Organoids Provide an Important Window on Inflammation in Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Organoid Technique
3. The Benefits of Using Organoids to Study Inflammation and Cancer
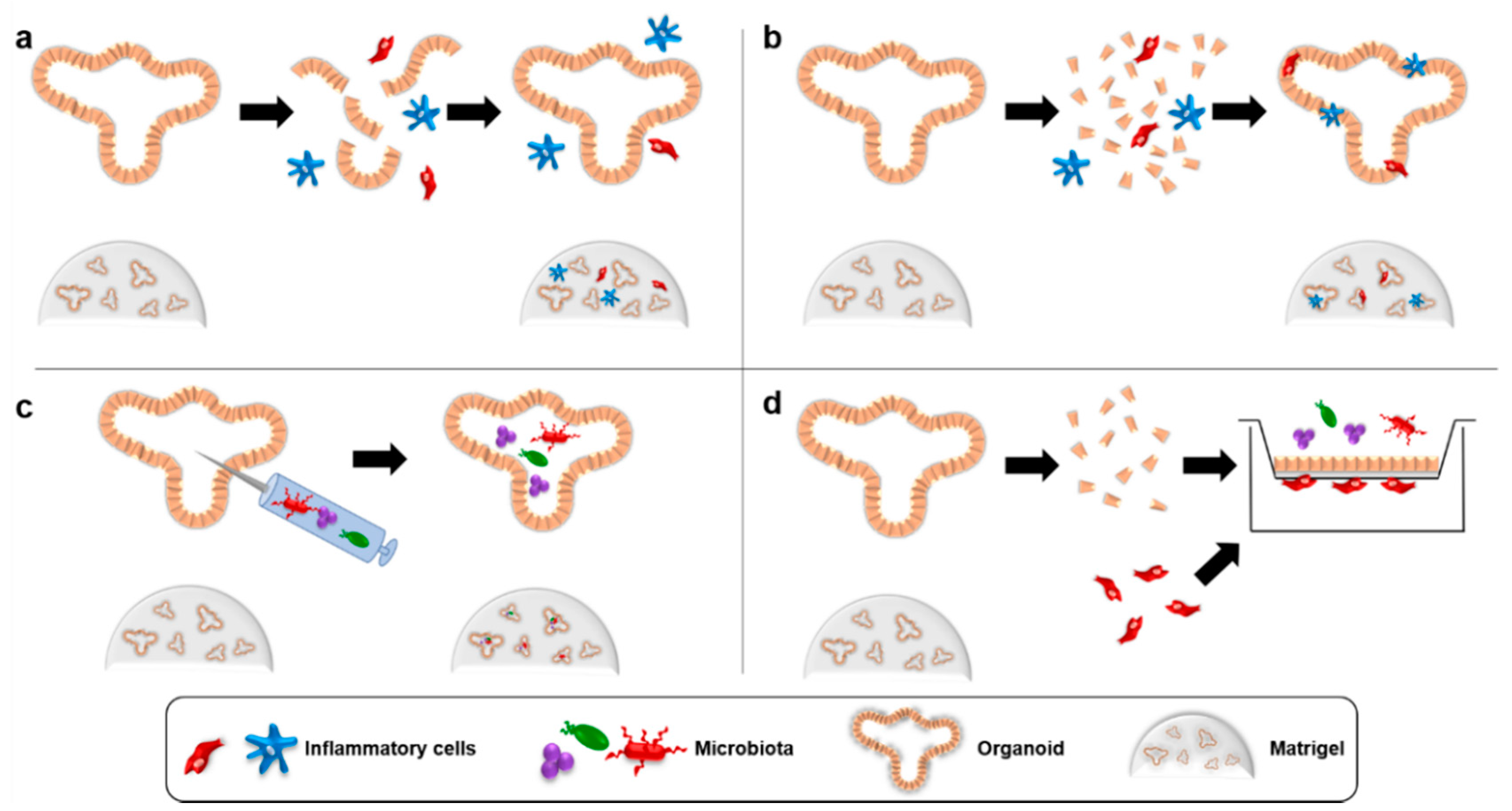
3.1. Organoids Can Be Cocultured with Inflammatory Cells
3.2. Organoids Enable Study of the Cancer-Initiating Mechanisms in Chronic Inflammatory Diseases
3.3. Organoids Can Shed Light on the Interaction between Inflammation and Cancer Stem Cells
4. Current Applications of Organoid Technology
4.1. Current Organoid Technology
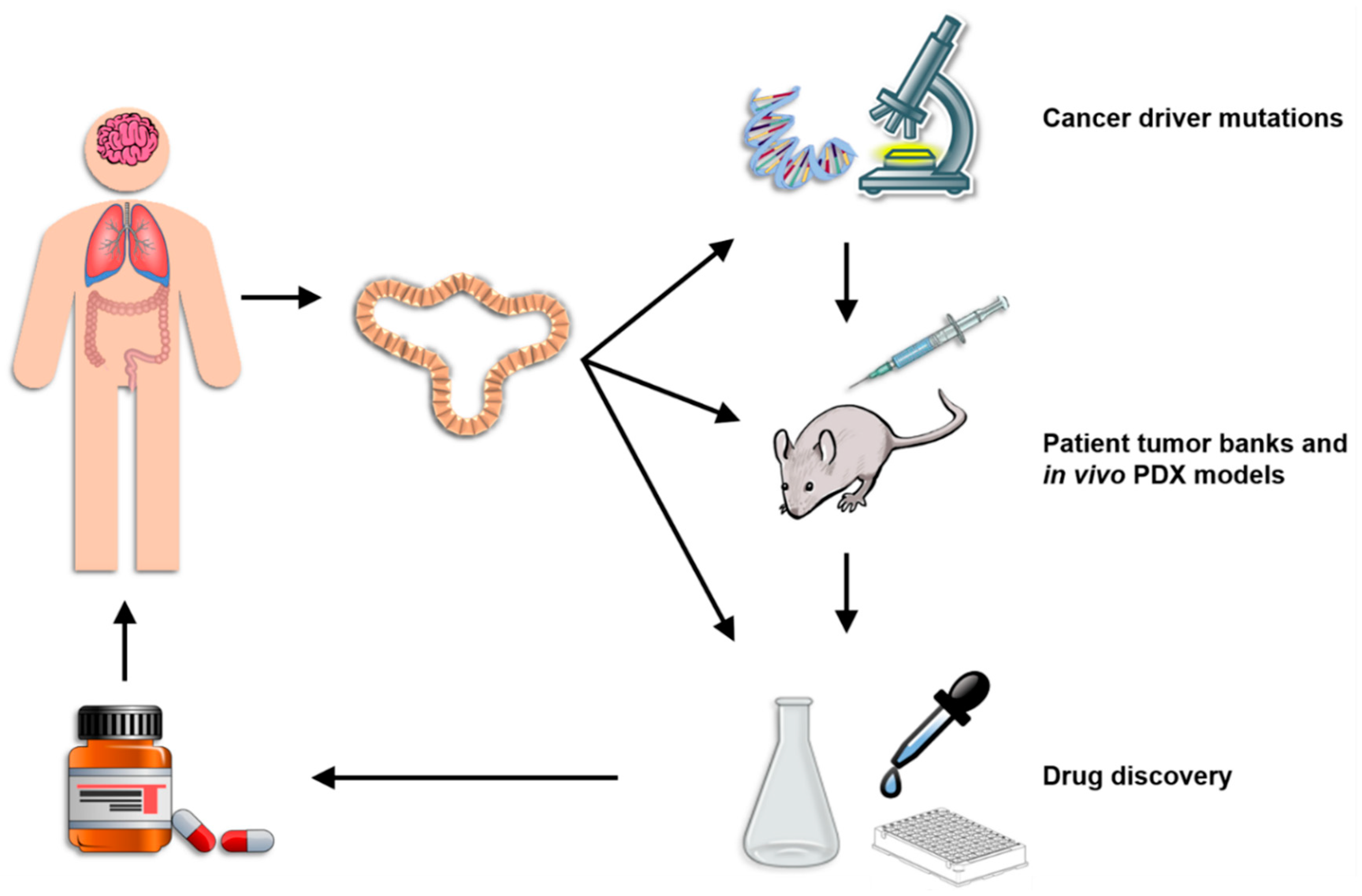
4.2. Identifying the Function and Immune Consequences of Driver Mutations
4.3. Patient Tumor Banks and In Vivo PDX Models
4.4. Platform for Drug Discovery
5. The Future of 3D Culture
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, inflammation and cancer. Nat. Immunol. 2017, 18, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Man, S.M.; Kanneganti, T.-D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Pitmon, E.; Wang, K. Microbiome, inflammation and colorectal cancer. Semin. Immunol. 2017, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Tang, Y.; Hua, S. Immunological Approaches towards Cancer and Inflammation: A Cross Talk. Front. Immunol. 2018, 9, 563. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Simian, M.; Bissell, M.J. Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Aboulkheyr, E.H.; Montazeri, L.; Aref, A.R.; Vosough, M.; Baharvand, H. Personalized Cancer Medicine: An Organoid Approach. Trends Biotechnol. 2018, 36, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; van Es, J.H.; van den Brink, S.; van Houdt, W.J.; Pronk, A.; van Gorp, J.; Siersema, P.D.; et al. Long-term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering Stem Cell Organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Lancaster, M.A.; Castanon, R.; Nery, J.R.; Knoblich, J.A.; Ecker, J.R. Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain. Cell Rep. 2016, 17, 3369–3384. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, S.L.; Ilieva, M.; Maria Michel, T. Epigenetics and cerebral organoids: Promising directions in autism spectrum disorders. Transl. Psychiatry 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Kraiczy, J.; Nayak, K.M.; Howell, K.J.; Ross, A.; Forbester, J.; Salvestrini, C.; Mustata, R.; Perkins, S.; Andersson-Rolf, A.; Leenen, E.; et al. DNA methylation defines regional identity of human intestinal epithelial organoids and undergoes dynamic changes during development. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.C.; Al-Jawadi, A.; Kishida, K.; Yu, S.; Hu, M.; Fritzky, L.F.; Edelblum, K.L.; Gao, N.; Ferraris, R.P. Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol. 2018, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Daillere, R.; Boneca, I.G.; Lepage, P.; Langella, P.; Chamaillard, M.; Pittet, M.J.; Ghiringhelli, F.; Trinchieri, G.; Goldszmid, R.; et al. Gut microbiome and anticancer immune response: Really hot Sh*t! Cell Death Differ. 