
Anti-Methanogenic Effect of Phytochemicals on Methyl-Coenzyme M Reductase—Potential: In Silico and Molecular Docking Studies for Environmental Protection


 ,
,  ,
,
Abstract
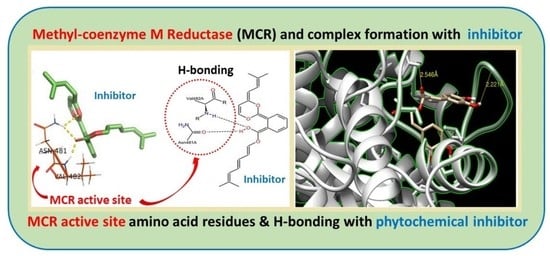
:
1. Introduction
2. Materials and Methods
2.1. Plant Selection
2.2. Primary Database Preparation
2.3. Virtual Screening of Ligands
2.4. Molecular Docking
2.4.1. Ligand Preparation and Target Protein
2.4.2. Protocol of Docking Studies
2.5. Molecular Dynamics Simulation Studies
3. Results and Discussion
3.1. Plant Selection and Database Preparation
3.2. Virtual Screening of Ligands
3.3. Molecular Docking of Compounds
3.4. Molecular Dynamics Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pachauri, R.K.; Allen, M.R.; Barros, V.R.; Broome, J.; Cramer, W.; Christ, R.; Church, J.A.; Clarke, L.; Dahe, Q.; Dasgupta, P.; et al. Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC (Intergovernmental Panel on Climate Change): Geneva, Switzerland, 2014. [Google Scholar]
- Hristov, A.N.; Oh, J.; Lee, C.; Meinen, R.; Montes, F.; Ott, T.; Firkins, J.; Rotz, A.; Dell, C.; Adesogan, A.; et al. Mitigation of Greenhouse Gas Emissions in Livestock Production: A Review of Technical Options for Non-CO2 Emissions, No. 177; Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2013. [Google Scholar]
- McAllister, T.A.; Meale, S.J.; Valle, E.; Guan, L.L.; Zhou, M.; Kelly, W.J.; Henderson, G.; Attwood, G.; Janssen, P. Ruminant nutrition symposium: Use of genomics and transcriptomics to identify strategies to lower ruminal methanogenesis. J. Anim. Sci. 2015, 93, 1431–1449. [Google Scholar] [CrossRef]
- Tapio, I.; Snelling, T.J.; Strozzi, F.; Wallace, R.J. The ruminal microbiome associated with methane emissions from ruminant livestock. J. Anim. Sci. Biotechnol. 2017, 8, 7. [Google Scholar] [CrossRef]
- Morgavi, D.P.; Forano, E.; Martin, C.; Newbold, C.J. Microbial ecosystem and methanogenesis in ruminants. Animal 2010, 4, 1024–1036. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.; Morgavi, D.P.; Doreau, M. Methane mitigation in ruminants: From microbe to the farm scale. Animal 2010, 4, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi, I.; Jami, E. The compositional variation of the rumen microbiome and its effect on host performance and methane emission. Animal 2018, 12, s220–s232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joblin, K.N. Methanogenic archaea. In Methods in Gut Microbial Ecology for Ruminants; Springer: Berlin/Heidelberg, Germany, 2005; pp. 47–53. [Google Scholar]
- Paynter, M.J.B.; Hungate, R.E. Characterization of Methanobacterium mobilis, sp. n., isolated from the bovine rumen. J. Bacteriol. 1968, 95, 1943–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, S.; Bowman, J.P.; Popovski, S.; Pimm, C.; Wright, A.-D.G. Methanobrevibacter millerae sp. nov. and Methanobrevibacter olleyae sp. nov., methanogens from the ovine and bovine rumen that can utilize formate for growth. Int. J. Syst. Evol. Microbiol. 2007, 57, 450–456. [Google Scholar] [CrossRef]
- Oppermann, R.A.; Nelson, W.O.; Brown, R.E. In vitro studies on methanogenic rumen bacteria. J. Dairy Sci. 1957, 40, 779–788. [Google Scholar] [CrossRef]
- Smith, P.H.; Hungate, R.E. Isolation and characterization of Methanobacterium ruminantium n. sp. J. Bacteriol. 1958, 75, 713. [Google Scholar] [CrossRef] [Green Version]
- Patra, A.K.; Kamra, D.N.; Agarwal, N. Effects of extracts of spices on rumen methanogenesis, enzyme activities and fermentation of feeds in vitro. J. Sci. Food Agric. 2010, 90, 511–520. [Google Scholar] [CrossRef]
- Patra, A.; Park, T.; Kim, M.; Yu, Z. Rumen methanogens and mitigation of methane emission by anti-methanogenic compounds and substances. J. Anim. Sci. Biotechnol. 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dr. Duke’s Phytochemical and Ethnobotanical Databases. Available online: https://phytochem.nal.usda.gov/phytochem/search (accessed on 17 October 2021).
