The Role of Chromosomal Instability in Cancer and Therapeutic Responses


Abstract
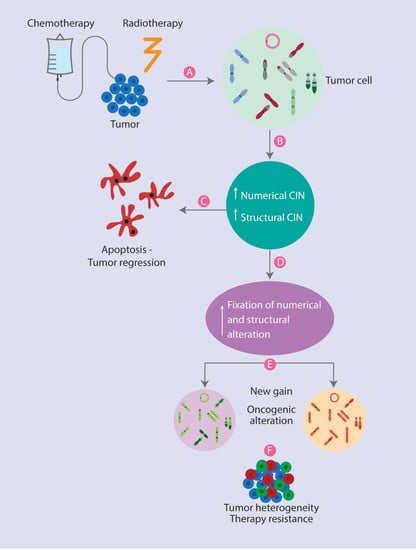
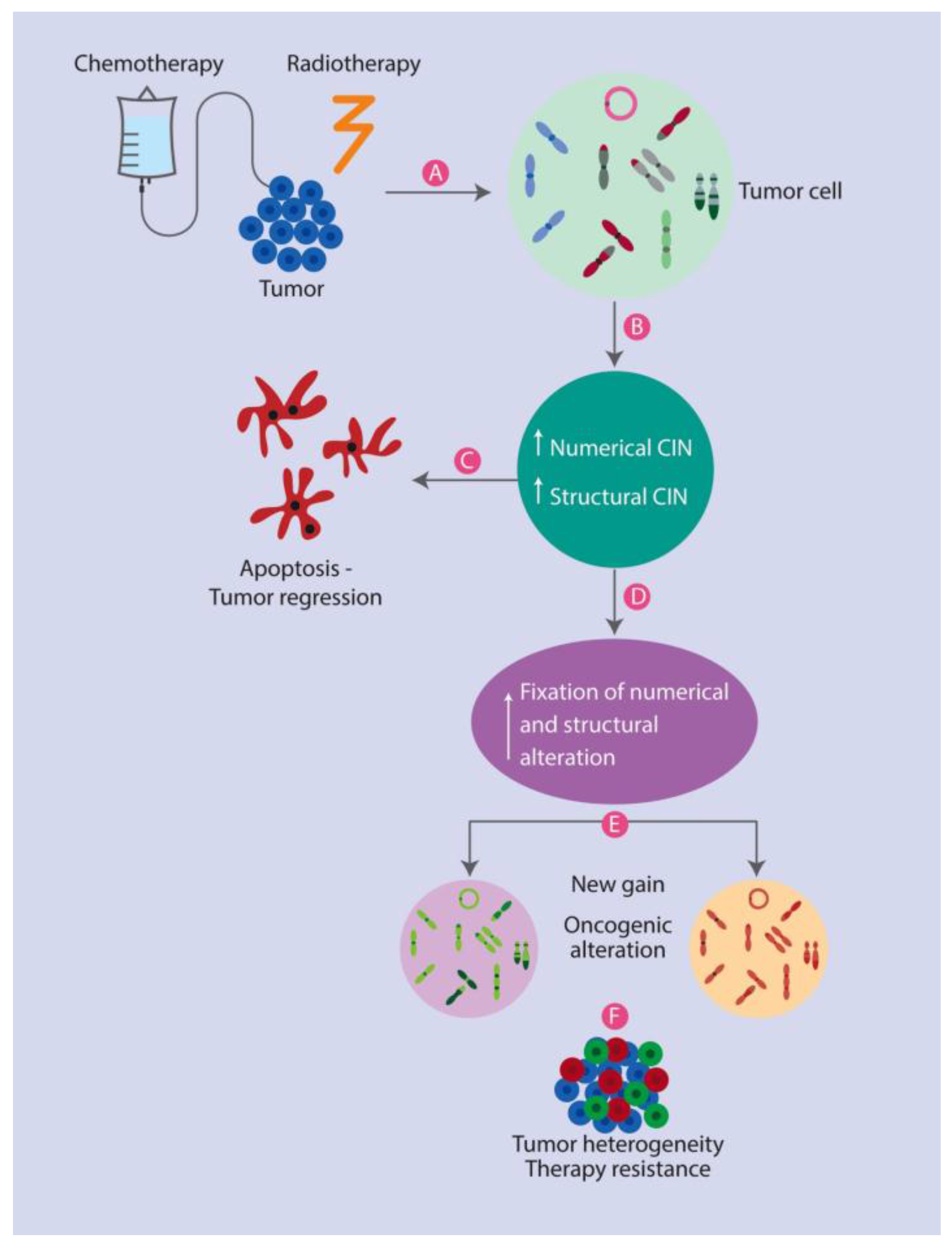
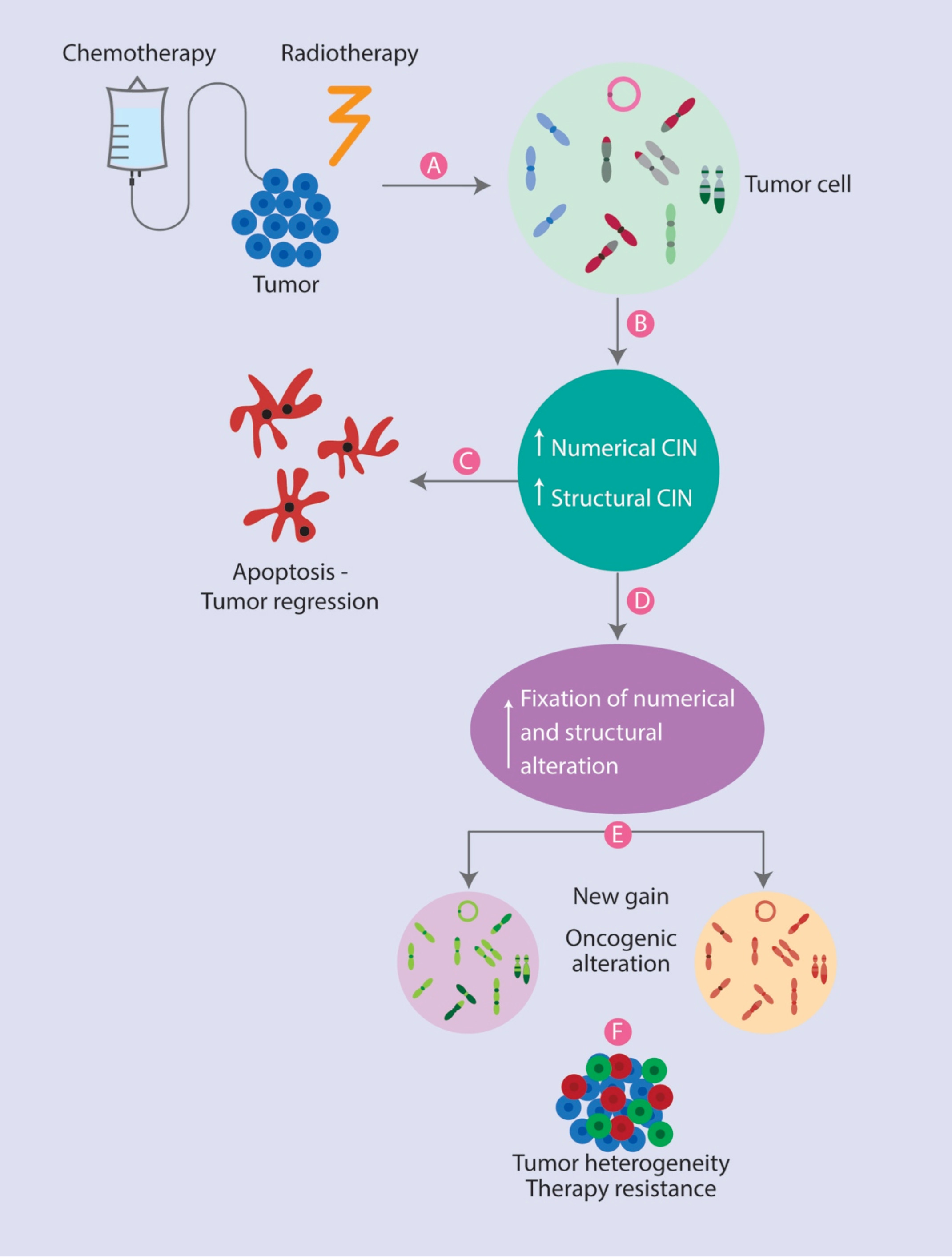
:
1. Introduction
2. CIN and Cancer
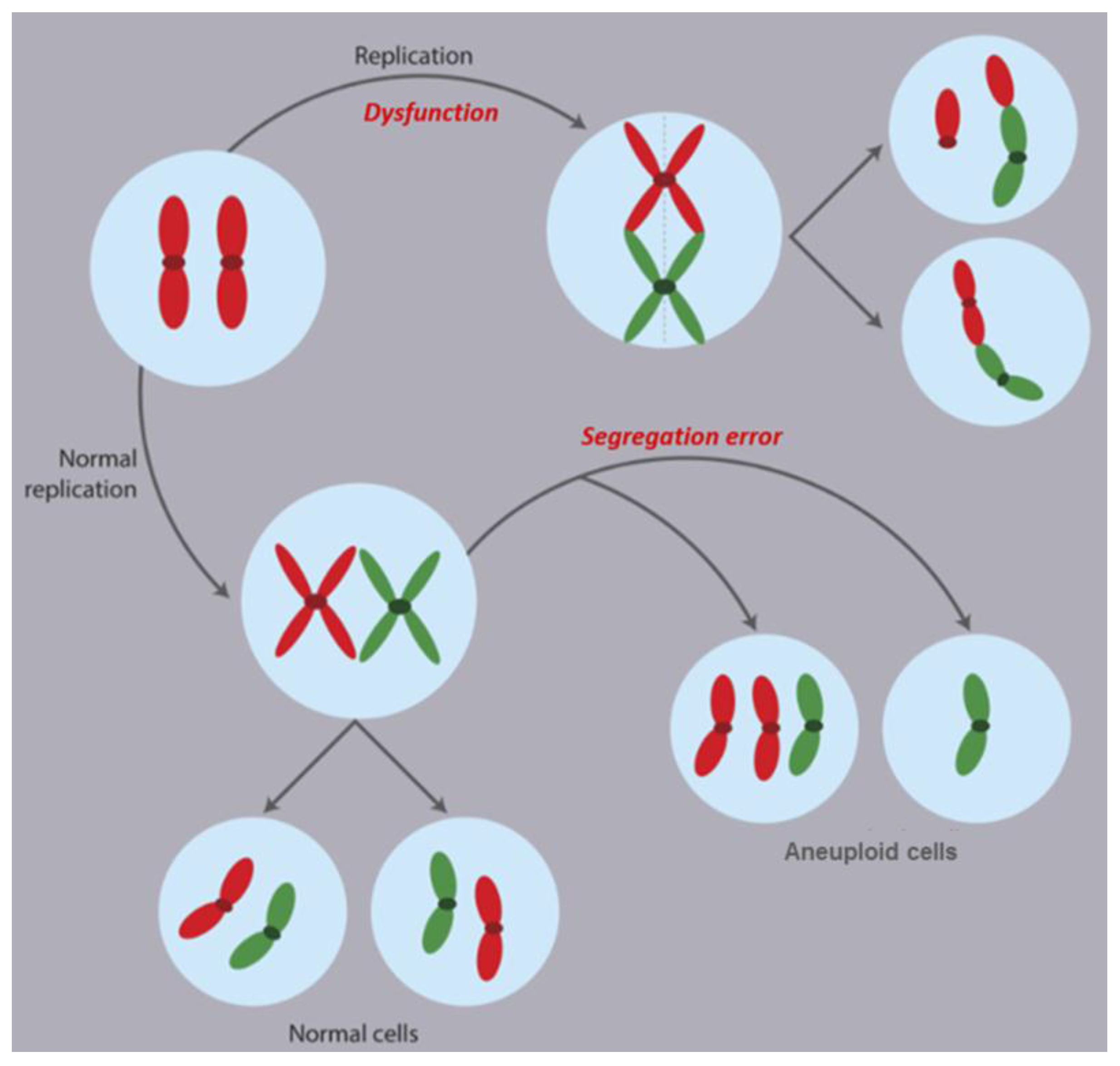
2.1. Mechanisms of CIN
2.2. The Role of CIN in Cancer Development and Progression
2.2.1. Breast Cancer (BC)
2.2.2. Prostate Cancer
2.2.3. Colorectal Cancer
2.2.4. Cervical Cancer
2.2.5. Endometrial Cancer
2.2.6. Bladder Cancer
2.2.7. Multiple Myeloma
2.2.8. High Hyperdiploid Acute Lymphoblastic Leukemia (HeH ALL)
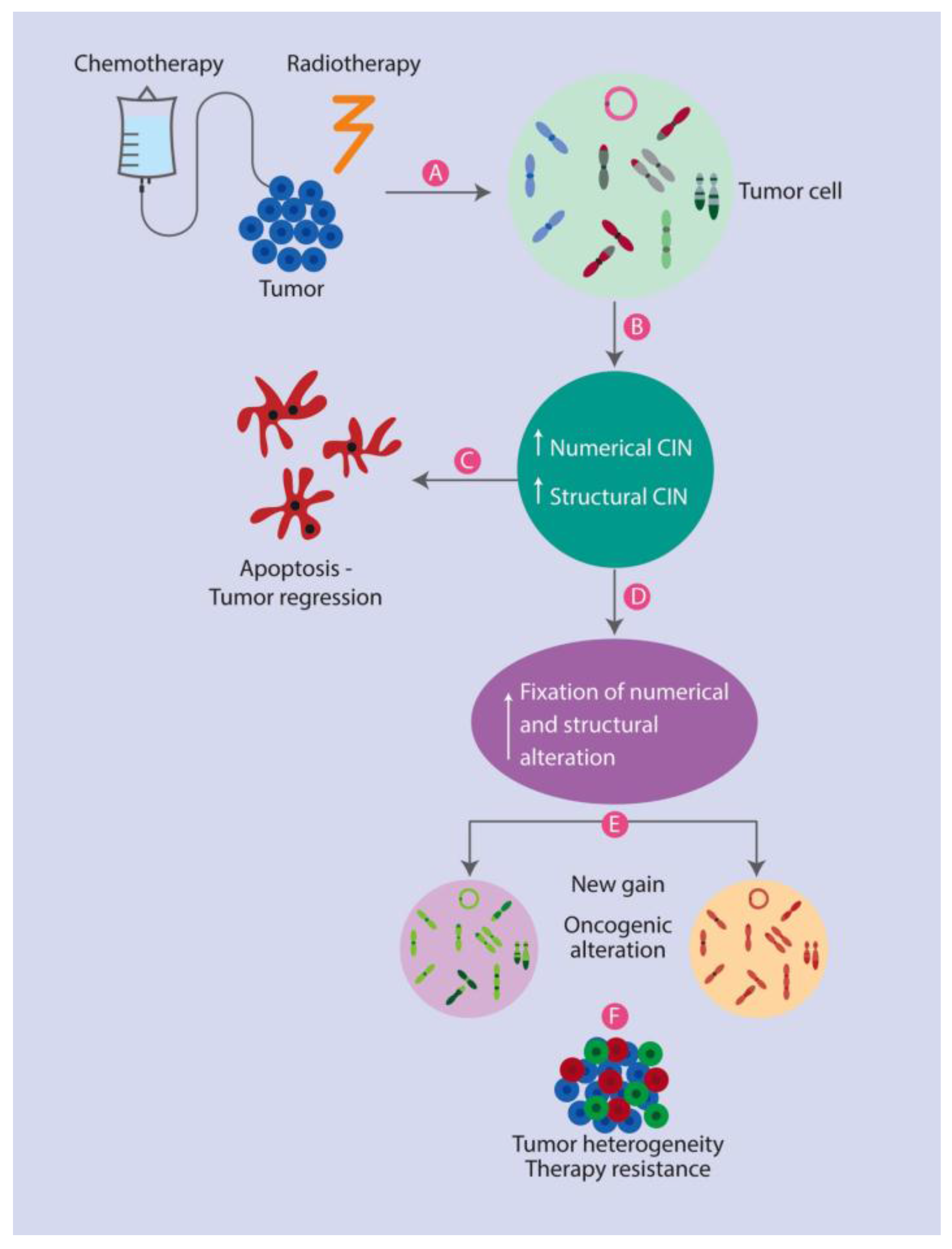
3. The Role of CIN in Anticancer Therapy Responses
3.1. Therapeutic Strategies Based on CIN
3.2. The Association between CIN and Poor Prognoses
3.3. CIN and Its Potential Beneficial Effects for Therapy
4. CIN in Naturally Occurring Congenital Aneuploidy of Non-Cancerous Origin
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nicholson, J.M.; Cimini, D. Cancer karyotypes: Survival of the fittest. Front. Oncol. 2013, 3, 148. [Google Scholar] [CrossRef] [PubMed]
- Soerjomataram, I.; Louwman, M.W.; Ribot, J.G.; Roukema, J.A.; Coebergh, J.W. An overview of prognostic factors for long-term survivors of breast cancer. Breast Cancer Res. Treat. 2008, 107, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Hirota, T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim. Biophys. Acta 2016, 1866, 64–75. [Google Scholar] [CrossRef] [PubMed]
- McClelland, S.E. Role of chromosomal instability in cancer progression. Endocr. Relat. Cancer 2017, 24, T23–T31. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Compton, D.A. Chromosomal instability and cancer: A complex relationship with therapeutic potential. J. Clin. Investig. 2012, 122, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Bayani, J.; Selvarajah, S.; Maire, G.; Vukovic, B.; Al-Romaih, K.; Zielenska, M.; Squire, J.A. Genomic mechanisms and measurement of structural and numerical instability in cancer cells. Semin. Cancer Biol. 2007, 17, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Gagos, S.; Irminger-Finger, I. Chromosome instability in neoplasia: Chaotic roots to continuous growth. Int. J. Biochem. Cell Biol. 2005, 37, 1014–1033. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Stevens, J.B.; Horne, S.D.; Abdallah, B.Y.; Ye, K.J.; Bremer, S.W.; Ye, C.J.; Chen, D.J.; Heng, H.H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Rangel, N.; Forero-Castro, M.; Rondon-Lagos, M. New Insights in the Cytogenetic Practice: Karyotypic Chaos, Non-Clonal Chromosomal Alterations and Chromosomal Instability in Human Cancer and Therapy Response. Genes (Basel) 2017, 8, E155. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Regan, S.M.; Liu, G.; Ye, C.J. Why it is crucial to analyze non clonal chromosome aberrations or NCCAs? Mol. Cytogenet. 2016, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Bremer, S.W.; Stevens, J.; Ye, K.J.; Miller, F.; Liu, G.; Ye, C.J. Cancer progression by non-clonal chromosome aberrations. J. Cell. Biochem. 2006, 98, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F. Recurrent chromosome aberrations in cancer. Mutat. Res. 2000, 462, 247–253. [Google Scholar] [CrossRef]
- Heng, H.H.; Liu, G.; Stevens, J.B.; Abdallah, B.Y.; Horne, S.D.; Ye, K.J.; Bremer, S.W.; Chowdhury, S.K.; Ye, C.J. Karyotype heterogeneity and unclassified chromosomal abnormalities. Cytogenet. Genome Res. 2013, 139, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Dereli-Oz, A.; Versini, G.; Halazonetis, T.D. Studies of genomic copy number changes in human cancers reveal signatures of DNA replication stress. Mol. Oncol. 2011, 5, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Tanzarella, C.; Degrassi, F. Differences in malsegregation rates obtained by scoring ana-telophases or binucleate cells. Mutagenesis 1999, 14, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 2008, 180, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The spindle assembly checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed]