2014, 22, 199–214. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.T.; Monteleone, I.; Fantini, M.C.; Monteleone, G. Regulation of Homeostasis and Inflammation in the Intestine. Gastroenterology 2011, 140, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Killion, J.J.; Radinsky, R.; Fidler, I.J. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998, 17, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Tammela, T.; Akkad, A.; Almeqdadi, M.; Santos, S.B.; Jacks, T.; Yilmaz, O.H. Colonoscopy-based colorectal cancer modeling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat. Protoc. 2018, 13, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Clevers, H. CRISPR/Cas9-Mediated Genome Editing of Mouse Small Intestinal Organoids. In Gastrointestinal Physiology and Diseases: Methods and Protocols; Ivanov, A.I., Ed.; Springer: New York, NY, USA, 2016; pp. 3–11. [Google Scholar]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Perianez, R.; Molina-Privado, I.; Rojo, F.; Guijarro-Munoz, I.; Alonso-Camino, V.; Zazo, S.; Compte, M.; Alvarez-Cienfuegos, A.; Cuesta, A.M.; Sanchez-Martin, D.; et al. Basement membrane-rich organoids with functional human blood vessels are permissive niches for human breast cancer metastasis. PLoS ONE 2013, 8, e72957. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Mochizuki, W.; Matsumoto, Y.; Matsumoto, T.; Fukuda, M.; Mizutani, T.; Watanabe, M.; Nakamura, T. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J. Gastroenterol. 2016, 51, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Fernando, E.H.; Dicay, M.; Stahl, M.; Gordon, M.H.; Vegso, A.; Baggio, C.; Alston, L.; Lopes, F.; Baker, K.; Hirota, S.; et al. A simple, cost-effective method for generating murine colonic 3D enteroids and 2D monolayers for studies of primary epithelial cell function. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G467–G475. [Google Scholar] [CrossRef] [PubMed]
- Koledova, Z.; Lu, P. A 3D Fibroblast-Epithelium Co-culture Model for Understanding Microenvironmental Role in Branching Morphogenesis of the Mammary Gland. In Mammary Gland Development: Methods and Protocols; Martin, F., Stein, T., Howlin, J., Eds.; Springer: New York, NY, USA, 2017; pp. 217–231. [Google Scholar]
- Hegab, A.E.; Arai, D.; Gao, J.; Kuroda, A.; Yasuda, H.; Ishii, M.; Naoki, K.; Soejima, K.; Betsuyaku, T. Mimicking the niche of lung epithelial stem cells and characterization of several effectors of their in vitro behavior. Stem Cell Res. 2015, 15, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Sekine, K.; Gerber, T.; Loeffler-Wirth, H.; Binder, H.; Gac, M.; Kanton, S.; Kageyama, J.; Damm, G.; Seehofer, D.; et al. Multilineage communication regulates human liver bud development from pluripotency. Nature 2017, 546, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Pastula, A.; Middelhoff, M.; Brandtner, A.; Tobiasch, M.; Hohl, B.; Nuber, A.H.; Demir, I.E.; Neupert, S.; Kollmann, P.; Mazzuoli-Weber, G.; et al. Three-Dimensional Gastrointestinal Organoid Culture in Combination with Nerves or Fibroblasts: A Method to Characterize the Gastrointestinal Stem Cell Niche. Stem Cells Int. 2016, 2016, 3710836. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Noel, G.; Baetz, N.W.; Staab, J.F.; Donowitz, M.; Kovbasnjuk, O.; Pasetti, M.F.; Zachos, N.C. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci. Rep. 2017, 7, 45270. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.R.; Yu, Q. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ihara, S.; Hayakawa, Y.; Konishi, M.; Hirata, Y.; Koike, K. 3d Co-Culture System of Intestinal Organoids and Dendritic Cells to Study Epithelial Differentiation. Gastroenterology 2017, 152, S13–S135. [Google Scholar] [CrossRef]
- Rogoz, A.; Reis, B.S.; Karssemeijer, R.A.; Mucida, D. A 3-D enteroid-based model to study T-cell and epithelial cell interaction. J. Immunol. Methods 2015, 421, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sato, S.; Kurashima, Y.; Lai, C.-Y.; Otsu, M.; Hayashi, M.; Yamaguchi, T.; Kiyono, H. Reciprocal Inflammatory Signaling Between Intestinal Epithelial Cells and Adipocytes in the Absence of Immune Cells. EBioMedicine 2017, 23, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Zitvogel, L. The breakthrough of the microbiota. Nat. Rev. Immunol. 2018, 18, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Oke, S.; Martin, A. Insights into the role of the intestinal microbiota in colon cancer. Therap. Adv. Gastroenterol. 2017, 10, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Scanu, T.; Spaapen, R.M.; Bakker, J.M.; Pratap, C.B.; Wu, L.E.; Hofland, I.; Broeks, A.; Shukla, V.K.; Kumar, M.; Janssen, H.; et al. Salmonella Manipulation of Host Signaling Pathways Provokes Cellular Transformation Associated with Gallbladder Carcinoma. Cell Host Microbe 2015, 17, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.; Peek, R.M. Pathobiology of Helicobacter pylori-induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S.; Bayram, T.; van de Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In Vitro Expansion of Human Gastric Epithelial Stem Cells and Their Responses to Bacterial Infection. Gastroenterology 2015, 148, 126–136.e126. [Google Scholar] [CrossRef] [PubMed]
- McCracken, K.W.; Catá, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.-H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.L.; Huang, S.; Opp, J.S.; Nagy, M.S.; Kobayashi, M.; Young, V.B.; Spence, J.R. Persistence and Toxin Production by Clostridium difficile within Human Intestinal Organoids Result in Disruption of Epithelial Paracellular Barrier Function. Infect. Immun. 