- IMPPAT: Indian Medicinal Plants, Phytochemistry and Therapeutics. Available online: https://cb.imsc.res.in/imppat/home (accessed on 19 October 2021).
- Super Natural II-A Database of Natural Products. Available online: http://bioinf-applied.charite.de/supernatural_new/index.php?site=home (accessed on 17 October 2021).
- Ajayi, E.O.; Sadimenko, A.P.; Afolayan, A.J. GC—MS evaluation of Cymbopogon citratus (DC) Stapf oil obtained using modified hydrodistillation and microwave extraction methods. Food Chem. 2016, 209, 262–266. [Google Scholar] [CrossRef]
- Al-Tameme, H.J.; Hameed, I.H.; Idan, S.A.; Hadi, M.Y. Biochemical analysis of Origanum vulgare seeds by Fourier-Transform Infrared (FT-IR) spectroscopy and gas chromatography-mass spectrometry (GC-MS). J. Pharmacogn. Phyther. 2015, 7, 221–237. [Google Scholar]
- Zagorcheva, T.; Stanev, S.; Rusanov, K.; Atanassov, I. Comparative GC/MS analysis of lavender (Lavandula angustifolia Mill.) inflorescence and essential oil volatiles. Agric. Sci. Technol. 2013, 5, 2013. [Google Scholar]
- Uma, B.; Prabhakar, K.; Rajendran, S.; Sarayu, Y.L. Studies on GC/MS spectroscopic analysis of some bioactive antimicrobial compounds from Cinnamomum zeylanicum. J. Med. Plants 2009, 8, 125–131. [Google Scholar]
- Deshpande, S.N.; Kadam, D.G. GCMS analysis and antibacterial activity of Piper beetle (Linn) leaves against Streptococcus mutans. Asian J. Pharm. Clin. Res. 2013, 6, 99–101. [Google Scholar]
- Al-Rubaye, A.F.; Kadhim, M.J.; Hameed, I.H. Phytochemical profiles of methanolic seeds extract of Cuminum cyminum using GC-MS technique. Int. J. Curr. Pharm. Rev. Res. 2017, 8, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.K. GC—MS analysis of the essential oil of Ocimum gratissimum L. growing desolately in South India. Acta Chromatogr. 2017, 29, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Lin, P.; Zhu, X.; Su, Q. Comparative analysis of clary sage (S. sclarea L.) oil volatiles by GC—FTIR and GC—MS. Food Chem. 2006, 99, 401–407. [Google Scholar] [CrossRef]
- Chekki, R.Z.; Snoussi, A.; Hamrouni, I.; Bouzouita, N. Chemical composition, antibacterial and antioxidant activities of Tunisian garlic (Allium sativum) essential oil and ethanol extract. Mediterr. J. Chem. 2014, 3, 947–956. [Google Scholar] [CrossRef]
- Martínez, A.L.; González-Trujano, M.E.; Pellicer, F.; López-Muñoz, F.J.; Navarrete, A. Antinociceptive effect and GC/MS analysis of Rosmarinus officinalis L. essential oil from its aerial parts. Planta Med. 2009, 75, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Hudaib, M.; Speroni, E.; di Pietra, A.M.; Cavrini, V. GC/MS evaluation of thyme (Thymus vulgaris L.) oil composition and variations during the vegetative cycle. J. Pharm. Biomed. Anal. 2002, 29, 691–700. [Google Scholar] [CrossRef]