- Dobles, M.; Liberal, V.; Scott, M.L.; Benezra, R.; Sorger, P.K. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell 2000, 101, 635–645. [Google Scholar] [CrossRef]
- Rieder, C.L.; Schultz, A.; Cole, R.; Sluder, G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J. Cell Biol. 1994, 127, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.L.; Cole, R.W.; Khodjakov, A.; Sluder, G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 1995, 130, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Khodjakov, A.; Cole, R.W.; McEwen, B.F.; Buttle, K.F.; Rieder, C.L. Chromosome fragments possessing only one kinetochore can congress to the spindle equator. J. Cell Biol. 1997, 136, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Fioravanti, D.; Salmon, E.D.; Degrassi, F. Merotelic kinetochore orientation versus chromosome mono-orientation in the origin of lagging chromosomes in human primary cells. J. Cell Sci. 2002, 115, 507–515. [Google Scholar] [PubMed]
- Gregan, J.; Polakova, S.; Zhang, L.; Tolic-Norrelykke, I.M.; Cimini, D. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 2011, 21, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.C.; van Wely, K.H. Are aneuploidy and chromosome breakage caused by a CINgle mechanism? Cell Cycle 2010, 9, 2275–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Iwasa, Y.; Vogelstein, B.; Lengauer, C.; Nowak, M.A. Can chromosomal instability initiate tumorigenesis? Semin. Cancer Biol. 2005, 15, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fu, L.; Zhang, L.Y.; Kwong, D.L.; Yan, L.; Guan, X.Y. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chin. J. Cancer 2012, 31, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.A.; Zhu, J.; Li, R. Aneuploidy and chromosomal instability: A vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013, 32, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Ye, K.J.; Ye, C.J. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Chandrakasan, S.; Ye, C.J.; Chitlur, M.; Mohamed, A.N.; Rabah, R.; Konski, A.; Heng, H.H.; Savasan, S. Malignant fibrous histiocytoma two years after autologous stem cell transplant for Hodgkin lymphoma: Evidence for genomic instability. Pediatr. Blood Cancer 2011, 56, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Chromosomes and cancer cells. Chromosome Res. Int. J. Mol. Supramol. Evolut. Asp. Chromosome Biol. 2011, 19, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Houlston, R.; Tomlinson, I. Association between chromosomal instability and prognosis in colorectal cancer: A meta-analysis. Gut 2008, 57, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Florl, A.R.; Schulz, W.A. Chromosomal instability in bladder cancer. Arch. Toxicol. 2008, 82, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr. The current clinical value of genomic instability. Semin. Cancer Biol. 2005, 15, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Orsetti, B.; Selves, J.; Bascoul-Mollevi, C.; Lasorsa, L.; Gordien, K.; Bibeau, F.; Massemin, B.; Paraf, F.; Soubeyran, I.; Hostein, I.; et al. Impact of chromosomal instability on colorectal cancer progression and outcome. BMC Cancer 2014, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Kwei, K.A.; Kung, Y.; Salari, K.; Holcomb, I.N.; Pollack, J.R. Genomic instability in breast cancer: Pathogenesis and clinical implications. Mol. Oncol. 2010, 4, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Tsarouha, H.; Pandis, N.; Bardi, G.; Teixeira, M.R.; Andersen, J.A.; Heim, S. Karyotypic evolution in breast carcinomas with i(1)(q10) and der(1;16)(q10;p10) as the primary chromosome abnormality. Cancer Genet. Cytogenet. 1999, 113, 156–161. [Google Scholar] [CrossRef]
- Cummings, M.C.; Aubele, M.; Mattis, A.; Purdie, D.; Hutzler, P.; Hofler, H.; Werner, M. Increasing chromosome 1 copy number parallels histological progression in breast carcinogenesis. Br. J. Cancer 2000, 82, 1204–1210. [Google Scholar] [PubMed]