2015, 83, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kotani, T.; Konno, T.; Setiawan, J.; Kitamura, Y.; Imada, S.; Usui, Y.; Hatano, N.; Shinohara, M.; Saito, Y.; et al. Promotion of Intestinal Epithelial Cell Turnover by Commensal Bacteria: Role of Short-Chain Fatty Acids. PLoS ONE 2016, 11, e0156334. [Google Scholar] [CrossRef] [PubMed]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 2014, 5, e01438-14. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Hemann, M.T. Drugs, Bugs, and Cancer: Fusobacterium nucleatum Promotes Chemoresistance in Colorectal Cancer. Cell 2017, 170, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal Bacteria Control Cancer Response to Therapy by Modulating the Tumor Microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2017, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, D.; Chen, Y. The potential of organoids in urological cancer research. Nat. Rev. Urol. 2017, 14, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling pancreatic cancer with organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 217–226, 229; discussion 212–230. [Google Scholar] [PubMed]
- Lee, Y.; Urbanska, A.M.; Hayakawa, Y.; Wang, H.; Au, A.S.; Luna, A.M.; Chang, W.; Jin, G.; Bhagat, G.; Abrams, J.A.; et al. Gastrin stimulates a cholecystokinin-2-receptor-expressing cardia progenitor cell and promotes progression of Barrett’s-like esophagus. Oncotarget 2017, 8, 203–214. [Google Scholar] [PubMed]
- Mazza, G.; Al-Akkad, W.; Rombouts, K. Engineering in vitro models of hepatofibrogenesis. Adv. Drug Deliv. Rev. 2017, 121, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Munera, J.O.; Wells, J.M. Stem Cells and Organoids to Study Epithelial Cell Biology in IBD. In Crohn’s Disease and Ulcerative Colitis: From Epidemiology and Immunobiology to a Rational Diagnostic and Therapeutic Approach; Baumgart, D.C., Ed.; Springer: Cham, Switzerland, 2017; pp. 167–172. [Google Scholar]
- Gunther, C.; Buchen, B.; He, G.W.; Hornef, M.; Torow, N.; Neumann, H.; Wittkopf, N.; Martini, E.; Basic, M.; Bleich, A.; et al. Caspase-8 controls the gut response to microbial challenges by TNF-α-dependent and independent pathways. Gut 2015, 64, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, A.; Lopes, F.; Poon, K.K.H.; McKay, D.M. Absence of the NOD2 protein renders epithelia more susceptible to barrier dysfunction due to mitochondrial dysfunction. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G26–G38. [Google Scholar] [CrossRef] [PubMed]
- Chiriac, M.T.; Buchen, B.; Wandersee, A.; Hundorfean, G.; Günther, C.; Bourjau, Y.; Doyle, S.E.; Frey, B.; Ekici, A.B.; Büttner, C.; et al. Activation of Epithelial Signal Transducer and Activator of Transcription 1 by Interleukin 28 Controls Mucosal Healing in Mice With Colitis and Is Increased in Mucosa of Patients With Inflammatory Bowel Disease. Gastroenterology 2017, 153, 123–138.e128. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Nakayama, M.; Han, T.