Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Idasanutlin Activates p53 and Induces Cell Cycle Arrest in Four Model p53wt Cancer Cell Lines
2.2. Dose-Dependent Response to Idasanutlin Is Biphasic, Revealing Two Inhibitory Effects on Cell Survival
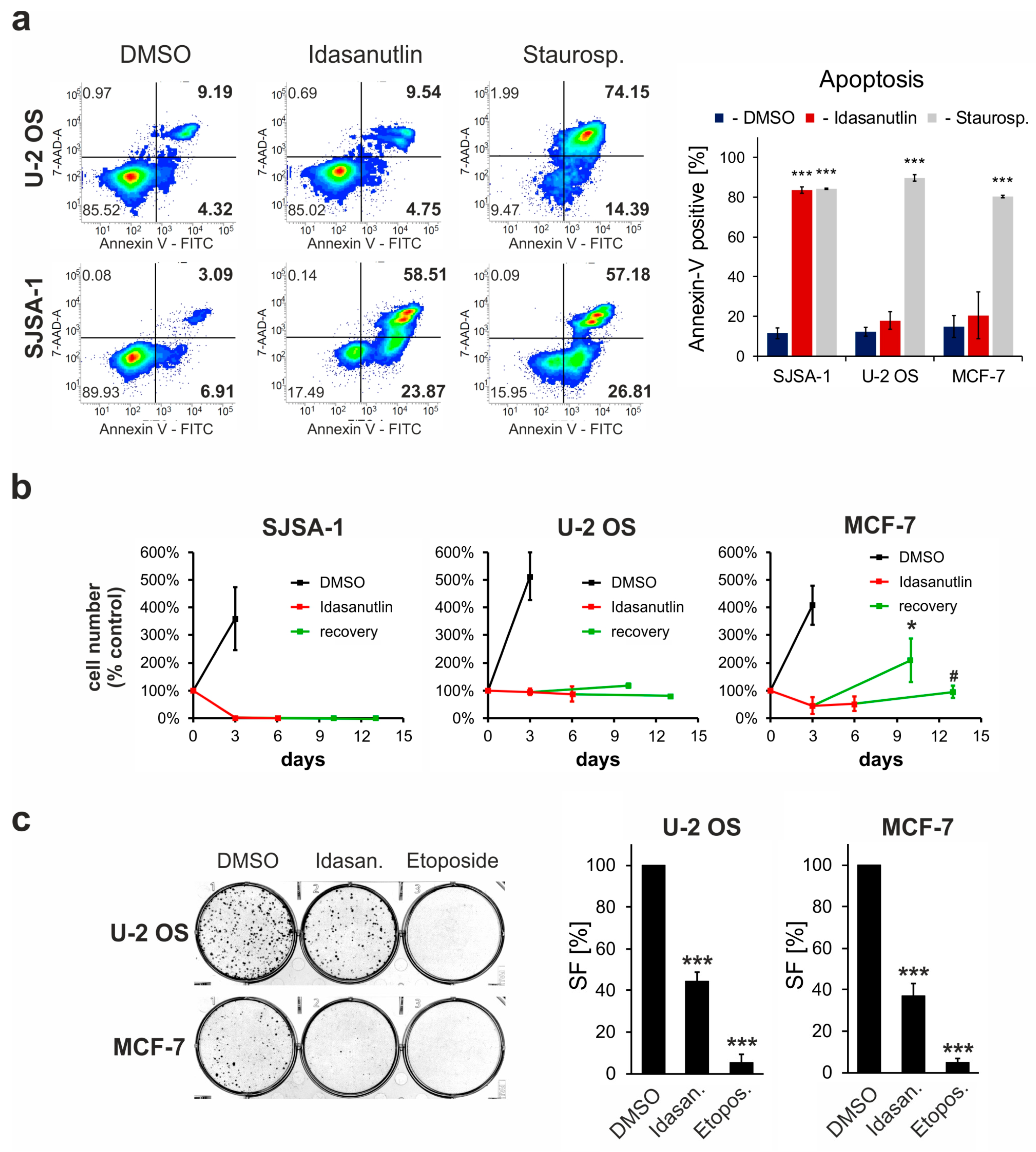
2.3. Idasanutlin Fails to Induce Apoptosis in U-2 OS and MCF-7 Cells
2.4. p53wt cells Remain Viable Following the Idasanutlin Treatment
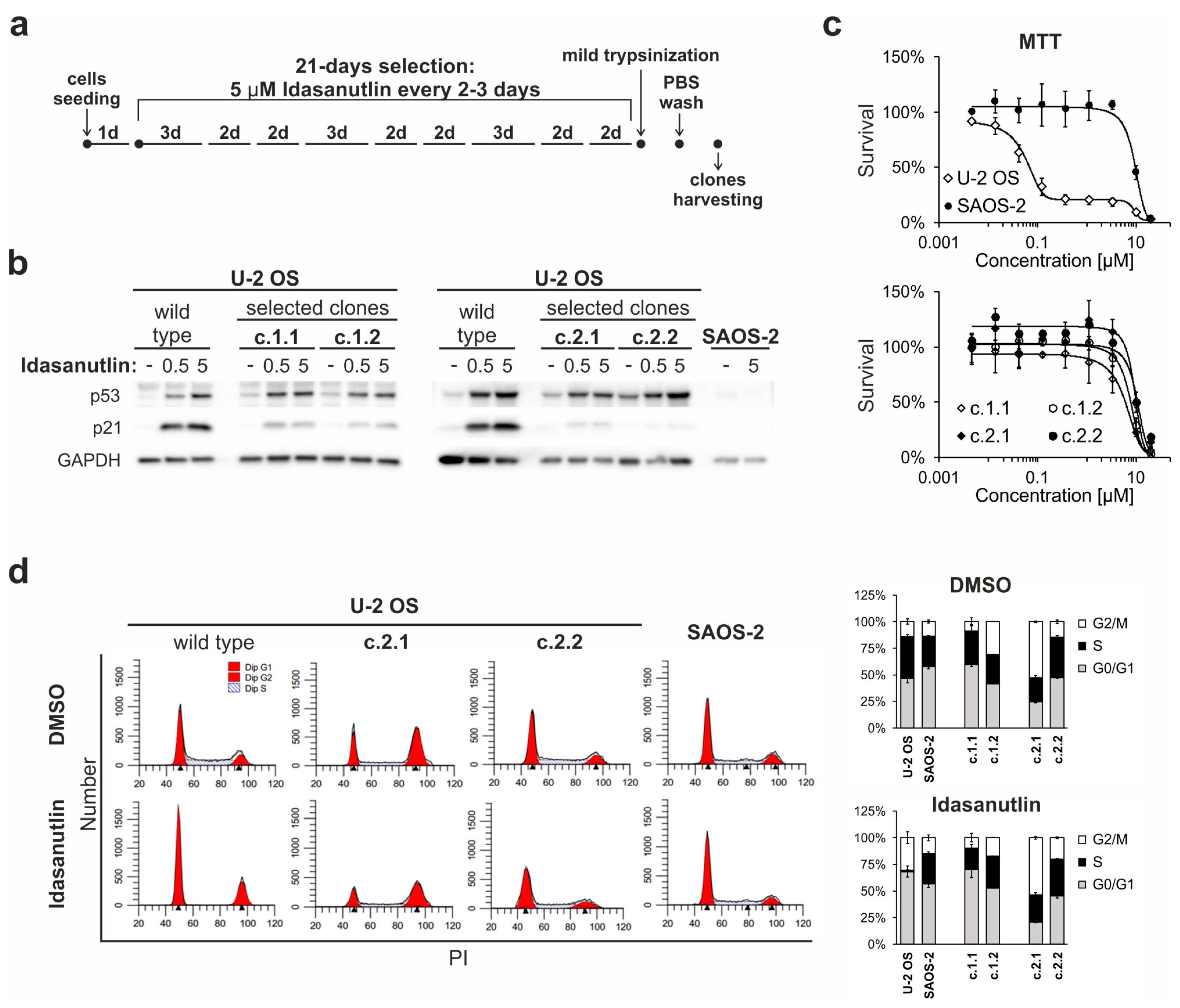
2.5. Continuous Treatment of U-2 OS Cells with Idasanutlin Leads to the Selection of p53-Mutated Drug-Resistant Clones
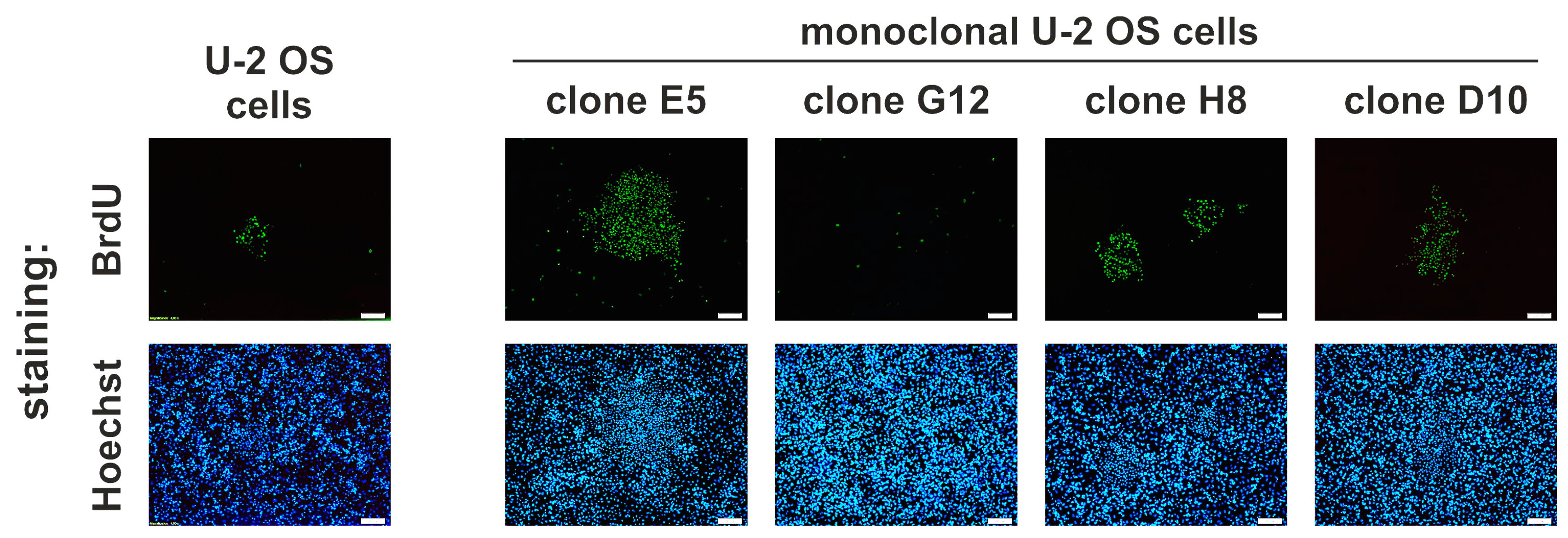
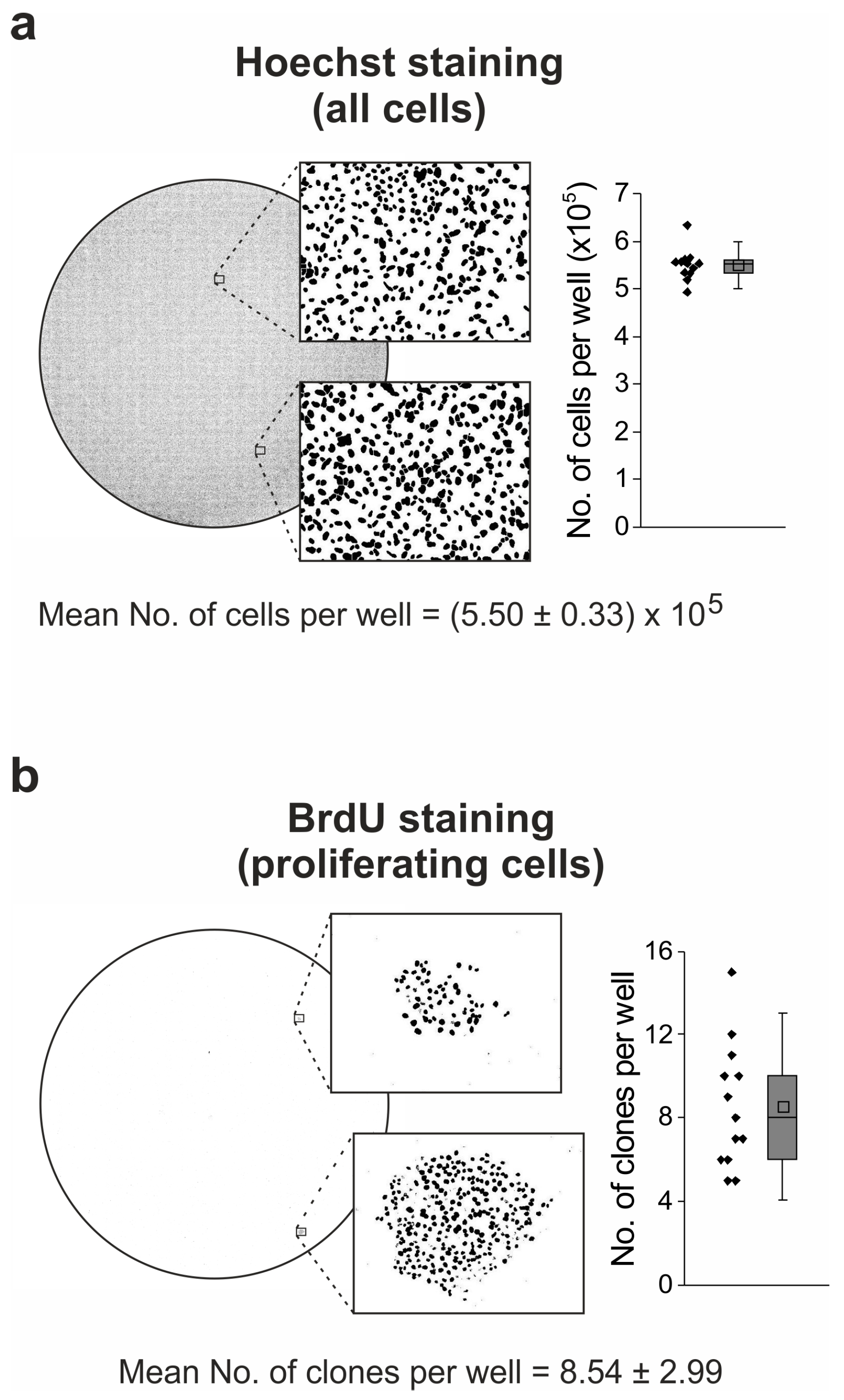
2.6. Idasanutlin Resistance Is Newly Developed from Initially Sensitive, Monoclonal Cell Populations
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Western Blotting
4.3. Cell Cycle Analysis
4.4. Annexin V/7-AAD Staining of Apoptotic Cells
4.5. Cell Viability MTT Assay and Dose-Response Curve Fitting
4.6. Treatment-Recovery Assay
4.7. Colony Formation Assay
4.8. Staining of Proliferating Cells
4.9. DNA Profiling (Cell Line Authentication)
4.10. DNA Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Wiman, K.G. Shaping Genetic Alterations in Human Cancer: The p53 Mutation Paradigm. Cancer Cell 2007, 12, 303–312. [Google Scholar] [CrossRef] [PubMed]
- París, R.; Henry, R.E.; Stephens, S.J.; McBryde, M.; Espinosa, J.M. Multiple p53-independent gene silencing mechanisms define the cellular response to p53 activation. Cell Cycle 2008, 7, 2427–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.Z.; Eksterowicz, J.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Fox, B.M.; Fu, J.; et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J. Med. Chem. 2014, 57, 2472–2488. