TGFβ Imprinting During Activation Promotes Natural Killer Cell Cytokine Hypersecretion

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Chronic Stimulation by TGFβ during NK Cell Expansion and Activation Generates NK Cells with Increased Cytokine Secretion
2.2. NK Cell Activation is Required for TGFβ Induced Cytokine Hypersecretion
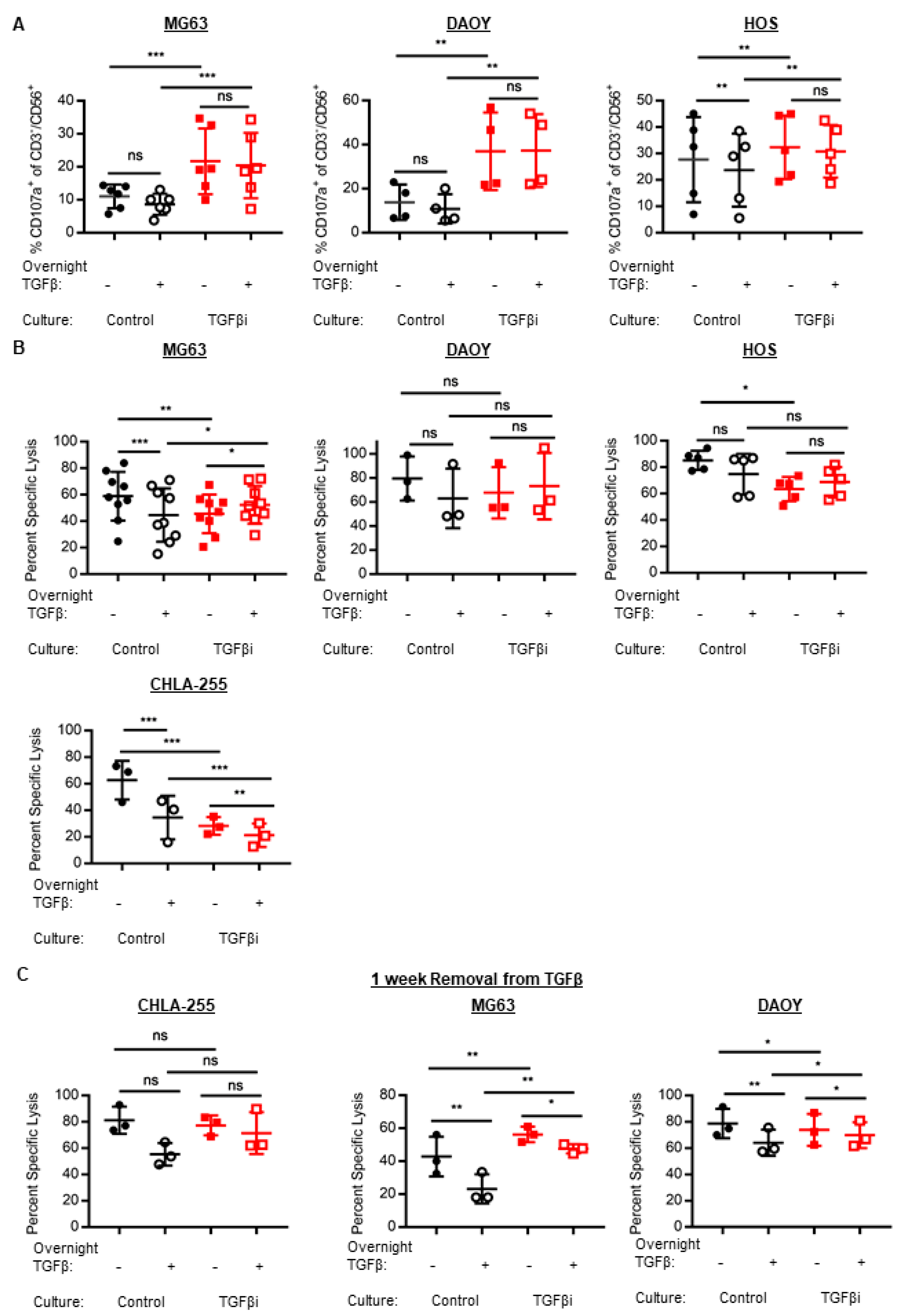
2.3. TGFβi generates NK Cells with Increased Degranulation but Transiently Impairs Cytotoxicity
2.4. TGFβi Remodels NK Cell Receptor Expression
2.5. TGFβi Decreases Granzyme A and Perforin Expression.
2.6. TGFβ Imprinting Modifies NK Cell IFNγ Regulation
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Lines and Culture Conditions
5.2. Flow Cytometry
5.3. Cytotoxicity Assay
5.4. Cytokine Secretion
5.5. qPCR
5.6. RNA-Seq Sample Preparation and Sequencing
5.7. ATAC-seq
5.8. Western Blotting
5.9. Statistical Analysis
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of trail and fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Takeda, K.; Akiba, H.; Tsutsui, H.; Okamura, H.; Nakanishi, K.; Okumura, K.; Yagita, H. Expression and function of tnf-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 1999, 163, 1906–1913. [Google Scholar] [PubMed]
- Ljunggren, H.G.; Karre, K. In search of the “missing self”: Mhc molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Rosario, M.; Romee, R.; Berrien-Elliott, M.M.; Schneider, S.E.; Leong, J.W.; Sullivan, R.P.; Jewell, B.A.; Becker-Hapak, M.; Schappe, T.; et al. Cd56bright NK cells exhibit potent antitumor responses following il-15 priming. J. Clin. Investig. 2017, 127, 4042–4058. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the cd56(bright) subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Michel, T.; Theresine, M.; Andres, E.; Hentges, F.; Zimmer, J. Cd56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Caligiuri, M.A. Interleukin 15: Biology and relevance to human disease. Blood 2001, 97, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Cooper, M.A.; Caligiuri, M.A. Interleukin-2 and interleukin-15: Immunotherapy for cancer. Cytokine Growth Factor Rev. 2002, 13, 169–183. [Google Scholar] [CrossRef]
- Zhu, S.; Phatarpekar, P.V.; Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Nguyen-Jackson, H.T.; Mace, E.M.; Freeman, A.F.; Watowich, S.S.; Orange, J.S.; et al. Transcription of the activating receptor nkg2d in natural killer cells is regulated by stat3 tyrosine phosphorylation. Blood 2014, 124, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Denman, C.; Senyukov, V.; Somanchi, S.; Phatarpekar, P.; Kopp, L.; Johnson, J.; Singh, H.; Hurton, L.; Maiti, S.; Huls, M.; et al. Membrane-bound il-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, e357. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Hippen, K.L.; Lemire, A.; Hying, S.; Luo, X.; Lenvik, T.; Curtsinger, J.; Davis, Z.; Zhang, B.; Cooley, S.; et al. Adaptive NK cells resist regulatory t-cell suppression driven by il37. Cancer Immunol. Res. 2018, 6, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of tgfbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.; Gold, L.I.; Klimstra, D.; Zeng, Z.S.; Winawer, S.; Cohen, A. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol. Biomark. Prev. 1995, 4, 549–554. [Google Scholar]
