8.71 |
| 10.0 | 101.6 | 4.44 | 5.33 | |
| 100.0 | 98.8 | 1.77 | 6.04 | |
| Ergotaminine | 0.5 | 100.5 | 3.30 | 7.19 |
| 10.0 | 97.6 | 2.88 | 8.93 | |
| 100.0 | 97.0 | 1.64 | 5.69 | |
| Ergot Alkaloid | Spike Level, (ng/g) | Dairy Feed | ||
|---|---|---|---|---|
| %Recovery, (%) | Intra-Day Precision, (%RSD) | Inter-Day Precision, (%RSD) | ||
| Ergometrine | 0.5 | 92.1 | 2.11 | 8.2 |
| 10.0 | 101.1 | 1.40 | 7.2 | |
| 100.0 | 97.4 | 4.77 | 6.8 | |
| Ergosine | 0.5 | 102.0 | 7.10 | 6.33 |
| 10.0 | 103.0 | 2.22 | 4.55 | |
| 100.0 | 101.0 | 1.95 | 3.08 | |
| Ergocornine | 0.5 | 99.6 | 2.98 | 7.78 |
| 10.0 | 99.4 | 4.18 | 5.49 | |
| 100.0 | 92.7 | 4.33 | 10.79 | |
| Ergocryptine | 0.5 | 101.9 | 2.67 | 6.14 |
| 10.0 | 98.2 | 2.97 | 5.01 | |
| 100.0 | 91.5 | 4.10 | 11.83 | |
| Ergocristine | 0.5 | 102.1 | 1.09 | 4.09 |
| 10.0 | 98.1 | 5.51 | 7.02 | |
| 100.0 | 90.6 | 3.24 | 13.29 | |
| Ergotamine | 0.5 | 102.4 | 4.13 | 7.85 |
| 10.0 | 103.0 | 5.19 | 5.54 | |
| 100.0 | 101.3 | 1.91 | 2.18 | |
| Ergosinine | 0.5 | 100.3 | 3.49 | 3.98 |
| 10.0 | 100.0 | 1.92 | 3.77 | |
| 100.0 | 101.4 | 1.70 | 4.47 | |
| Ergocorninine | 0.5 | 95.7 | 3.21 | 9.92 |
| 10.0 | 97.7 | 0.97 | 2.32 | |
| 100.0 | 98.1 | 2.02 | 3.46 | |
| Ergocryptinine | 0.5 | 97.1 | 2.57 | 8.42 |
| 10.0 | 99.1 | 1.13 | 2.85 | |
| 100.0 | 100.4 | 1.82 | 3.34 | |
| Ergocristinine | 0.5 | 102.7 | 4.51 | 6.73 |
| 10.0 | 95.9 | 3.84 | 2.93 | |
| 100.0 | 98.9 | 1.72 | 3.55 | |
| Ergotaminine | 0.5 | 101.3 | 1.39 | 4.39 |
| 10.0 | 100.0 | 3.13 | 5.79 | |
| 100.0 | 100.3 | 2.05 | 6.73 | |
| Ergot Alkaloid | Swine Feed (n = 100) | ||
|---|---|---|---|
| Number of Positive Samples | Range (ng/g) | Mean (ng/g) | |
| Ergosinine | 37 | 0.53–9.72 | 2.06 |
| Ergosine | 30 | 0.40–4.99 | 1.57 |
| Ergocorninine | 26 | 0.46–25.25 | 4.99 |
| Ergocornine | 23 | 0.29–4.82 | 1.83 |
| Ergocryptinine | 40 | 0.25–100.55 | 7.64 |
| Ergocryptine | 17 | 0.63–17.22 | 4.41 |
| Ergotaminine | 37 | 0.27–13.46 | 2.96 |
| Ergotamine | 33 | 0.31–18.5 | 3.14 |
| Ergocristinine | 28 | 0.67–77.6 | 16.15 |
| Ergocristine | 28 | 0.57–48.00 | 9.16 |
| Ergometrine | 20 | 0.52–10.87 | 3.06 |
| Ergot Alkaloid | Dairy Feed (n = 100) | ||
|---|---|---|---|
| Number of Positive Samples | Range (ng/g) | Mean (ng/g) | |
| Ergosinine | 48 | 0.52–16.61 | 2.69 |
| Ergosine | 36 | 0.45–12.17 | 2.00 |
| Ergocorninine | 46 | 0.38–43.60 | 6.25 |
| Ergocornine | 35 | 0.31–11.47 | 2.44 |
| Ergocryptinine | 50 | 0.44–31.57 | 8.25 |
| Ergocryptine | 33 | 0.58–13.19 | 3.52 |
| Ergotaminine | 49 | 0.46–52.11 | 6.04 |
| Ergotamine | 48 | 0.34–43.02 | 5.34 |
| Ergocristinine | 49 | 0.62–210.53 | 26.75 |
| Ergocristine | 47 | 0.26–98.19 | 13.05 |
| Ergometrine | 36 | 0.26–31.67 | 2.89 |