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Masuya, K.; Kallen, J.; Stachyra-Valat, T.; Ruetz, S.; Guagnano, V.; Holzer, P.; Mah, R.; Stutz, S.; Vaupel, A.; et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg. Med. Chem. Lett. 2016, 26, 4837–4841. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.; Keir, S.T.; Maris, J.M.; Wu, J.; Lyalin, D.; et al. Initial Testing (Stage 1) of MK-8242-A Novel MDM2 Inhibitor-by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2016, 63, 1744–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wünschel, J.; Toedling, J.; Deubzer, H.E.; Künkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Shen, H.; Maki, C.G. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 2011, 30, 4678–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.J.; Joseph, T.; Sim, A.Y.L.; Yurlova, L.; Zolghadr, K.; Lane, D.; Verma, C.; Ghadessy, F. In vitro selection of mutant HDM2 resistant to Nutlin inhibition. PLoS ONE 2013, 8, e62564. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Luca, C.G.; Ziazadeh, D.; McEachern, D.; Zhao, Y.; Sun, W.; Debussche, L.; Wang, S. Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia In Vitro and In Vivo. Clin. Cancer Res. 2015, 21, 2558–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Lee, J.S.; Dickson, M.A.; Schwartz, G.K.; Le Cesne, A.; Varga, A.; Bahleda, R.; Wagner, A.J.; Choy, E.; de Jonge, M.J.; et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat. Commun. 