- Rye, I.H.; Lundin, P.; Maner, S.; Fjelldal, R.; Naume, B.; Wigler, M.; Hicks, J.; Borresen-Dale, A.L.; Zetterberg, A.; Russnes, H.G. Quantitative multigene FISH on breast carcinomas identifies der(1;16)(q10;p10) as an early event in luminal A tumors. Genes Chromosomes Cancer 2015, 54, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; DeVries, S.; Fridlyand, J.; Spellman, P.T.; Roydasgupta, R.; Kuo, W.L.; Lapuk, A.; Neve, R.M.; Qian, Z.; Ryder, T.; et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006, 10, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.T.; Gale, T.; Reis-Filho, J.S.; Jones, C.; Parry, S.; Sloane, J.P.; Hanby, A.; Pinder, S.E.; Lee, A.H.; Humphreys, S.; et al. Columnar cell lesions of the breast: The missing link in breast cancer progression? A morphological and molecular analysis. Am. J. Surg. Pathol. 2005, 29, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Courjal, F.; Cuny, M.; Simony-Lafontaine, J.; Louason, G.; Speiser, P.; Zeillinger, R.; Rodriguez, C.; Theillet, C. Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: Definition of phenotypic groups. Cancer Res. 1997, 57, 4360–4367. [Google Scholar] [PubMed]
- Smid, M.; Hoes, M.; Sieuwerts, A.M.; Sleijfer, S.; Zhang, Y.; Wang, Y.; Foekens, J.A.; Martens, J.W. Patterns and incidence of chromosomal instability and their prognostic relevance in breast cancer subtypes. Breast Cancer Res. Treat. 2011, 128, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.; Stanford, J.L.; McIndoe, R.A.; Jarvik, G.P.; Kolb, S.; Goode, E.L.; Chakrabarti, L.; Schuster, E.F.; Buckley, V.A.; Miller, E.L.; et al. Evidence for a rare prostate cancer-susceptibility locus at chromosome 1p36. Am. J. Hum. Genet. 1999, 64, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.R.; Jeronimo, C.; Henrique, R.; Fonseca, D.; Oliveira, J.; Lothe, R.A.; Teixeira, M.R. 8q gain is an independent predictor of poor survival in diagnostic needle biopsies from prostate cancer suspects. Clin. Cancer Res. 2006, 12, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Wolter, H.; Trijic, D.; Gottfried, H.W.; Mattfeldt, T. Chromosomal changes in incidental prostatic carcinomas detected by comparative genomic hybridization. Eur. Urol. 2002, 41, 328–334. [Google Scholar] [CrossRef]
- Al-Maghrabi, J.; Vorobyova, L.; Chapman, W.; Jewett, M.; Zielenska, M.; Squire, J.A. p53 Alteration and chromosomal instability in prostatic high-grade intraepithelial neoplasia and concurrent carcinoma: Analysis by immunohistochemistry, interphase in situ hybridization, and sequencing of laser-captured microdissected specimens. Mod. Pathol. 2001, 14, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- El-Zein, R.; Gu, Y.; Sierra, M.S.; Spitz, M.R.; Strom, S.S. Chromosomal instability in peripheral blood lymphocytes and risk of prostate cancer. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cospons. Am. Soc. Prev. Oncol. 2005, 14, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Bonassi, S.; Hagmar, L.; Stromberg, U.; Montagud, A.H.; Tinnerberg, H.; Forni, A.; Heikkila, P.; Wanders, S.; Wilhardt, P.; Hansteen, I.L.; et al. Chromosomal aberrations in lymphocytes predict human cancer independently of exposure to carcinogens. European Study Group on Cytogenetic Biomarkers and Health. Cancer Res. 2000, 60, 1619–1625. [Google Scholar] [PubMed]
- Holcomb, I.N.; Grove, D.I.; Kinnunen, M.; Friedman, C.L.; Gallaher, I.S.; Morgan, T.M.; Sather, C.L.; Delrow, J.J.; Nelson, P.S.; Lange, P.H.; et al. Genomic alterations indicate tumor origin and varied metastatic potential of disseminated cells from prostate cancer patients. Cancer Res. 2008, 68, 5599–5608. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Kallioniemi, A.H.; Syvanen, A.C.; Hyytinen, E.R.; Karhu, R.; Tammela, T.; Isola, J.J.; Kallioniemi, O.P. Genetic changes in primary and recurrent prostate cancer by comparative genomic hybridization. Cancer Res. 1995, 55, 342–347. [Google Scholar] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Keino-Masu, K.; Masu, M.; Hinck, L.; Leonardo, E.D.; Chan, S.S.; Culotti, J.G.; Tessier-Lavigne, M. Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell 1996, 87, 175–185. [Google Scholar] [CrossRef]