-S.; Naoi, K.; Ju, X.; Maeda, Y.; Robine, S.; Tsuchiya, K.; Sato, T.; Sato, H.; et al. Suppressing TGFβ Signaling in Regenerating Epithelia in an Inflammatory Microenvironment Is Sufficient to Cause Invasive Intestinal Cancer. Cancer Res. 2015, 75, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Kabat, A.M.; Maloy, K.J. Intestinal Epithelial Cell Autophagy Is Required to Protect against TNF-Induced Apoptosis during Chronic Colitis in Mice. Cell Host Microbe 2018, 23, 191–202.e194. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single LGR5 stem cells build Crypt–villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.-Z.; Zheng, Y.-W.; Ogawa, M.; Miyagi, E.; Taniguchi, H. Human liver organoids generated with single donor-derived multiple cells rescue mice from acute liver failure. Stem Cell Res. Ther. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Li, Y.; Bhattacharya, S.; O’Connor, M.; Pu, C.; Lin, J.; Wang, T.; Xiang, D.; Kong, L.; Wei, M.Q.; et al. Inflammation and cancer stem cells. Cancer Lett. 2014, 345, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.; Farabaugh, S.; Ford, H. Epithelial-Mesenchymal Transition in Cancer: Parallels between Normal Development and Tumor Progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Zhang, M.; Chu, Q.; Gan, Y.; Ren, H.; Zhang, L.; Wang, L.; Li, X.; Wang, W. The Role of p-STAT3 as a Prognostic and Clinicopathological Marker in Colorectal Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0160125. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, Y.; Shi, Q.; Yu, Q.; Liu, C.; Feng, J.; Deng, J.; Evers, B.M.; Zhou, B.P.; Wu, Y. Stabilization of the transcription factors slug and twist by the deubiquitinase DUB3 is a key requirement for tumor metastasis. Oncotarget 2017, 8, 75127–75140. [Google Scholar] [CrossRef] [PubMed]
- Sakaki-Yumoto, M.; Katsuno, Y.; Derynck, R. TGF-β family signaling in stem cells. Biochim. Biophys. Acta 2013, 1830, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Liu, J.; Tang, Y.; Liang, X. Inflammation linking EMT and cancer stem cells. Oral Oncol. 2012, 48, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Bartels, K.; Grenz, A.; Eltzschig, H.K. Hypoxia and inflammation are two sides of the same coin. Proc. Natl. Acad. Sci. USA 2013, 110, 18351–18352. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Zhao, E.; Liu, Y.; Wang, Y.; Vatan, L.; Szeliga, W.; Moyer, J.; Klimczak, A.; Lange, A.; Zou, W. Human TH17 cells are long-lived effector memory cells. Sci. Transl. Med. 2011, 3, 104ra100. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Wen, H.; Corsa, C.A.S.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Sotomayor, E.M.; Seto, E. Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene 2009, 29, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Bayarsaihan, D. Epigenetic mechanisms in inflammation. J. Dent. Res. 2011, 90, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, S.; Wu, N.; Cho, K.