- Mishra, S.; Dahima, R. In vitro ADME studies of TUG-891, a GPR-120 inhibitor using SWISS ADME predictor. J. Drug Deliv. Ther. 2019, 9, 366–369. [Google Scholar]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [Green Version]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Shinoda, W.; Mikami, M. Rigid-body dynamics in the isothermal-isobaric ensemble: A test on the accuracy and computational efficiency. J. Comput. Chem. 2003, 24, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, C.R.; Paidesetty, S.K.; Dehury, B.; Padhy, R.N. Molecular dynamics and computational study of Mannich-based coumarin derivatives: Potent tyrosine kinase inhibitor. J. Biomol. Struct. Dyn. 2020, 38, 5419–5428. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Formula | MW | Lipinski Violations | Lead Likeness Violations |
|---|---|---|---|---|
| Cinnamic-acid | C9H8O2 | 148.16 | 0 | 1 |
| Ferulic-acid | C10H10O4 | 194.18 | 0 | 1 |
| Niacin | C6H5NO2 | 123.11 | 0 | 1 |
| p-Hydroxy-benzoic acid | C7H6O3 | 138.12 | 0 | 1 |
| Vanillic-acid | C8H8O4 | 168.15 | 0 | 1 |
| Biotin | C10H16N2O3S | 244.31 | 0 | 1 |
| Caffeic-acid | C9H8O4 | 180.16 | 0 | 1 |
| Gallic-acid | C7H6O5 | 170.12 | 0 | 1 |
| Rosmarinic-acid | C18H16O8 | 360.31 | 0 | 1 |
| Spathulenol | C15H24O | 220.35 | 0 | 1 |
| 1,3,4-Eugenol | C10H12O2 | 164.2 | 0 | 1 |
| 1,3,8-p-Menthatriene | C10H14 | 134.22 | 0 | 1 |
| 1,3-Cyclopentadiene | C5H6 | 66.1 | 0 | 1 |
| 3,7-dimethyl-endo-borneol | C10H18O | 154.25 | 0 | 1 |
| Methyl trans-geranylacetate | C13H20O3 | 224.3 | 0 | 1 |
| 2,4,7,9-tetramethyl-5-decyn-4,7diol | C14H26O2 | 226.36 | 0 | 1 |
| 2,6-Bis(3,4methylenedioxyphenyl)-3,7-dioxabicyclo (3.3.0)octane | C20H16O7 | 368.34 | 0 | 1 |
| 2-Methyl-5-(1-propenyl)pyrazine | C8H10N2 | 134.18 | 0 | 1 |
| α-cadinol | C15H26O | 222.37 | 0 | 1 |
| Diallyl tetrasulfide | C6H10S4 | 210.4 | 0 | 1 |
| epi-Cubebol | C15H24O | 220.35 | 0 | 1 |
| Eugenol | C10H12O2 | 164.2 | 0 | 1 |
| linalool | C10H18O | 154.25 | 0 | 1 |
| Pinacol | C6H14O2 | 118.17 | 0 | 1 |
| Pulegone | C10H16O | 152.23 | 0 | 1 |
| Chain | Residues Selected |
|---|---|
| Chain A | 37 LYS, 62 THR, 64 LEU, 65 GLY, 67 ARG, 69 LEU, 70 MET, 72 TYR, 82 GLU, 83 GLY, 84 ASP, 87 HIS, 90 ASN, 267 LEU, 268 PRO, 269 VAL, 270 ARG, 272 ALA, 319 TRP, 320 LEU, 324 MET, 328 VAL, 329 GLY, 330 PHE, 331 THR, 332 GLN, 333 TYR, 336 ALA, 394 ASP, 395 GLN, 396 PHE, 397 GLY, 399 SER, 401 ARG, 403 ALA, 443 PHE, 444 TYR, 473 PRO, 474 ASN, 479 ALA, 480 MET, 481 ASN, 482 VAL |
| Chain B | 49 ASN, 51 GLU, 52 GLY, 55 ASN, 111 ARG, 131 PRO, 169 GLU, 170 TYR, 173 ALA, 174 ASN, 175 ILE, 176 ALA, 177 THR, 178 MET, 179 LEU, 180 ASP, 181 ILE, 184 LYS, 186 GLU, 194 ASN, 196 MET, 199 HIS, 361 PHE, 362 PHE, 364 HIS, 365 SER, 366 ILE, 367 TYR, 368 GLY, 369 GLY, 374 ILE, 376 ASN, 378 ASN, 379 HIS, 380 ILE, 409 GLU, 410 ALA, 411 THR, 413 GLY, 414 LEU, 415 ILE, 417 GLU |