- Fazeli, A.; Dickinson, S.L.; Hermiston, M.L.; Tighe, R.V.; Steen, R.G.; Small, C.G.; Stoeckli, E.T.; Keino-Masu, K.; Masu, M.; Rayburn, H.; et al. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature 1997, 386, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Kohmura, H.; Futamura, M.; Kida, H.; Tanemura, H.; Shimokawa, K.; Saji, S. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology 1996, 111, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Koumura, H.; Futamura, M.; Aoki, S.; Ymaguchi, K.; Kida, H.; Tanemura, H.; Shimokawa, K.; Saji, S. Somatic alterations of the SMAD-2 gene in human colorectal cancers. Br. J. Cancer 1998, 78, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.M.; Zhou, W.; Goodman, S.N.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001, 61, 818–822. [Google Scholar] [PubMed]
- Cardoso, J.; Molenaar, L.; de Menezes, R.X.; van Leerdam, M.; Rosenberg, C.; Moslein, G.; Sampson, J.; Morreau, H.; Boer, J.M.; Fodde, R. Chromosomal instability in MYH- and APC-mutant adenomatous polyps. Cancer Res. 2006, 66, 2514–2519. [Google Scholar] [CrossRef] [PubMed]
- Gutenberg, A.; Gerdes, J.S.; Jung, K.; Sander, B.; Gunawan, B.; Bock, H.C.; Liersch, T.; Bruck, W.; Rohde, V.; Fuzesi, L. High chromosomal instability in brain metastases of colorectal carcinoma. Cancer Genet. Cytogenet. 2010, 198, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Marani, M.; Pardo, O.; Warne, P.H.; Kelly, G.; Sahai, E.; Elustondo, F.; Chang, J.; Temple, J.; Ahmed, A.A.; et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 2007, 11, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzi, S.; Lindsay, C.R.; Morozov, V.M.; Escobar-Cabrera, E.; Summers, M.K.; Han, H.S.; McIntosh, L.P.; Ishov, A.M. Regulation of mitosis and taxane response by Daxx and Rassf1. Oncogene 2012, 31, 13–26. [Google Scholar] [CrossRef] [PubMed]
- How, C.; Bruce, J.; So, J.; Pintilie, M.; Haibe-Kains, B.; Hui, A.; Clarke, B.A.; Hedley, D.W.; Hill, R.P.; Milosevic, M.; et al. Chromosomal instability as a prognostic marker in cervical cancer. BMC Cancer 2015, 15, 361. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Mehrotra, S.; Kalra, N.; Singh, U.; Shukla, Y. Correlation of DNA ploidy with progression of cervical cancer. J. Cancer Epidemiol. 2008, 2008, 298495. [Google Scholar] [CrossRef] [PubMed]
- Olaharski, A.J.; Sotelo, R.; Solorza-Luna, G.; Gonsebatt, M.E.; Guzman, P.; Mohar, A.; Eastmond, D.A. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 2006, 27, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Gutierrez, E.I.; Davila-Rodriguez, M.I.; Muraira-Rodriguez, M.; Said-Fernandez, S.; Cerda-Flores, R.M. Association between the stages of cervical cancer and chromosome 1 aneusomy. Cancer Genet. Cytogenet. 2005, 159, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, B.; Zhang, R.R.; Li, S.; Chen, X.; Mulvihill, J.J.; Lu, X.; Pang, H.; Liu, H. Automated analysis of fluorescent in situ hybridization (FISH) labeled genetic biomarkers in assisting cervical cancer diagnosis. Technol. Cancer Res. Treat. 2010, 9, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Murty, V.V.; Mitra, A.B.; Luthra, U.K. Spontaneous chromosomal aberrations in patients with precancerous and cancerous lesions of the cervix uteri. Cancer Genet. Cytogenet. 1985, 17, 347–353. [Google Scholar] [CrossRef]
- Susini, T.; Olivieri, S.; Molino, C.; Amunni, G.; Rapi, S.; Taddei, G.; Scarselli, G. DNA ploidy is stronger than lymph node metastasis as prognostic factor in cervical carcinoma: 10-Year results of a prospective study. Int. J. Gynecol. Cancer 2011, 21, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Luzhna, L.; Kathiria, P.; Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 2013, 4, 131. [Google Scholar] [CrossRef] [PubMed]
