Profiling Immune Escape in Hodgkin’s and Diffuse large B-Cell Lymphomas Using the Transcriptome and Immunostaining

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
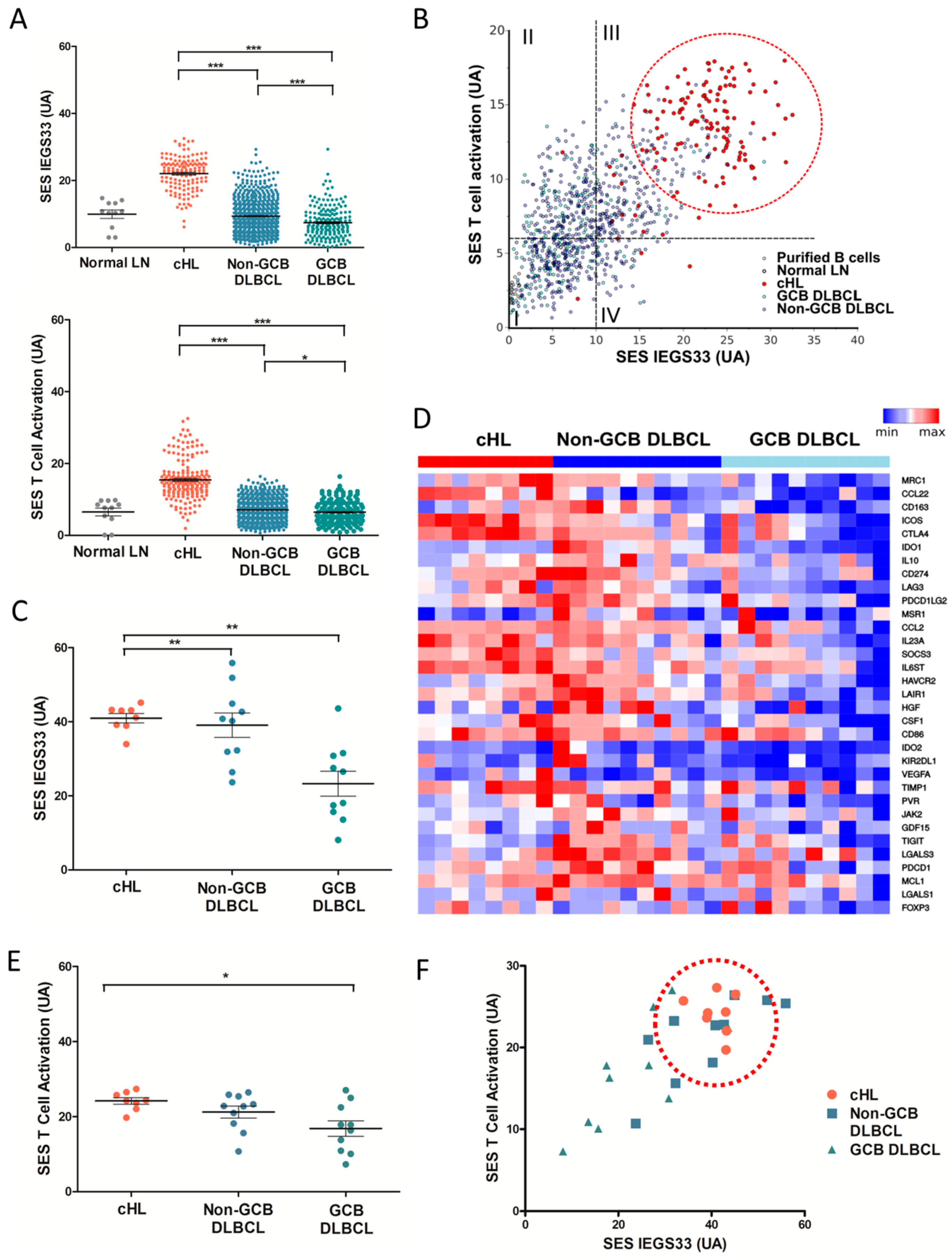
2.1. Molecular Profiling of Immune Escape in cHL and DLBCL
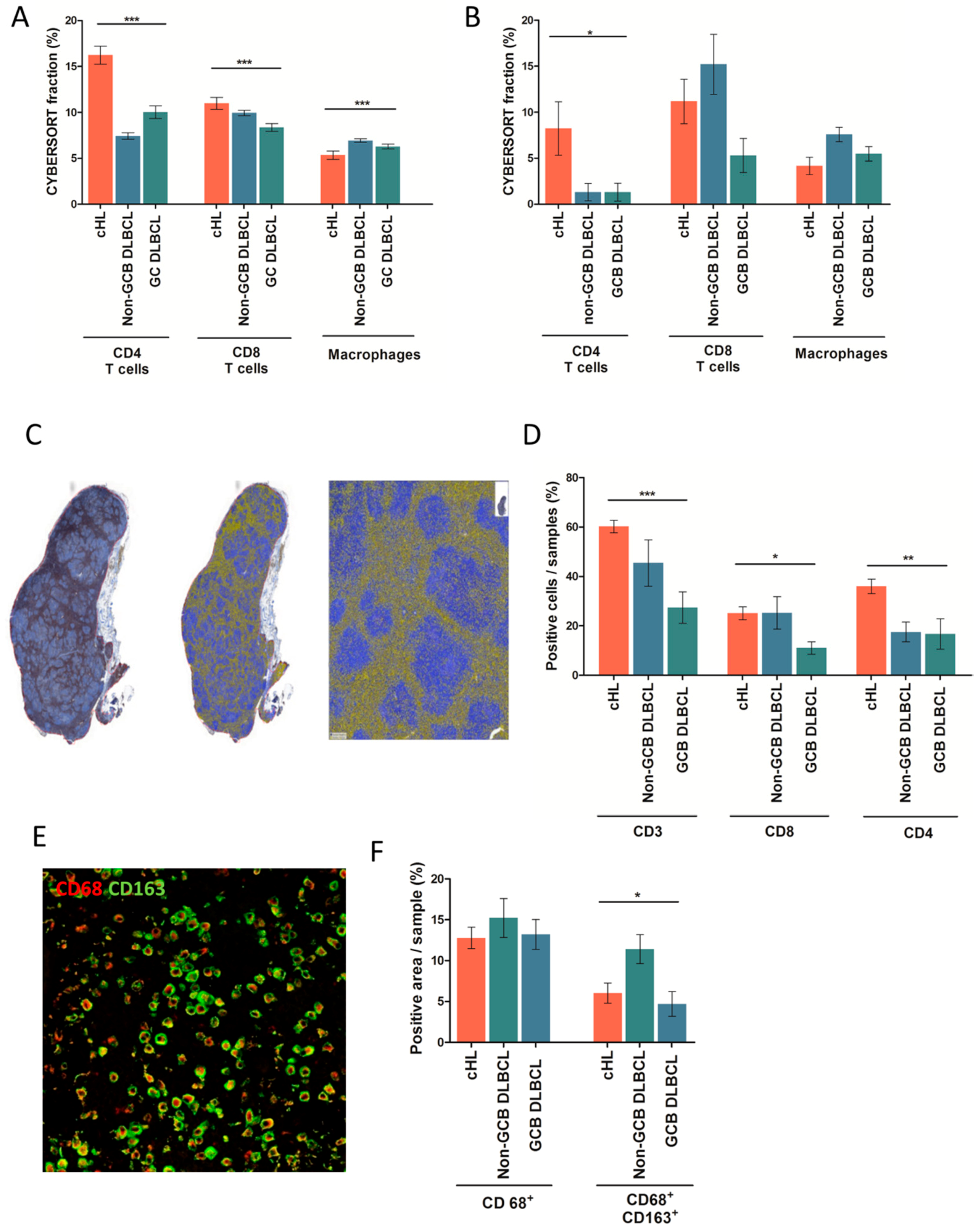
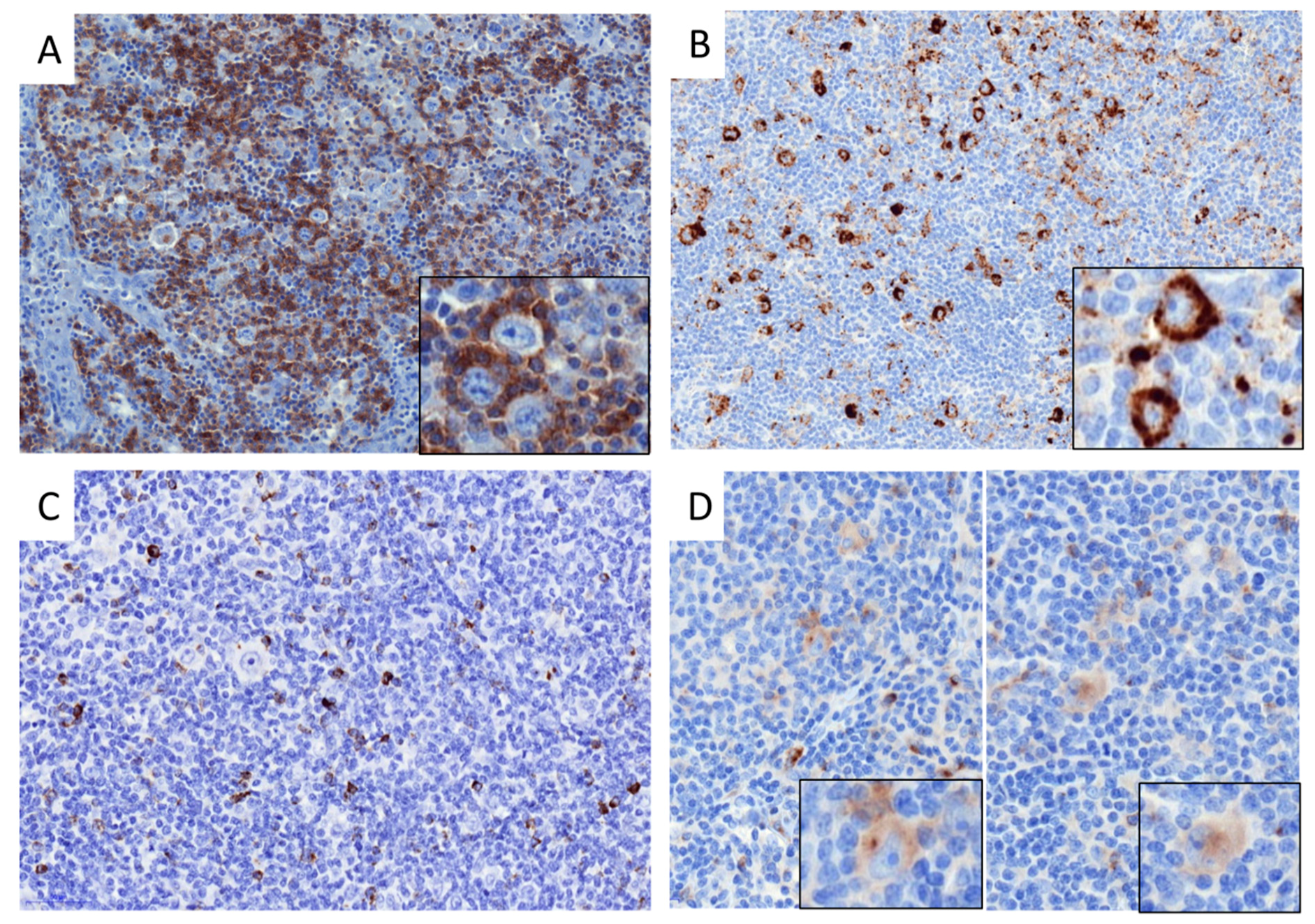
2.2. Distribution of Immune Cells in cHL and DLBCL Tissue Samples
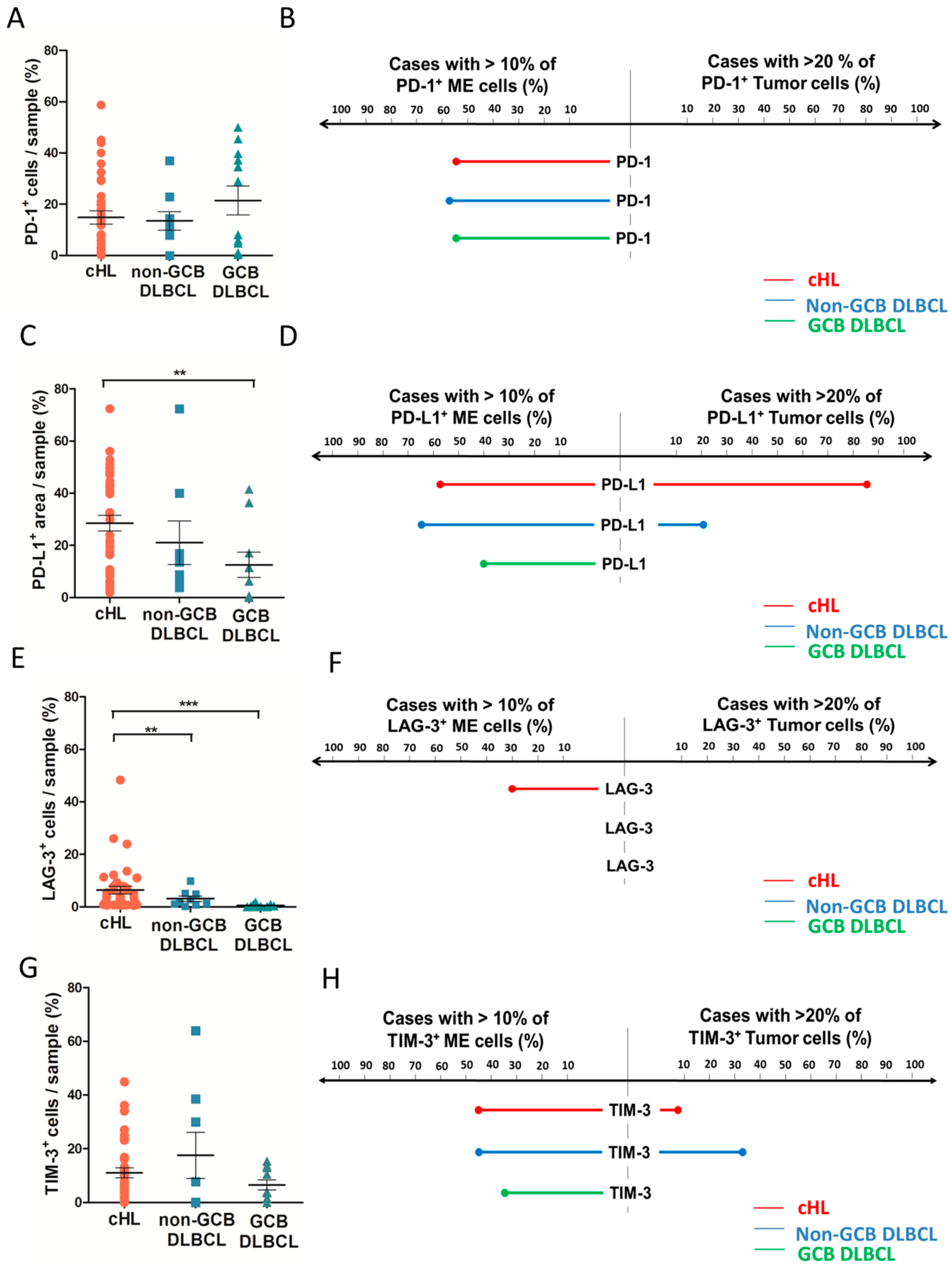
2.3. Distribution of Inhibitory Immune Checkpoint in HL and DLBCL Samples
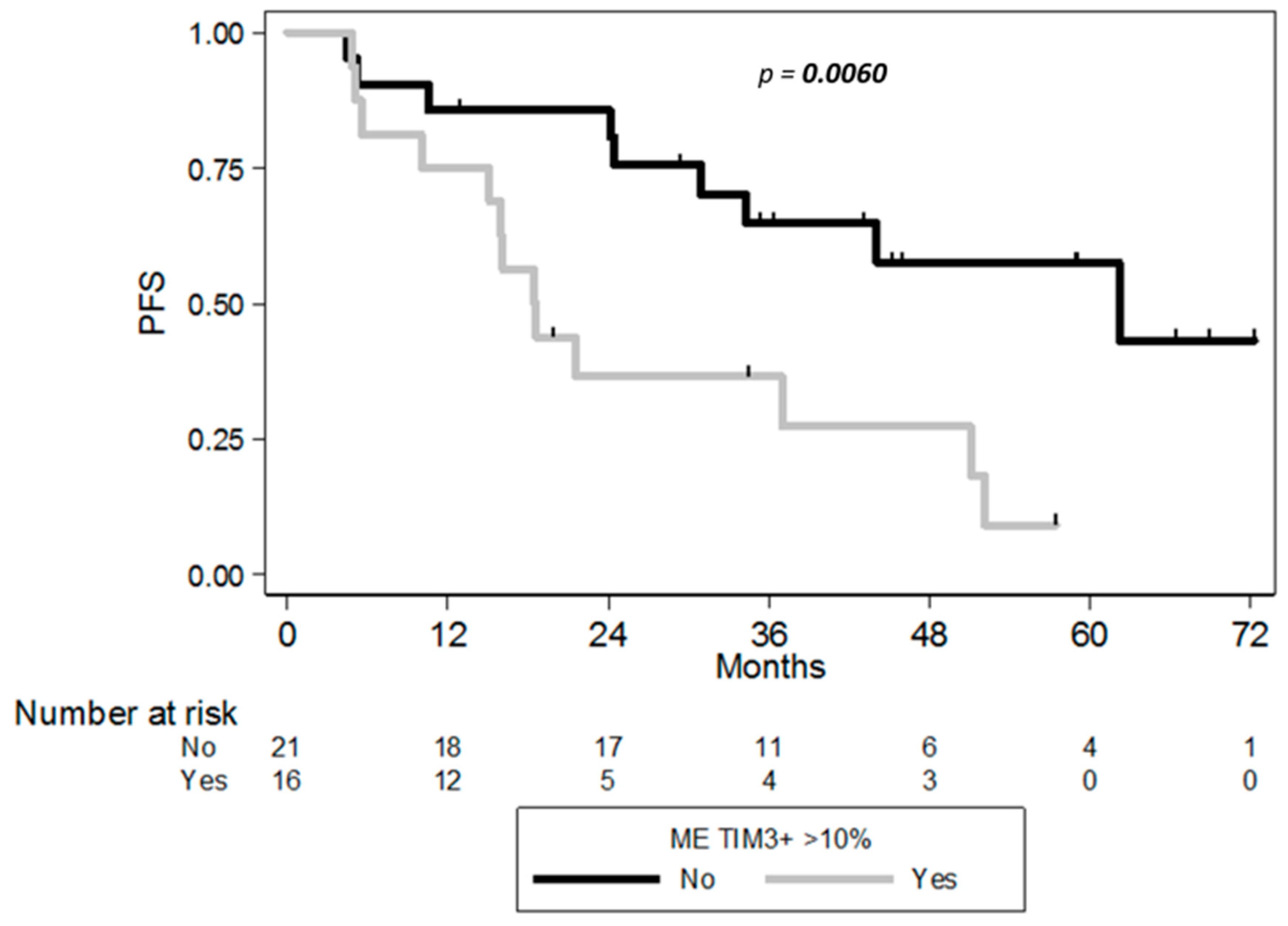
2.4. Clinical Correlates of Immune Status
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Microarray Analysis
4.3. Immunohistochemistry
4.4. Immunofluorescence
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Charmpi, K.; Gravelle, P.; Tosolini, M.; Franchet, C.; Ysebaert, L.; Brousset, P.; Bidaut, A.; Ycart, B.; Fournié, J.-J. Several immune escape patterns in non-Hodgkin’s lymphomas. Oncoimmunology 2015, 4, e1026530. [Google Scholar] [CrossRef] [PubMed]
- Swaika, A.; Hammond, W.A.; Joseph, R.W. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol. Immunol. 2015, 67, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef]