2016, 7, 12609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, C.J.; Esfandiari, A.; Liu, J.; Lu, X.; Hutton, C.; Jackson, J.; Bennaceur, K.; Xu, Q.; Makimanejavali, A.R.; Del Bello, F.; et al. TP53 mutant MDM2-amplified cell lines selected for resistance to MDM2-p53 binding antagonists retain sensitivity to ionizing radiation. Oncotarget 2016, 7, 46203–46218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf-/- mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef] [PubMed]
- Skalniak, L.; Smejda, M.; Cierniak, A.; Adamczyk, A.; Konieczny, P.; Madej, E.; Wolnicka-Glubisz, A. p38 but not p53 is responsible for UVA-induced MCPIP1 expression. Mech. Ageing Dev. 2018, 172, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Di Veroli, G.Y.; Fornari, C.; Goldlust, I.; Mills, G.; Koh, S.B.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. An automated fitting procedure and software for dose-response curves with multiphasic features. Sci. Rep. 2015, 5, 14701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, E.J. Paradigm lost, paradigm found: The re-emergence of hormesis as a fundamental dose response model in the toxicological sciences. Environ. Pollut. 2005, 138, 379–411. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Deo, D.; Xia, M.; Vassilev, L.T. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol. Cancer Res. 2009, 7, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Jeay, S.; Gaulis, S.; Ferretti, S.; Bitter, H.; Ito, M.; Valat, T.; Murakami, M.; Ruetz, S.; Guthy, D.A.; Rynn, C.; et al. A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. eLife 2015, 4, e06498. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: P53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Ishizawa, J.; Ruvolo, V.; Dilip, A.; Quintás-Cardama, A.; McDonnell, T.J.; Neelapu, S.S.; Kwak, L.W.; Shacham, S.; Kauffman, M.; et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma. Cancer Sci. 2014, 105, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Marine, J.