The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia


Abstract
:1. Introduction
2. Results
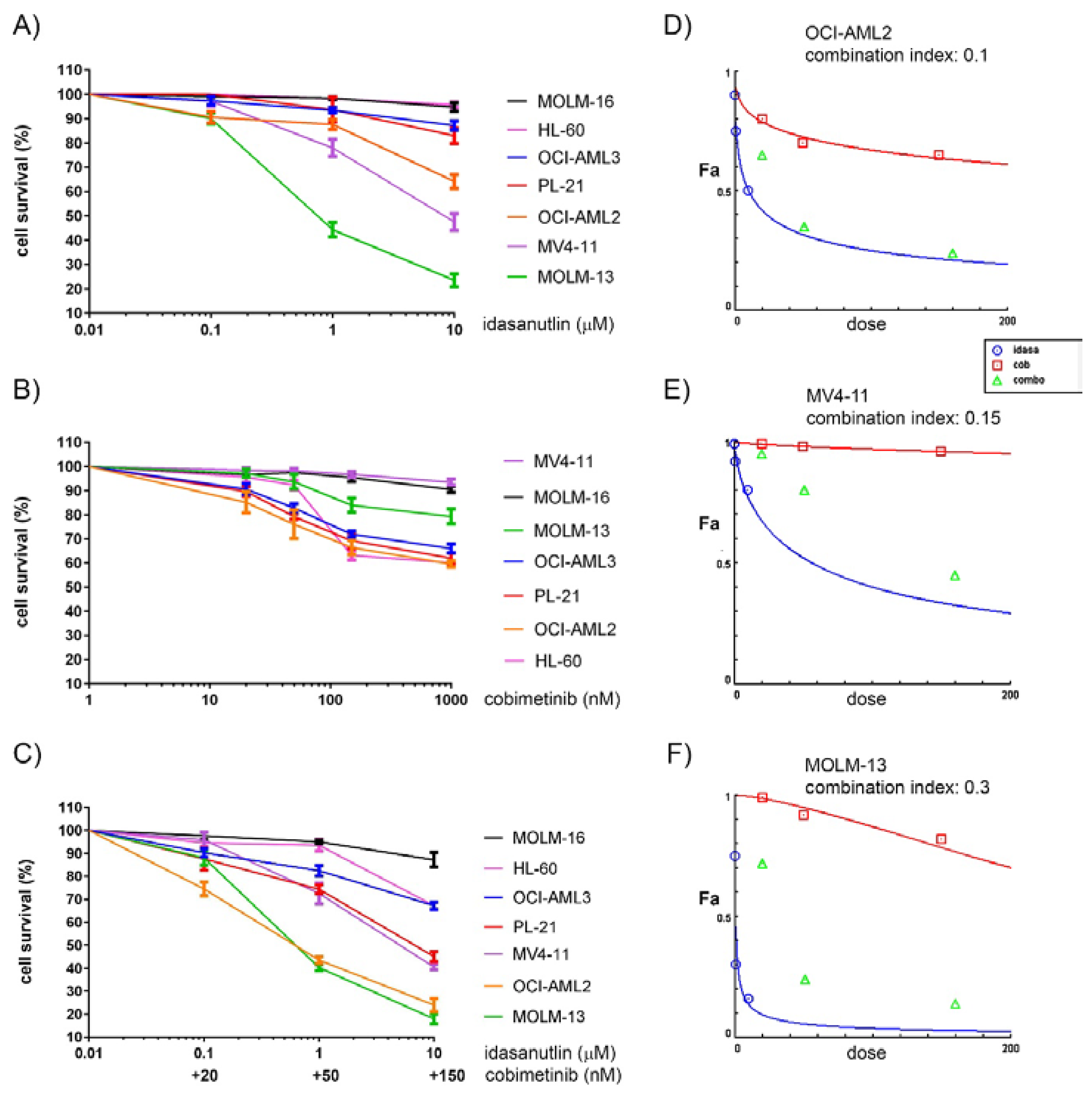
2.1. Synergistic Effects on Cell Viability in AML Cell Lines Treated with the MDM2 Inhibitor Idasanutlin and the MEK Inhibitor Cobimetinib
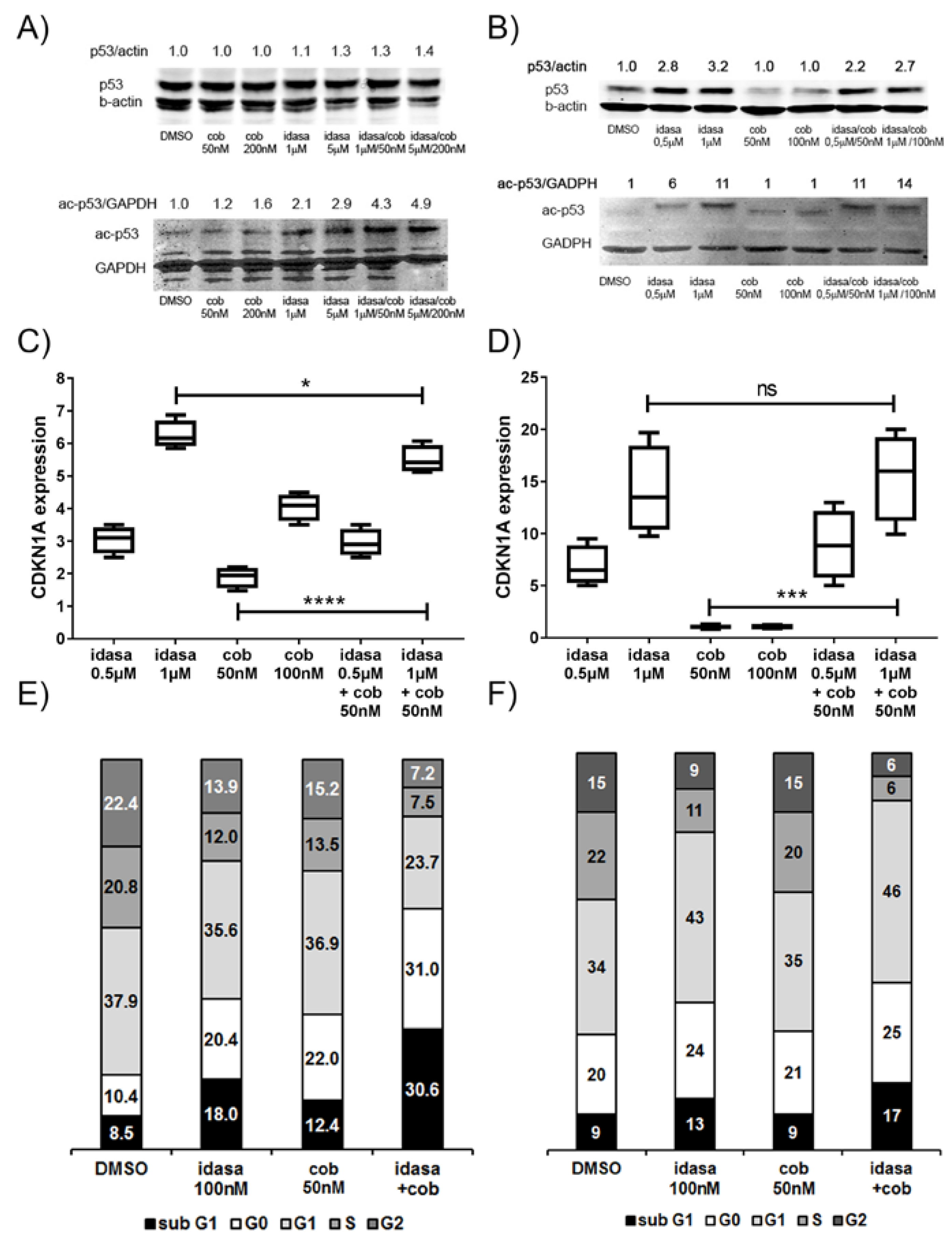
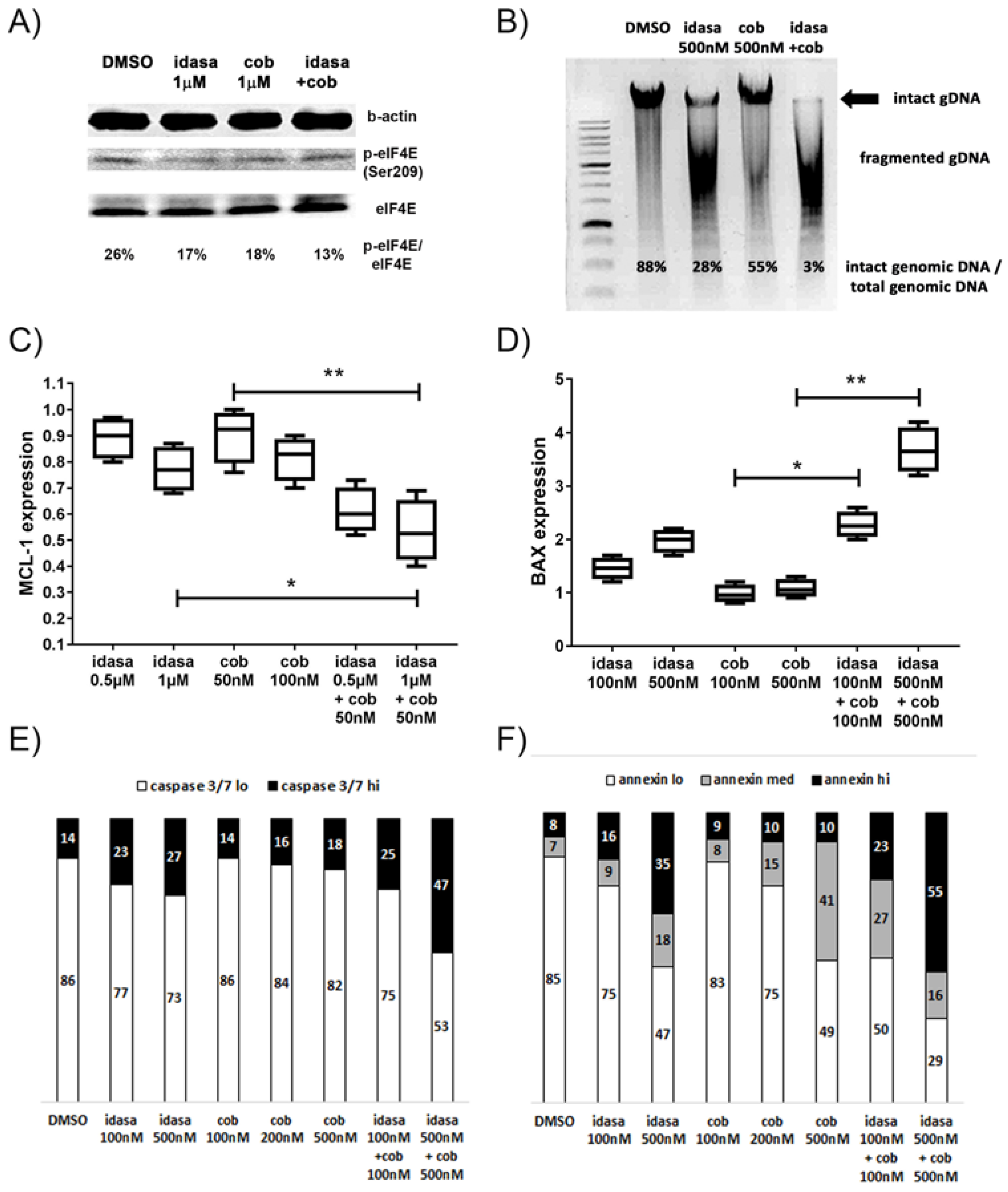
2.2. Inhibition of Cell Cycling and Induction of Apoptosis by Idasanutlin and Cobimetinib
2.3. Varying Sensitivity of AML Patient Cells to Treatment with the MDM2 Inhibitor Idasanutlin and the MEK Inhibitor Cobimetinib