- Kiyasu, J.; Miyoshi, H.; Hirata, A.; Arakawa, F.; Ichikawa, A.; Niino, D.; Sugita, Y.; Yufu, Y.; Choi, I.; Abe, Y.; et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015, 126, 2193–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenst, S.; Hoeller, S.; Willi, N.; Dirnhofera, S.; Tzankov, A. Diagnostic and prognostic utility of PD-1 in B cell lymphomas. Dis. Markers 2010, 29, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, K.; Chen, L.; Berglund, M.; Ren, W.; de Miranda, N.F.C.C.; Lisboa, S.; Fangazio, M.; Zhu, S.; Hou, Y.; et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood 2016, 127, 3026–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017, 130, 2420–2430. [Google Scholar] [CrossRef] [PubMed]
- Gravelle, P.; Do, C.; Franchet, C.; Mueller, S.; Oberic, L.; Ysebaert, L.; Larocca, L.M.; Hohaus, S.; Calmels, M.-N.; Frenois, F.-X.; et al. Impaired functional responses in follicular lymphoma CD8+TIM-3+ T lymphocytes following TCR engagement. Oncoimmunology 2016, 5, e1224044. [Google Scholar] [CrossRef] [PubMed]
- Hollander, P.; Kamper, P.; Smedby, K.E.; Enblad, G.; Ludvigsen, M.; Mortensen, J.; Amini, R.-M.; Hamilton-Dutoit, S.; d’Amore, F.; Molin, D.; et al. High proportions of PD-1+ and PD-L1+ leukocytes in classical Hodgkin lymphoma microenvironment are associated with inferior outcome. Blood Adv. 2017, 1, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravelle, P.; Burroni, B.; Péricart, S.; Rossi, C.; Bezombes, C.; Tosolini, M.; Damotte, D.; Brousset, P.; Fournié, J.