-S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Pandey, R.; Byun, J.S.; Gardner, K.; Qin, Z.; Dou, Y. The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell 2012, 11, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Bender, S.; Raddatz, G.; Ansari, I.; Cohen, D.; Gutekunst, J.; Musch, T.; Linhart, H.; Breiling, A.; Pikarsky, E.; et al. Chronic Inflammation Induces a Novel Epigenetic Program That Is Conserved in Intestinal Adenomas and in Colorectal Cancer. Cancer Res. 2015, 75, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Orihashi, K.; Hippo, Y. Lentivirus-Based Stable Gene Delivery into Intestinal Organoids. In Gastrointestinal Physiology and Diseases: Methods and Protocols; Ivanov, A.I., Ed.; Springer: New York, NY, USA, 2016; pp. 13–21. [Google Scholar]
- Nadauld, L.D.; Garcia, S.; Natsoulis, G.; Bell, J.M.; Miotke, L.; Hopmans, E.S.; Xu, H.; Pai, R.K.; Palm, C.; Regan, J.F.; et al. Metastatic tumor evolution and organoid modeling implicate TGFBR2 as a cancer driver in diffuse gastric cancer. Genome Biol. 2014, 15, 428. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Clevers, H. Imaging organoids: A bright future ahead. Nat. Methods 2018, 15, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [PubMed]
- Boehnke, K.; Iversen, P.W.; Schumacher, D.; Lallena, M.J.; Haro, R.; Amat, J.; Haybaeck, J.; Liebs, S.; Lange, M.; Schafer, R.; et al. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. J. Biomol. Screen 2016, 21, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2017, 172, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ootani, A.; Kuo, C. An Air–Liquid Interface Culture System for 3D Organoid Culture of Diverse Primary Gastrointestinal Tissues. In Gastrointestinal Physiology and Diseases: Methods and Protocols; Ivanov, A.I., Ed.; Springer: New York, NY, USA, 2016; pp. 33–40. [Google Scholar]
- Zhang, Z.; Christin, J.R.; Wang, C.; Ge, K.; Oktay, M.H.; Guo, W. Mammary Stem Cell Based Somatic Mouse Models Reveal Breast Cancer Drivers Causing Cell Fate Dysregulation. Cell Rep. 2016, 16, 3146–3156. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Fennemann, F.L.; Weren, R.D.A.; Bisseling, T.M.; Ligtenberg, M.J.L.; Figdor, C.G.; Schreibelt, G.; Hoogerbrugge, N.; Wimmers, F.; de Vries, I.J.M. Opportunities for immunotherapy in microsatellite instable colorectal cancer. Cancer Immunol. Immunother. 2016, 65, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; de Visser, K.E. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol. 2015, 36, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Godzik, A. Mutation Drivers of Immunological Responses to Cancer. Cancer Immunol. Res. 2016, 4, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology 2011, 25, 400–410, 413. [Google Scholar] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/γ(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Nervi, B.; Rettig, M.P.; Ritchey, J.K.; Wang, H.L.; Bauer, G.; Walker, J.; Bonyhadi, M.L.; Berenson, R.J.; Prior, J.L.; Piwnica-Worms, D.; et al. Factors affecting human T cell engraftment, trafficking, and associated xenogeneic graft-vs-host disease in NOD/SCID beta2mnull mice. Exp. Hematol. 