- Yang, R.S.; Wu, C.T.; Lin, K.H.; Hong, R.L.; Liu, T.K.; Lin, K.S. Relation between histological intensity of transforming growth factor-beta isoforms in human osteosarcoma and the rate of lung metastasis. Tohoku J. Exp. Med. 1998, 184, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Picon, A.; Gold, L.I.; Wang, J.; Cohen, A.; Friedman, E. A subset of metastatic human colon cancers expresses elevated levels of transforming growth factor beta1. Cancer Epidemiol. Biomark. Prev. 1998, 7, 497–504. [Google Scholar]
- Trotta, R.; Dal Col, J.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J., 2nd; Wei, M.; Mao, H.; Byrd, J.C.; et al. Tgf-beta utilizes smad3 to inhibit cd16-mediated ifn-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wei, M.; Becknell, B.; Trotta, R.; Liu, S.; Boyd, Z.; Jaung, M.S.; Blaser, B.W.; Sun, J.; Benson, D.M., Jr.; et al. Pro- and antiinflammatory cytokine signaling: Reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity 2006, 24, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.; Zhou, S.; Meng, X.M.; Wang, Q.M.; Li, C.J.; Lian, G.Y.; Huang, X.R.; Tang, Y.J.; Guan, X.Y.; Yan, B.P.; et al. Smad3 promotes cancer progression by inhibiting e4bp4-mediated NK cell development. Nat. Commun. 2017, 8, e14677. [Google Scholar] [CrossRef] [PubMed]
- Bellone, G.; Aste-Amezaga, M.; Trinchieri, G.; Rodeck, U. Regulation of NK cell functions by tgf-beta 1. J. Immunol. 1995, 155, 1066–1073. [Google Scholar] [PubMed]
- Donatelli, S.S.; Zhou, J.M.; Gilvary, D.L.; Eksioglu, E.A.; Chen, X.; Cress, W.D.; Haura, E.B.; Schabath, M.B.; Coppola, D.; Wei, S.; et al. Tgf-beta-inducible microrna-183 silences tumor-associated natural killer cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4203–4208. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Allan, D.S.; Rybalov, B.; Andzelm, M.M.; Stern, J.N.; Kopcow, H.D.; Koopman, L.A.; Strominger, J.L. Tgfbeta promotes conversion of cd16+ peripheral blood NK cells into cd16- NK cells with similarities to decidual NK cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3378–3383. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, K.M.; Kim, D.W.; Heo, D.S. Elevated tgf-beta1 secretion and down-modulation of nkg2d underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.C.; Wan, Z.; Sheard, M.A.; Sun, J.; Jackson, J.R.; Malvar, J.; Xu, Y.; Wang, L.; Sposto, R.; Kim, E.S.; et al. Tgfbetar1 blockade with galunisertib (ly2157299) enhances anti-neuroblastoma activity of the anti-gd2 antibody dinutuximab (ch14.18) with natural killer cells. Clin. Cancer Res. 2017, 23, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; El-Jawhari, J.J.; Neilson, A.L.; Hall, G.D.; Melcher, A.A.; Meade, J.L.; Cook, G.P. Human tumour immune evasion via tgf-beta blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS ONE 2011, 6, e22842. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, J.; Sheard, M.A.; Tran, H.C.; Wan, Z.; Liu, W.Y.; Asgharzadeh, S.; Sposto, R.; Wu, H.W.; Seeger, R.C. Lenalidomide overcomes suppression of human natural killer cell anti-tumor functions by neuroblastoma microenvironment-associated il-6 and tgfbeta1. Cancer Immunol. Immunother CII 2013, 62, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of nkp30 and nkg2d receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.P.; Choi, S.C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human nkg2d-dap10 cell surface expression: Opposing roles of the gammac cytokines and tgf-beta1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Jochems, C.; Tritsch, S.R.; Wong, H.C.; Schlom, J.; Hodge, J.W. An il-15 superagonist/il-15ralpha fusion complex protects and rescues NK cell-cytotoxic function from tgf-beta1-mediated immunosuppression. Cancer Immunol. Immunother CII 2018, 67, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting tgf-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in aml and colon cancer models. PLoS ONE 2018, 13, e0191358. [Google Scholar]
- Allan, D.S.; Rybalov, B.; Awong, G.; Zuniga-Pflucker, J.C.; Kopcow, H.D.; Carlyle, J.R.; Strominger, J.L. Tgf-beta affects development and differentiation of human natural killer cell subsets. Eur. J. Immunol. 2010, 40, 2289–2295. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. Smad4 impedes the conversion of NK cells into ilc1-like cells by curtailing non-canonical tgf-beta signaling. Nat. Immunol. 2017, 18, 995–1003. [Google Scholar] [PubMed]
- Cortez, V.S.; Cervantes-Barragan, L.; Robinette, M.L.; Bando, J.K.; Wang, Y.; Geiger, T.L.; Gilfillan, S.; Fuchs, A.; Vivier, E.; Sun, J.C.; et al. Transforming growth factor-beta signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity 2016, 44, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Hemmers, S.; Firth, M.A.; Le Floc’h, A.; Huse, M.; Sun, J.C.; Rudensky, A.Y. Il-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory t cells. J. Exp. Med. 2013, 210, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- McKarns, S.C.; Schwartz, R.H. Distinct effects of tgf-beta 1 on cd4+ and cd8+ t cell survival, division, and il-2 production: A role for t cell intrinsic smad3. J. Immunol. 2005, 174, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Dahmani, A.; Delisle, J.S. Tgf-beta in t cell biology: Implications for cancer immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Gwalani, L.A.; Orange, J.S. Single degranulations in NK cells can mediate target cell killing. J. Immunol. 2018, 200, 3231–3243. [Google Scholar] [CrossRef] [PubMed]
- Samten, B.; Townsend, J.C.; Weis, S.E.; Bhoumik, A.; Klucar, P.; Shams, H.; Barnes, P.F. Creb, atf, and ap-1 transcription factors regulate ifn-gamma secretion by human t cells in response to mycobacterial antigen. J. Immunol. 2008, 181, 2056–2064. [Google Scholar] [CrossRef] [PubMed]
- Penix, L.A.; Sweetser, M.T.; Weaver, W.M.; Hoeffler, J.P.; Kerppola, T.K.; Wilson, C.B. The proximal regulatory element of the interferon-gamma promoter mediates selective expression in t cells. J. Biol. Chem. 1996, 271, 31964–31972. [Google Scholar] [CrossRef] [PubMed]