- Nersesyan, A.K. Possible role of the micronucleus assay in diagnostics and secondary prevention of cervix cancer: A minireview. Tsitol. Genet. 2007, 41, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Guzman, P.; Sotelo-Regil, R.C.; Mohar, A.; Gonsebatt, M.E. Positive correlation between the frequency of micronucleated cells and dysplasia in Papanicolaou smears. Environ. Mol. Mutagen. 2003, 41, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Gutierrez, E.I.; D’Avila-Rodriguez, M.I.; Cerda-Flores, R.M. Chromosomal damage as prognosis marker in cervical carcinogenesis. Tsitol. Genet. 2014, 48, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Weiderpass, E.; Labreche, F. Malignant tumors of the female reproductive system. Saf. Health Work 2012, 3, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, G.; du Manoir, S.; Godwin, A.K.; Bell, D.W.; Liu, Z.; Hogan, M.; Yakushiji, M.; Testa, J.R. Detection of DNA gains and losses in primary endometrial carcinomas by comparative genomic hybridization. Genes Chromosomes Cancer 1997, 18, 115–125. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997, 386. [Google Scholar] [CrossRef] [PubMed]
- Peiffer-Schneider, S.; Noonan, F.C.; Mutch, D.G.; Simpkins, S.B.; Herzog, T.; Rader, J.; Elbendary, A.; Gersell, D.J.; Call, K.; Goodfellow, P.J. Mapping an endometrial cancer tumor suppressor gene at 10q25 and development of a bacterial clone contig for the consensus deletion interval. Genomics 1998, 52, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Sirchia, S.M.; Pariani, S.; Rossella, F.; Garagiola, I.; De Andreis, C.; Bulfamante, G.; Zannoni, E.; Radaelli, U.; Simoni, G. Cytogenetic abnormalities and microsatellite instability in endometrial adenocarcinoma. Cancer Genet. Cytogenet. 1997, 94, 113–119. [Google Scholar] [CrossRef]
- Muresu, R.; Sini, M.C.; Cossu, A.; Tore, S.; Baldinu, P.; Manca, A.; Pisano, M.; Loddo, C.; Dessole, S.; Pintus, A.; et al. Chromosomal abnormalities and microsatellite instability in sporadic endometrial cancer. Eur. J. Cancer 2002, 38, 1802–1809. [Google Scholar] [CrossRef]
- Peterson, L.M.; Kipp, B.R.; Halling, K.C.; Kerr, S.E.; Smith, D.I.; Distad, T.J.; Clayton, A.C.; Medeiros, F. Molecular characterization of endometrial cancer: A correlative study assessing microsatellite instability, MLH1 hypermethylation, DNA mismatch repair protein expression, and PTEN, PIK3CA, KRAS, and BRAF mutation analysis. Int. J. Gynecol. Pathol. 2012, 31, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, N.P.; Chebotarev, A.N.; Katosova, L.D.; Platonova, V.I. The database for analysis of quantitative characteristics of chromosome aberration frequencies in the culture of human peripheral blood lymphocytes. Genetika 2001, 37, 549–557. [Google Scholar] [PubMed]
- Nesina, I.P.; Iurchenko, N.P.; Nespryadko, S.V.; Buchinska, L.G. The study of chromosomal instability in patients with endometrial cancer. Exp. Oncol. 2014, 36, 202–206. [Google Scholar] [PubMed]
- Pilinska, M.A.; Dybsky, S.S.; Dybska, O.B.; Shvayko, L.I.; Sushko, V.O. Peculiarities of induction and persistence of hidden chromosome instability in peripheral blood lymphocytes of persons occupationally exposed to ionizing radiation. Problemy Radiatsiinoi Medytsyny ta Radiobiolohii 2014, 19, 321–333. [Google Scholar] [PubMed]
- Abraham, R.; Pagano, F.; Gomella, L.G.; Baffa, R. Chromosomal deletions in bladder cancer: Shutting down pathways. Front. Biosci. 2007, 12, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Cairns, P.; Shaw, M.E.; Knowles, M.A. Preliminary mapping of the deleted region of chromosome 9 in bladder cancer. Cancer Res. 1993, 53, 1230–1232. [Google Scholar] [PubMed]
- Sauter, G.; Moch, H.; Wagner, U.; Bubendorf, L.; Gasser, T.C.; Mihatsch, M.J. Genomic changes in urinary bladder cancer. Verh. Dtsch. Ges. Pathol. 1997, 81, 287–296. [Google Scholar] [PubMed]
- Sauter, G.; Simon, R.; Bubendorf, L.; Mihatsch, M. Molecular genetics of urinary bladder cancer progression. Verh. Dtsch. Ges. Pathol. 2002, 86, 49–56. [Google Scholar] [PubMed]
- Kimura, F.; Florl, A.R.; Seifert, H.H.; Louhelainen, J.; Maas, S.; Knowles, M.A.; Schulz, W.A. Destabilization of chromosome 9 in transitional cell carcinoma of the urinary bladder. Br. J. Cancer 2001, 85, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Bernues, M.; Casadevall, C.; Caballin, M.R.; Miro, R.; Ejarque, M.J.; Chechile, G.; Gelabert, A.; Egozcue, J. Study of allelic losses on 3p, 6q, and 17p in human urothelial cancer. Cancer Genet. Cytogenet. 1999, 112, 42–45. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.F.; Reuter, V.E.; Cordon-Cardo, C. Chromosome 3 allelic losses and microsatellite alterations in transitional cell carcinoma of the urinary bladder. Am. J. Pathol. 1996, 149, 229–235. [Google Scholar] [PubMed]