-C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat. 2009, 117, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Chang, H. Molecular mechanisms of nutlin-induced apoptosis in multiple myeloma: Evidence for p53-transcription-dependent and -independent pathways. Cancer Biol. Ther. 2010, 10, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Flørenes, V.A.; Maelandsmo, G.M.; Forus, A.; Andreassen, A.; Myklebost, O.; Fodstad, O. MDM2 gene amplification and transcript levels in human sarcomas: Relationship to TP53 gene status. J. Natl. Cancer Inst. 1994, 86, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Konopleva, M.; McQueen, T.; O’Brien, S.; Plunkett, W.; Andreeff, M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006, 108, 993–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caboche, M. Comparison of the frequencies of spontaneous and chemically-induced 5-bromodeoxyuridine-resistance mutations in wild-type and revertant BHK-21-13 cells. Genetics 1974, 77, 309–322. [Google Scholar] [PubMed]
- Phelps, D.; Bondra, K.; Seum, S.; Chronowski, C.; Leasure, J.; Kurmasheva, R.T.; Middleton, S.; Wang, D.; Mo, X.; Houghton, P.J. Inhibition of MDM2 by RG7388 confers hypersensitivity to X-radiation in xenograft models of childhood sarcoma. Pediatr. Blood Cancer 2015, 62, 1345–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Rousseau, R.F.; Middleton, S.A.; Nichols, G.L.; Newell, D.R.; Lunec, J.; Tweddle, D.A. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 2015, 6, 10207–10221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, C.; Friess, T.; Birzele, F.; Kiialainen, A.; Dangl, M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J. Hematol. Oncol. 