2007, 35, 1823–1838. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Smietana, K.; Siatkowski, M.; Møller, M. Trends in clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2018. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Stein, W.D.; Litman, T.; Fojo, T.; Bates, S.E. A Serial Analysis of Gene Expression (SAGE) database analysis of chemosensitivity: Comparing solid tumors with cell lines and comparing solid tumors from different tissue origins. Cancer Res. 2004, 64, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; Schneider, M.A.; Meister, M.; et al. Screening drug effects in patient-derived cancer cells links organoid responses to genome alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer–derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Smadar, C.; Dvir-Ginzberg, M. Recent patents in organoids. Nat. Biotechnol. 2016, 34, 619. [Google Scholar]
- Sinha, G. The organoid architect. Science 2017, 357, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Boussommier-Calleja, A.; Li, R.; Chen, M.B.; Wong, S.C.; Kamm, R.D. Microfluidics: A new tool for modeling cancer-immune interactions. Trends Cancer 2016, 2, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Morales, R.-T.T.; Qian, W.; Wang, H.; Gagner, J.-P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Agliari, E.; Biselli, E.; De Ninno, A.; Schiavoni, G.; Gabriele, L.; Gerardino, A.; Mattei, F.; Barra, A.; Businaro, L. Cancer-driven dynamics of immune cells in a microfluidic environment. Sci. Rep. 2014, 4, 6639. [Google Scholar] [CrossRef] [PubMed]
- Businaro, L.; De Ninno, A.; Schiavoni, G.; Lucarini, V.; Ciasca, G.; Gerardino, A.; Belardelli, F.; Gabriele, L.; Mattei, F. Cross talk between cancer and immune cells: Exploring complex dynamics in a microfluidic environment. Lab Chip 2013, 13, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Bhutia, S.K.; Nagrath, S.; Bitting, R.L.; Deep, G. Circulating tumor cell-derived organoids: Current challenges and promises in medical research and precision medicine. Biochim. Biophys. Acta 2018, 1869, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, V.R.; Schumacher, T.N.M.; Busch, D.H. T Cell Fate at the Single-Cell Level. Annu. Rev. Immunol. 2016, 34, 65–92. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baker, K. Organoids Provide an Important Window on Inflammation in Cancer. Cancers 2018, 10, 151. https://doi.org/10.3390/cancers10050151
Baker K. Organoids Provide an Important Window on Inflammation in Cancer. Cancers. 2018; 10(5):151. https://doi.org/10.3390/cancers10050151
Chicago/Turabian StyleBaker, Kristi. 2018. "Organoids Provide an Important Window on Inflammation in Cancer" Cancers 10, no. 5: 151. https://doi.org/10.3390/cancers10050151
APA StyleBaker, K. (2018). Organoids Provide an Important Window on Inflammation in Cancer. Cancers, 10(5), 151. https://doi.org/10.3390/cancers10050151