-J.; Laurent, C. Mechanisms of PD-1/PD-L1 expression and prognostic relevance in non-Hodgkin lymphoma: A summary of immunohistochemical studies. Oncotarget 2017, 8, 44960–44975. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Han, J.J.; Stenson, M.; Maurer, M.; Wellik, L.; Hu, G.; Ziesmer, S.; Dogan, A.; Witzig, T.E. Elevated serum IL-10 levels in diffuse large B-cell lymphoma: A mechanism of aberrant JAK2 activation. Blood 2012, 119, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Van Roosbroeck, K.; Ferreiro, J.F.; Tousseyn, T.; van der Krogt, J.-A.; Michaux, L.; Pienkowska-Grela, B.; Theate, I.; De Paepe, P.; Dierickx, D.; Doyen, C.; et al. Genomic alterations of the JAK2 and PDL loci occur in a broad spectrum of lymphoid malignancies. Genes Chromosomes Cancer 2016, 55, 428–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twa, D.D.W.; Chan, F.C.; Ben-Neriah, S.; Woolcock, B.W.; Mottok, A.; Tan, K.L.; Slack, G.W.; Gunawardana, J.; Lim, R.S.; McPherson, A.W.; et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood 2014, 123, 2062–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Nagler, A.; Weller, E.A.; Devine, S.M.; Avigan, D.E.; Chen, Y.