2016, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, A.; Yigit, N.; Moreno-Smith, M.; Vasudevan, S.A.; Barbieri, E.; Speleman, F.; Shohet, J.; Vandesompele, J.; Van Maerken, T. Dual targeting of MDM2 and BCL2 as a therapeutic strategy in neuroblastoma. Oncotarget 2017, 8, 57047–57057. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, A.; Hawthorne, T.A.; Nakjang, S.; Lunec, J. Chemical Inhibition of Wild-Type p53-Induced Phosphatase 1 (WIP1/PPM1D) by GSK2830371 Potentiates the Sensitivity to MDM2 Inhibitors in a p53-Dependent Manner. Mol. Cancer Ther. 2016, 15, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-E.; Esfandiari, A.; Ho, Y.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Herting, F.; Herter, S.; Friess, T.; Muth, G.; Bacac, M.; Sulcova, J.; Umana, P.; Dangl, M.; Klein, C. Antitumour activity of the glycoengineered type II anti-CD20 antibody obinutuzumab (GA101) in combination with the MDM2-selective antagonist idasanutlin (RG7388). Eur. J. Haematol. 2016, 97, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Zanjirband, M.; Curtin, N.; Edmondson, R.J.; Lunec, J. Combination treatment with rucaparib (Rubraca) and MDM2 inhibitors, Nutlin-3 and RG7388, has synergistic and dose reduction potential in ovarian cancer. Oncotarget 2017, 8, 69779–69796. [Google Scholar] [CrossRef] [PubMed]
- Laroche-Clary, A.; Chaire, V.; Algeo, M.; Derieppe, M.; Loarer, F.L.; Italiano, A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J. Hematol. Oncol. 2017, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Laroche, A.; Chaire, V.; Algeo, M.; Karanian, M.; Fourneaux, B.; Italiano, A. MDM2 antagonists synergize with PI3K/mTOR inhibition in well-differentiated/dedifferentiated liposarcomas. Oncotarget 2017, 8, 53968–53977. [Google Scholar] [CrossRef] [PubMed]