| Analyte | m/z | Dwell Time (ms) | Q1 Pre Bias (V) | CE (V) | Q3 Pre Bias (V) | Retention Time (Min) |
|---|---|---|---|---|---|---|
| Ergocorninine | 562.40 > 223.30 | 70.0 | −22.0 | −34.0 | −15.0 | |
| 562.40 > 277.30 | 70.0 | −26.0 | −29.0 | −19.0 | 9.4 | |
| Ergocornine | 562.35 > 223.30 | 60.0 | −22.0 | −37.0 | −24.0 | |
| 562.35 > 208.20 | 60.0 | −22.0 | −45.0 | −23.0 | 6.15 | |
| Ergocryptine | 576.40 > 223.30 | 60.0 | −22.0 | −35.0 | −25.0 | |
| 576.40 > 208.30 | 60.0 | −22.0 | −49.0 | −22.0 | 6.93 | |
| Ergocryptinine | 576.35 > 223.30 | 80.0 | −22.0 | −37.0 | −16.0 | |
| 576.35 > 208.20 | 80.0 | −22.0 | −52.0 | −23.0 | 10.99 | |
| Ergotaminine | 582.30 > 223.30 | 70.0 | −22.0 | −34.0 | −16.0 | |
| 582.30 > 277.25 | 70.0 | −22.0 | −26.0 | −20.0 | 8.57 | |
| Ergotamine | 582.30 > 223.30 | 60.0 | −22.0 | −33.0 | −16.0 | |
| 582.30 > 208.20 | 60.0 | −22.0 | −44.0 | −23.0 | 5.38 | |
| Ergocristine | 610.40 > 223.30 | 60.0 | −24.0 | −36.0 | −25.0 | |
| 610.40 > 208.25 | 60.0 | −24.0 | −47.0 | −22.0 | 7.29 | |
| Ergocristinine | 610.40 > 223.30 | 60.0 | −28.0 | −36.0 | −16.0 | |
| 610.40 > 325.30 | 60.0 | −24.0 | −28.0 | −22.0 | 11.85 | |
| Ergosine | 548.45 > 223.10 | 60.0 | −40.0 | −33.0 | −16.0 | |
| 548.45 > 208.25 | 60.0 | −40.0 | −40.0 | −14.0 | 5.05 | |
| Ergosinine | 548.35 > 223.30 | 80.0 | −20.0 | −32.6 | −16.0 | |
| 548.35 > 263.10 | 80.0 | −20.0 | −27.8 | −19.0 | 7.6 | |
| Ergometrine | 326.30 > 223.30 | 60.0 | −24.0 | −25.0 | −25.0 | |
| 326.30 > 208.20 | 60.0 | −24.0 | −30.0 | −22.0 | 3.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poapolathep, S.; Klangkaew, N.; Zhang, Z.; Giorgi, M.; Logrieco, A.F.; Poapolathep, A. Simultaneous Determination of Ergot Alkaloids in Swine and Dairy Feeds Using Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry. Toxins 2021, 13, 724. https://doi.org/10.3390/toxins13100724
Poapolathep S, Klangkaew N, Zhang Z, Giorgi M, Logrieco AF, Poapolathep A. Simultaneous Determination of Ergot Alkaloids in Swine and Dairy Feeds Using Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry. Toxins. 2021; 13(10):724. https://doi.org/10.3390/toxins13100724
Chicago/Turabian StylePoapolathep, Saranya, Narumol Klangkaew, Zhaowei Zhang, Mario Giorgi, Antonio Francesco Logrieco, and Amnart Poapolathep. 2021. "Simultaneous Determination of Ergot Alkaloids in Swine and Dairy Feeds Using Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry" Toxins 13, no. 10: 724. https://doi.org/10.3390/toxins13100724
APA StylePoapolathep, S., Klangkaew, N., Zhang, Z., Giorgi, M., Logrieco, A. F., & Poapolathep, A. (2021). Simultaneous Determination of Ergot Alkaloids in Swine and Dairy Feeds Using Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry. Toxins, 13(10), 724. https://doi.org/10.3390/toxins13100724