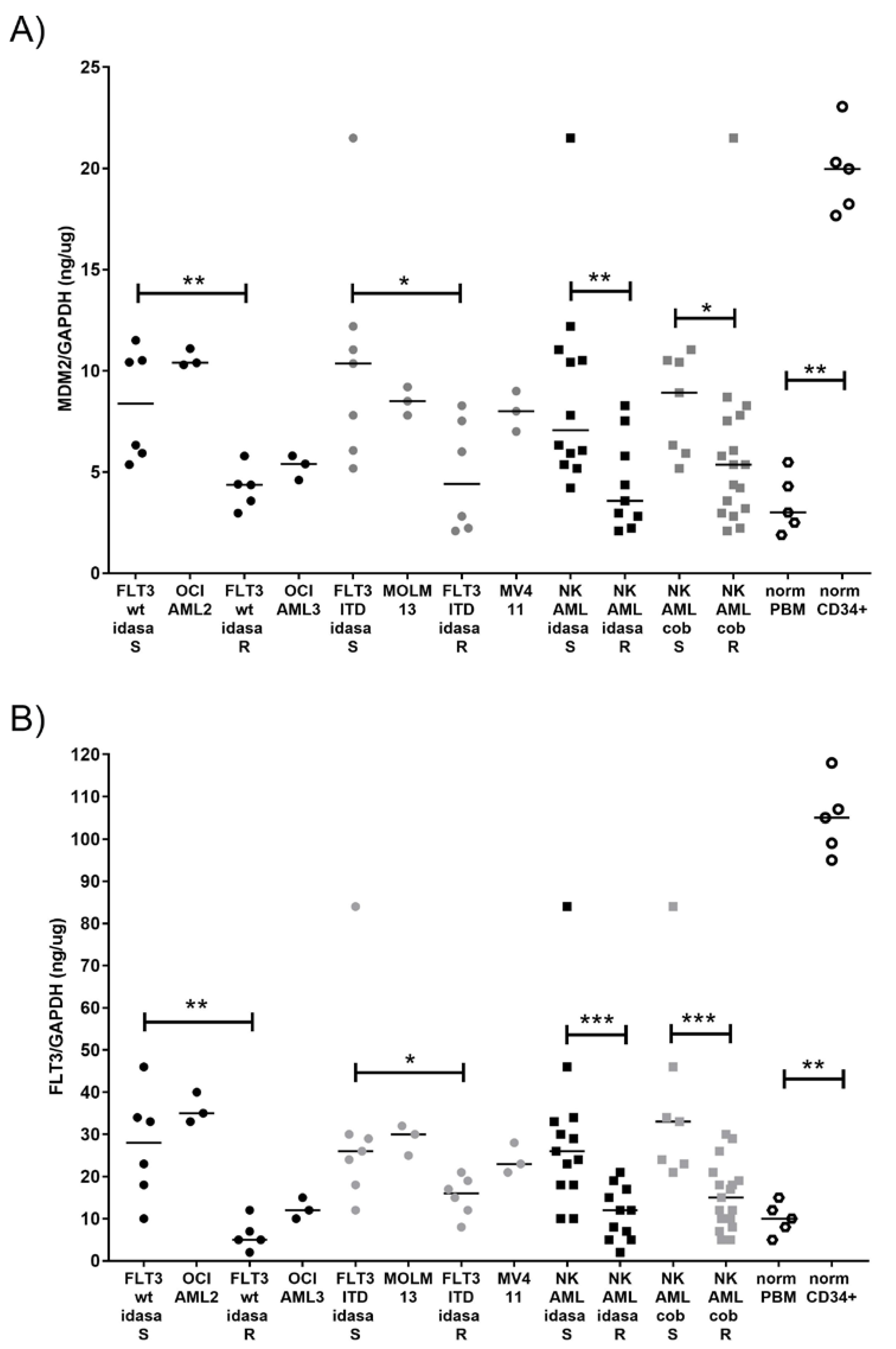
2.4. Correlation of Cellular FLT3 and MDM2 Protein Levels with Susceptibility to Idasanutlin and Cobimetinib
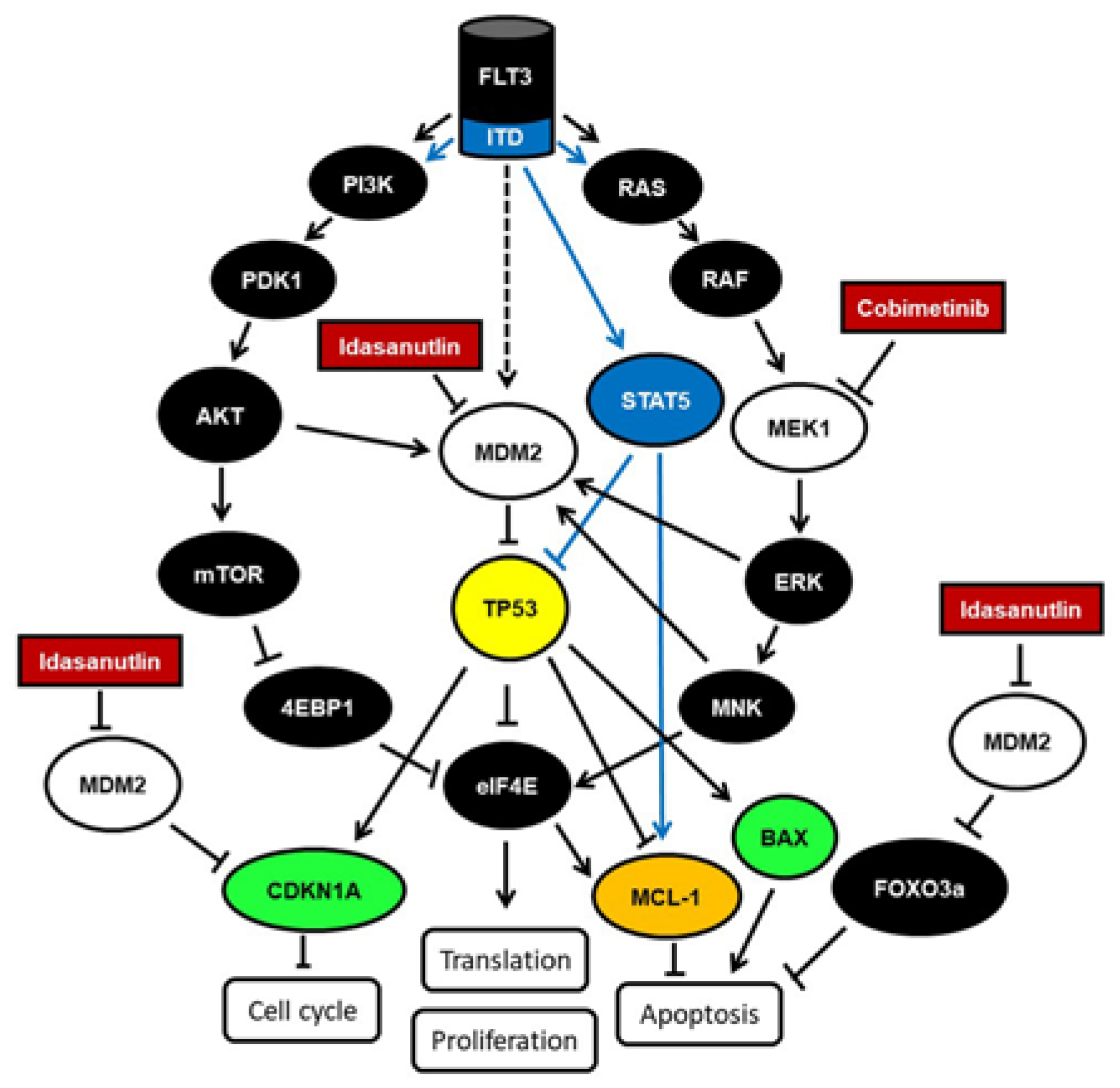
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Patient Samples
4.3. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| FLT3 | FMS like tyrosine kinase 3 |
| ITD | internal tandem duplication |
| TP53 | tumor suppressor p53 |
| MDM2 | mouse double minute 2 homolog |