- van Kuppeveld, F.J.; van der Logt, J.T.; Angulo, A.F.; van Zoest, M.J.; Quint, W.G.; Niesters, H.G.; Galama, J.M.; Melchers, W.J. Genus- and species-specific identification of mycoplasmas by 16S rRNA amplification. Appl. Environ. Microbiol. 1992, 58, 2606–2615. [Google Scholar] [PubMed]
- Magiera, K.; Tomala, M.; Kubica, K.; De Cesare, V.; Trost, M.; Zieba, B.J.; Kachamakova-Trojanowska, N.; Les, M.; Dubin, G.; Holak, T.A.; et al. Lithocholic Acid Hydroxyamide Destabilizes Cyclin D1 and Induces G0/G1 Arrest by Inhibiting Deubiquitinase USP2a. Cell Chem. Biol. 2017, 24, 458.e18–470.e18. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skalniak, L.; Kocik, J.; Polak, J.; Skalniak, A.; Rak, M.; Wolnicka-Glubisz, A.; Holak, T.A. Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells. Cancers 2018, 10, 396. https://doi.org/10.3390/cancers10110396
Skalniak L, Kocik J, Polak J, Skalniak A, Rak M, Wolnicka-Glubisz A, Holak TA. Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells. Cancers. 2018; 10(11):396. https://doi.org/10.3390/cancers10110396
Chicago/Turabian StyleSkalniak, Lukasz, Justyna Kocik, Justyna Polak, Anna Skalniak, Monika Rak, Agnieszka Wolnicka-Glubisz, and Tad A. Holak. 2018. "Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells" Cancers 10, no. 11: 396. https://doi.org/10.3390/cancers10110396
APA StyleSkalniak, L., Kocik, J., Polak, J., Skalniak, A., Rak, M., Wolnicka-Glubisz, A., & Holak, T. A. (2018). Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of p53-Mutated Cells. Cancers, 10(11), 396. https://doi.org/10.3390/cancers10110396