-B.; Kaminski, M.S.; Holland, H.K.; Winter, J.N.; Mason, J.R.; et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: Results of an international phase II trial. J. Clin. Oncol. 2013, 31, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosolini, M.; Algans, C.; Pont, F.; Ycart, B.; Fournié, J.-J. Large-scale microarray profiling reveals four stages of immune escape in non-Hodgkin lymphomas. Oncoimmunology 2016, 5, e1188246. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Loo, K.; Pauli, M.L.; Sanchez-Rodriguez, R.; Sandoval, P.M.; Taravati, K.; Tsai, K.; Nosrati, A.; Nardo, L.; Alvarado, M.D.; et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Investig. 2016, 126, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined genetic inactivation of β2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015, 125, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4+ regulatory T-cell-rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Greaves, P.; Clear, A.; Owen, A.; Iqbal, S.; Lee, A.; Matthews, J.; Wilson, A.; Calaminici, M.; Gribben, J.G. Defining characteristics of classical Hodgkin lymphoma microenvironment T-helper cells. Blood 2013, 122, 2856–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gaetano, R.; Gasparetto, V.; Padoan, A.; Callegari, B.; Candiotto, L.; Sanzari, M.C.; Scapinello, A.; Tagariello, G. Flow cytometry CD4(+)CD26(-)CD38(+) lymphocyte subset in the microenvironment of Hodgkin lymphoma-affected lymph nodes. Ann. Hematol. 