- Lougaris, V.; Patrizi, O.; Baronio, M.; Tabellini, G.; Tampella, G.; Damiati, E.; Frede, N.; van der Meer, J.W.M.; Fliegauf, M.; Grimbacher, B.; et al. Nfkb1 regulates human NK cell maturation and effector functions. Clin. Immunol. 2017, 175, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Krantz, S.B. Interferon gamma induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood 1999, 93, 3309–3316. [Google Scholar] [PubMed]
- Inaba, H.; Glibetic, M.; Buck, S.; Ravindranath, Y.; Kaplan, J. Interferon-gamma sensitizes osteosarcoma cells to fas-induced apoptosis by up-regulating fas receptors and caspase-8. Pediatr. Blood Cancer 2004, 43, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Meister, N.; Shalaby, T.; von Bueren, A.O.; Rivera, P.; Patti, R.; Oehler, C.; Pruschy, M.; Grotzer, M.A. Interferon-gamma mediated up-regulation of caspase-8 sensitizes medulloblastoma cells to radio- and chemotherapy. Eur. J. Cancer 2007, 43, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, Q.; Peng, H.; Cheng, R.; Sun, Z.; Ye, Z. Ifn-gamma enhances hos and u2os cell lines susceptibility to gammadelta t cell-mediated killing through the fas/fas ligand pathway. Int. Immunopharmacol. 2011, 11, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jaw, J.J.; Stutzman, N.C.; Zou, Z.; Sun, P.D. Natural killer cell-produced ifn-gamma and tnf-alpha induce target cell cytolysis through up-regulation of icam-1. J. Leukoc. Biol. 2012, 91, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (trail) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Screpanti, V.; Wallin, R.P.; Grandien, A.; Ljunggren, H.G. Impact of fasl-induced apoptosis in the elimination of tumor cells by NK cells. Mol. Immunol. 2005, 42, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Screpanti, V.; Wallin, R.P.; Ljunggren, H.G.; Grandien, A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J. Immunol. 2001, 167, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Somanchi, S.S.; Senyukov, V.V.; Denman, C.J.; Lee, D.A. Expansion, purification, and functional assessment of human peripheral blood NK cells. J. Vis. Exp. 2011, 48, e2540. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. Clustvis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foltz, J.A.; Moseman, J.E.; Thakkar, A.; Chakravarti, N.; Lee, D.A. TGFβ Imprinting During Activation Promotes Natural Killer Cell Cytokine Hypersecretion. Cancers 2018, 10, 423. https://doi.org/10.3390/cancers10110423
Foltz JA, Moseman JE, Thakkar A, Chakravarti N, Lee DA. TGFβ Imprinting During Activation Promotes Natural Killer Cell Cytokine Hypersecretion. Cancers. 2018; 10(11):423. https://doi.org/10.3390/cancers10110423
Chicago/Turabian StyleFoltz, Jennifer A., Jena E. Moseman, Aarohi Thakkar, Nitin Chakravarti, and Dean A. Lee. 2018. "TGFβ Imprinting During Activation Promotes Natural Killer Cell Cytokine Hypersecretion" Cancers 10, no. 11: 423. https://doi.org/10.3390/cancers10110423
APA StyleFoltz, J. A., Moseman, J. E., Thakkar, A., Chakravarti, N., & Lee, D. A. (2018). TGFβ Imprinting During Activation Promotes Natural Killer Cell Cytokine Hypersecretion. Cancers, 10(11), 423. https://doi.org/10.3390/cancers10110423