- Wagner, U.; Bubendorf, L.; Gasser, T.C.; Moch, H.; Gorog, J.P.; Richter, J.; Mihatsch, M.J.; Waldman, F.M.; Sauter, G. Chromosome 8p deletions are associated with invasive tumor growth in urinary bladder cancer. Am. J. Pathol. 1997, 151, 753–759. [Google Scholar] [PubMed]
- Choi, C.; Kim, M.H.; Juhng, S.W.; Oh, B.R. Loss of heterozygosity at chromosome segments 8p22 and 8p11.2–21.1 in transitional-cell carcinoma of the urinary bladder. Int. J. Cancer 2000, 86, 501–505. [Google Scholar] [CrossRef]
- Seripa, D.; Parrella, P.; Gallucci, M.; Gravina, C.; Papa, S.; Fortunato, P.; Alcini, A.; Flammia, G.; Lazzari, M.; Fazio, V.M. Sensitive detection of transitional cell carcinoma of the bladder by microsatellite analysis of cells exfoliated in urine. Int. J. Cancer 2001, 95, 364–369. [Google Scholar] [PubMed]
- Panani, A.D.; Babanaraki, A.; Malianga, E.; Roussos, C. Numerical aberrations of chromosomes 9 and 11 detected by FISH in Greek bladder cancer patients. Anticancer Res. 2004, 24, 3857–3861. [Google Scholar] [PubMed]
- Lokeshwar, V.B.; Habuchi, T.; Grossman, H.B.; Murphy, W.M.; Hautmann, S.H.; Hemstreet, G.P., 3rd; Bono, A.V.; Getzenberg, R.H.; Goebell, P.; Schmitz-Drager, B.J.; et al. Bladder tumor markers beyond cytology: International Consensus Panel on bladder tumor markers. Urology 2005, 66, 35–63. [Google Scholar] [CrossRef] [PubMed]
- Bonberg, N.; Taeger, D.; Gawrych, K.; Johnen, G.; Banek, S.; Schwentner, C.; Sievert, K.D.; Wellhausser, H.; Kluckert, M.; Leng, G.; et al. Chromosomal instability and bladder cancer: The UroVysion(TM) test in the UroScreen study. BJU Int. 2013, 112, E372–E382. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Reiman, T.; Belch, A.R.; Pilarski, L.M. Ten years and counting: So what do we know about t(4;14)(p16;q32) multiple myeloma. Leuk Lymphoma 2006, 47, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.D.; Ross, F.M.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gonzalez, D.; Walker, B.A.; Hockley, S.L.; Wardell, C.P.; et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes Chromosomes Cancer 2011, 50, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [PubMed]
- Harousseau, J.L.; Avet-Loiseau, H.; Attal, M.; Charbonnel, C.; Garban, F.; Hulin, C.; Michallet, M.; Facon, T.; Garderet, L.; Marit, G.; et al. Achievement of at least very good partial response is a simple and robust prognostic factor in patients with multiple myeloma treated with high-dose therapy: Long-Term analysis of the IFM 99-02 and 99-04 Trials. J. Clin. Oncol. 2009, 27, 5720–5726. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.H.; Mulligan, G.; Fonseca, R.; Chng, W.J. A novel measure of chromosome instability can account for prognostic difference in multiple myeloma. PLoS ONE 2013, 8, e66361. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, K.; Johansson, B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 2009, 48, 637–660. [Google Scholar] [CrossRef] [PubMed]
- Heerema, N.A.; Raimondi, S.C.; Anderson, J.R.; Biegel, J.; Camitta, B.M.; Cooley, L.D.; Gaynon, P.S.; Hirsch, B.; Magenis, R.E.; McGavran, L.; et al. Specific extra chromosomes occur in a modal number dependent pattern in pediatric acute lymphoblastic leukemia. Genes Chromosomes Cancer 2007, 46, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Clark, R.; Farrell, D.M.; Hawkins, J.M.; Martineau, M.; Secker-Walker, L.M. Probes for hidden hyperdiploidy in acute lymphoblastic leukaemia. Genes Chromosomes Cancer 1996, 16, 40–45. [Google Scholar] [CrossRef]