2014, 93, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- El Halabi, L.; Adam, J.; Marty, V.; Bosq, J.; Lazarovici, J.; Danu, A.; Ribrag, V.; Ghez, D. Strong Expression of the Immune Checkpoint Regulators LAG3 and Tim3 in Hodgkin Lymphoma. ASH Abstr. Blood J. 2016, 128, 2952. [Google Scholar]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. Baltim. Md 1950 2002, 169, 5392–5395. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Jiang, B.; Zhao, H.; Huang, Q. Prognostic implication of TIM-3 in clear cell renal cell carcinoma. Neoplasma 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.; Huang, Q.; Wang, J.; Fang, C.; Yang, L.; Zhu, M.; Chen, J.; Chen, L.; Dong, M. Up-regulation of Tim-3 is associated with poor prognosis of patients with colon cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8018–8027. [Google Scholar] [PubMed]
- Piao, Y.-R.; Piao, L.-Z.; Zhu, L.-H.; Jin, Z.-H.; Dong, X.-Z. Prognostic value of T cell immunoglobulin mucin-3 in prostate cancer. Asian Pac. J. Cancer Prev. 2013, 14, 3897–3901. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yu, Q.; Liu, J.; Fu, W.; Cao, Y.; Yu, L.; Shao, S.; Wang, X.; Niu, H.; Wang, Y. T-cell immunoglobulin mucin-3 expression in bladder urothelial carcinoma: Clinicopathologic correlations and association with survival. J. Surg. Oncol. 