| Chain C | 83 ARG, 84 TYR, 86 GLN, 117 LEU, 118 SER, 119 GLY, 120 ARG, 122 ILE, 124 GLU, 152 GLY, 153 LYS, 154 SER, 155 VAL, 156 HIS, 158 HIS, 171MET, 191 ILE |
| Sample Number | Ligand | Binding Energy (kcal/mol) | Ki | |
|---|---|---|---|---|
| AD | ADV | |||
| 1 | Cinnamic-acid | −4.44 | −6.6 | 557.1 µM |
| 2 | Ferulic-acid | −6.70 | −6.3 | 12.27 µM |
| 3 | Niacin | −6.19 | −5.2 | 28.85 µM |
| 4 | p-Hydroxy-benzoic acid | −6.83 | −5.9 | 9.82 µM |
| 5 | Vanillic-acid | −6.44 | −6.0 | 18.91 µM |
| 6 | Biotin | −9.38 | −6.0 | 0.132 µM |
| 7 | Caffeic-acid | −7.34 | −6.3 | 4.17 µM |
| 8 | Gallic-acid | −7.83 | −5.8 | 1.81 µM |
| 9 | Rosmarinic-acid | −10.71 | −8.5 | 0.014 µM |
| 10 | 1,3,8-p-Menthatriene | −6.90 | −5.8 | 8.73 µM |
| 11 | 1,3-Cyclopentadiene | −4.12 | −4.4 | 954.6 µM |
| 12 | 3,7-Dimethyl-endo-borneol | −6.81 | −5.8 | 10.22 µM |
| 13 | Methyl trans-geranylacetate | −7.58 | −7.4 | 2.78 µM |
| 14 | 2,4,7,9-Tetramethyl-5decyn-4,7-diol | −9.02 | −5.9 | 0.2465 µM |
| 15 | (3R,3aS,6R,6aR)-3-(2H-1,3-benzodioxol-4-yl)-6-(2H-1,3-benzodioxol-5-yl)-hexahydrofuro[3,4-c]furan-1-one | −12.21 | −8.6 | 0.0012 µM |
| 16 | 2-Methyl-5-(1-propenyl)pyrazine | −5.19 | −5.4 | 158.1 µM |
| 17 | α-cadinol | −8.16 | −6.6 | 1.04 µM |
| 18 | Diallyl tetrasulfide | −4.84 | −3.7 | 283.6 µM |
| 19 | epi-Cubebol | −7.83 | −6.3 | 1.82 µM |
| 20 | Eugenol | −7.08 | −5.6 | 6.47 µM |
| 21 | Linalool | −6.80 | −6.5 | 10.3 µM |
| 22 | Pulegone | −7.45 | −6.1 | 3.49 µM |
| 23 | Pinacol | −6.33 | −4.6 | 22.9 µM |
| 24 | Spathulenol | −4.45 | −7.0 | 31.4 µM |
| 25 | 2,6-Bis(3,4-methylenedi-oxyphenyl)-3,7-dioxabi-cyclo(3.3.0)octane | −5.18 | −8.4 | 158.9 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinakarkumar, Y.; Rajabathar, J.R.; Arokiyaraj, S.; Jeyaraj, I.; Anjaneyulu, S.R.; Sandeep, S.; Karthik, C.S.; Appaturi, J.N.; Wilson, L.D. Anti-Methanogenic Effect of Phytochemicals on Methyl-Coenzyme M Reductase—Potential: In Silico and Molecular Docking Studies for Environmental Protection. Micromachines 2021, 12, 1425. https://doi.org/10.3390/mi12111425
Dinakarkumar Y, Rajabathar JR, Arokiyaraj S, Jeyaraj I, Anjaneyulu SR, Sandeep S, Karthik CS, Appaturi JN, Wilson LD. Anti-Methanogenic Effect of Phytochemicals on Methyl-Coenzyme M Reductase—Potential: In Silico and Molecular Docking Studies for Environmental Protection. Micromachines. 2021; 12(11):1425. https://doi.org/10.3390/mi12111425
Chicago/Turabian StyleDinakarkumar, Yuvaraj, Jothi Ramalingam Rajabathar, Selvaraj Arokiyaraj, Iyyappan Jeyaraj, Sai Ramesh Anjaneyulu, Shadakshari Sandeep, Chimatahalli Shanthakumar Karthik, Jimmy Nelson Appaturi, and Lee D. Wilson. 2021. "Anti-Methanogenic Effect of Phytochemicals on Methyl-Coenzyme M Reductase—Potential: In Silico and Molecular Docking Studies for Environmental Protection" Micromachines 12, no. 11: 1425. https://doi.org/10.3390/mi12111425