- Paulsson, K.; Morse, H.; Fioretos, T.; Behrendtz, M.; Strombeck, B.; Johansson, B. Evidence for a single-step mechanism in the origin of hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 2005, 44, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Forestier, E.; Johansson, B.; Borgstrom, G.; Kerndrup, G.; Johansson, J.; Heim, S. Cytogenetic findings in a population-based series of 787 childhood acute lymphoblastic leukemias from the Nordic countries. The NOPHO Leukemia Cytogenetic Study Group. Eur. J. Haematol. 2000, 64, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Richards, S.M.; Martineau, M.; Cheung, K.L.; Robinson, H.M.; Jalali, G.R.; Broadfield, Z.J.; Harris, R.L.; Taylor, K.E.; Gibson, B.E.; et al. Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 2003, 102, 2756–2762. [Google Scholar] [CrossRef] [PubMed]
- Griffith, M. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. In Dictionary of Bioinformatics and Computational Biology; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Blandin, A.T.; Muhlematter, D.; Bougeon, S.; Gogniat, C.; Porter, S.; Beyer, V.; Parlier, V.; Beckmann, J.S.; van Melle, G.; Jotterand, M. Automated four-color interphase fluorescence in situ hybridization approach for the simultaneous detection of specific aneuploidies of diagnostic and prognostic significance in high hyperdiploid acute lymphoblastic leukemia. Cancer Genet. Cytogenet. 2008, 186, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Betts, D.R.; Riesch, M.; Grotzer, M.A.; Niggli, F.K. The investigation of karyotypic instability in the high-hyperdiploidy subgroup of acute lymphoblastic leukemia. Leuk. Lymphoma 2001, 42, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Talamo, A.; Chalandon, Y.; Marazzi, A.; Jotterand, M. Clonal heterogeneity and chromosomal instability at disease presentation in high hyperdiploid acute lymphoblastic leukemia. Cancer Genet. Cytogenet. 2010, 203, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Heerema, N.A.; Sather, H.N.; Sensel, M.G.; Zhang, T.; Hutchinson, R.J.; Nachman, J.B.; Lange, B.J.; Steinherz, P.G.; Bostrom, B.C.; Reaman, G.H.; et al. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (>50 chromosomes). J. Clin. Oncol. 2000, 18, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Dayal, J.; Albergant, L.; Newman, T.; South, A. Quantitation of multiclonality in control and drug-treated tumour populations using high-throughput analysis of karyotypic heterogeneity. Converg. Sci. Phys. Oncol. 2015, 1. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Wargo, J.A.; Flaherty, K.T.; Messina, J.L.; Smalley, K.S. BRAF Inhibition Generates a Host-Tumor Niche that Mediates Therapeutic Escape. J. Investig. Dermatol. 2015, 135, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, E151. [Google Scholar] [CrossRef] [PubMed]
- Horne, S.; Wexler, M.; Stevens, J.; Heng, H.H. Insights on processes of evolutionary tumor growth. Atlas Genet. Cytogenet. Oncol. Haematol. 2015. [Google Scholar] [CrossRef]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Penner-Goeke, S.; Lichtensztejn, Z.; Neufeld, M.; Ali, J.L.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. The temporal dynamics of chromosome instability in ovarian cancer cell lines and primary patient samples. PLoS Genet. 2017, 13, e1006707. [Google Scholar] [CrossRef] [PubMed]
- Cyll, K.; Ersvaer, E.; Vlatkovic, L.; Pradhan, M.; Kildal, W.; Avranden Kjaer, M.; Kleppe, A.; Hveem, T.S.; Carlsen, B.; Gill, S.; et al. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br. J. Cancer 2017, 117, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qin, T.; Li, J.; Wang, X.; Gao, C.; Xu, C.; Hao, J.; Liu, J.; Gao, S.; Ren, H. Detection of Circulating Tumor Cells Using Negative Enrichment Immunofluorescence and an In Situ Hybridization System in Pancreatic Cancer. Int. J. Mol. Sci. 2017, 18, 622. [Google Scholar] [CrossRef] [PubMed]
- Stomornjak-Vukadin, M.; Kurtovic-Basic, I.; Mehinovic, L.; Konjhodzic, R. Combined use of cytogenetic and molecular methods in prenatal diagnostics of chromosomal abnormalities. Acta Inform. Med. 2015, 23, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Roylance, R.; Endesfelder, D.; Gorman, P.; Burrell, R.A.; Sander, J.; Tomlinson, I.; Hanby, A.M.; Speirs, V.; Richardson, A.L.; Birkbak, N.J.; et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cospons. Am. Soc. Prev. Oncol. 2011, 20, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Nicke, B.; Schuett, M.; Eklund, A.C.; Ng, C.; Li, Q.; Hardcastle, T.; Lee, A.; Roy, R.; East, P.; et al. Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. USA 2009, 106, 8671–8676. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Foltz, D.R.; Cleveland, D.W. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA 2004, 101, 8699–8704. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Kops, G.J.; Medema, R.H. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19108–19113. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.D.; Zasadil, L.M.; Holland, A.J.; Vitre, B.; Cleveland, D.W.; Weaver, B.A. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA 2013, 110, E4134–E4141. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, V.; Kovacs, K.; Castro, A.V.; Zonis, S.; Melmed, S. Pituitary hypoplasia in Pttg−/− mice is protective for Rb+/− pituitary tumorigenesis. Mol. Endocrinol. 2005, 19, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, N.; Rancati, G.; Li, R. Dr Jekyll and Mr Hyde: Role of aneuploidy in cellular adaptation and cancer. Curr. Opin. Cell Biol. 2010, 22, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Cleveland, D.W. Aneuploidy: Instigator and inhibitor of tumorigenesis. Cancer Res. 2007, 67, 10103–10105. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Malinge, S.; Crispino, J. Myeloid leukemia in Down syndrome. Crit. Rev. Oncogen. 2011, 16, 25–36. [Google Scholar] [CrossRef]
- Schoemaker, M.J.; Swerdlow, A.J.; Higgins, C.D.; Wright, A.F.; Jacobs, P.A. UK Clinical Cytogenetics Group. Cancer incidence in women with Turner syndrome in Great Britain: A national cohort study. Lancet Oncol. 2008, 9, 239–246. [Google Scholar] [CrossRef]
- Swerdlow, A.J.; Schoemaker, M.J.; Higgins, C.D.; Wright, A.F.; Jacobs, P.A. UK Clinical Cytogenetics Group. Cancer incidence and mortality in men with Klinefelter syndrome: A cohort study. J. Natl. Cancer Inst. 2005, 97, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.D.; Swerdlow, A.J.; Schoemaker, M.J.; Wright, A.F.; Jacobs, P.A. UK Clinical Cytogenetics Group. Mortality and cancer incidence in males with Y polysomy in Britain: A cohort study. Hum. Genet. 2007, 121, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Pinson, L.; Mannini, L.; Willems, M.; Cucco, F.; Sirvent, N.; Frebourg, T.; Quarantotti, V.; Collet, C.; Schneider, A.; Sarda, P.; et al. CEP57 mutation in a girl with mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. Part A 2014, 164A, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Castillo, H.; Vasquez-Velasquez, A.I.; Rivera, H.; Barros-Nunez, P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: Delineation of clinical subtypes. Am. J. Med. Genet. Part A 2008, 146A, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; Fitzpatrick, D.; Kidd, A.; Mehes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, N.; Tohyama, J.; Ogawa, A.; Takachi, T.; Watanabe, A.; Asami, K. Refractory infantile spasms associated with mosaic variegated aneuploidy syndrome. Pediatr. Neurol. 2013, 49, 364–367. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Chromosomal Alterations | Number of Cases |
|---|---|
| t(9;22)(q34;q11) | 991 |
| t(12;21)(p13;q22) | 367 |
| der(19)t(1;19)(q23;p13) | 263 |
| i(9)(q10) | 183 |
| i(17)(q10) | 158 |
| i(7)(q10) | 155 |
| t(11;19)(q23;p13) | 138 |
| del(9)(p21) | 134 |
| del(12)(p12) | 123 |
| del(11)(q23) | 114 |
| del(12)(p13) | 77 |
| i(21)(q10) | 68 |
| add(19)(p13) | 60 |
| dic(9;20)(p11;q11) | 52 |
| dic(9;20)(p13;q11) | 50 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2018, 10, 4. https://doi.org/10.3390/cancers10010004
Vargas-Rondón N, Villegas VE, Rondón-Lagos M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers. 2018; 10(1):4. https://doi.org/10.3390/cancers10010004
Chicago/Turabian StyleVargas-Rondón, Natalia, Victoria E. Villegas, and Milena Rondón-Lagos. 2018. "The Role of Chromosomal Instability in Cancer and Therapeutic Responses" Cancers 10, no. 1: 4. https://doi.org/10.3390/cancers10010004
APA StyleVargas-Rondón, N., Villegas, V. E., & Rondón-Lagos, M. (2018). The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers, 10(1), 4. https://doi.org/10.3390/cancers10010004