2015, 112, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jin, M.-S.; Kong, F.; Cao, D.; Ma, H.-X.; Jia, Z.; Wang, Y.-P.; Suo, J.; Cao, X. Decreased galectin-9 and increased Tim-3 expression are related to poor prognosis in gastric cancer. PLoS ONE 2013, 8, e81799. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.L.; Smyth, M.J. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Compaci, G.; Rueter, M.; Lamy, S.; Oberic, L.; Recher, C.; Lapeyre-Mestre, M.; Laurent, G.; Despas, F. Ambulatory Medical Assistance-After Cancer (AMA-AC): A model for an early trajectory survivorship survey of lymphoma patients treated with anthracycline-based chemotherapy. BMC Cancer 2015, 15, 781. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. Gene Ontology Consortium The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Pont, F.; Poupot, M.; Vergez, F.; Nicolau-Travers, M.-L.; Vermijlen, D.; Sarry, J.-E.; Dieli, F.; Fournié, J.-J. Assessment of tumor-infiltrating TCRVγ9Vδ2 γδ lymphocyte abundance by deconvolution of human cancers microarrays. Oncoimmunology 2017, 6, e1284723. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Péricart, S.; Tosolini, M.; Gravelle, P.; Rossi, C.; Traverse-Glehen, A.; Amara, N.; Franchet, C.; Martin, E.; Bezombes, C.; Laurent, G.; et al. Profiling Immune Escape in Hodgkin’s and Diffuse large B-Cell Lymphomas Using the Transcriptome and Immunostaining. Cancers 2018, 10, 415. https://doi.org/10.3390/cancers10110415
Péricart S, Tosolini M, Gravelle P, Rossi C, Traverse-Glehen A, Amara N, Franchet C, Martin E, Bezombes C, Laurent G, et al. Profiling Immune Escape in Hodgkin’s and Diffuse large B-Cell Lymphomas Using the Transcriptome and Immunostaining. Cancers. 2018; 10(11):415. https://doi.org/10.3390/cancers10110415
Chicago/Turabian StylePéricart, Sarah, Marie Tosolini, Pauline Gravelle, Cédric Rossi, Alexandra Traverse-Glehen, Nadia Amara, Camille Franchet, Elodie Martin, Christine Bezombes, Guy Laurent, and et al. 2018. "Profiling Immune Escape in Hodgkin’s and Diffuse large B-Cell Lymphomas Using the Transcriptome and Immunostaining" Cancers 10, no. 11: 415. https://doi.org/10.3390/cancers10110415
APA StylePéricart, S., Tosolini, M., Gravelle, P., Rossi, C., Traverse-Glehen, A., Amara, N., Franchet, C., Martin, E., Bezombes, C., Laurent, G., Brousset, P., Fournié, J.-J., & Laurent, C. (2018). Profiling Immune Escape in Hodgkin’s and Diffuse large B-Cell Lymphomas Using the Transcriptome and Immunostaining. Cancers, 10(11), 415. https://doi.org/10.3390/cancers10110415