| MEK | mitogen-activated protein kinase kinase |
| NPM1 | nucleophosmin |
| RAS | rat sarcoma protein |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| wt | wild type |
| mut | mutated |
| del | deleted |
| fs | frame shift |
| idasa | idasanutlin |
| cob | cobimetinib |
| NK | normal karyotype |
References
- Colombo, E.; Marine, J.-C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Xu, L.; Massagué, J. Nucleocytoplasmic shuttling of signal transducers. Nat. Rev. Mol. Cell Biol. 2004, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.Y.; Li, M.; Puskas, N.; Qin, J.; Gu, W. Parc: A cytoplasmic anchor for p53. Cell 2003, 112, 29–40. [Google Scholar] [CrossRef]
- Seipel, K.; Marques, M.T.; Bozzini, M.-A.; Meinken, C.; Mueller, B.U.; Pabst, T. Inactivation of the p53-KLF4-CEBPA Axis in Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Carow, C.E.; Levenstein, M.; Kaufmann, S.H.; Chen, J.; Amin, S.; Rockwell, P.; Witte, L.; Borowitz, M.J.; Civin, C.I.; Small, D. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood 1996, 87, 1089–1096. [Google Scholar] [PubMed]
- Swords, R.; Freeman, C.; Giles, F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia 2012, 26, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, K.; Bagrintseva, K.; Schwab, R.; Schmieja, K.; Hiddemann, W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin. Cancer Res. 2003, 9, 2140–2150. [Google Scholar] [PubMed]
- Bowen, D.T.; Frew, M.E.; Hills, R.; Gale, R.E.; Wheatley, K.; Groves, M.J.; Langabeer, S.E.; Kottaridis, P.D.; Moorman, A.V.; Burnett, A.K.; et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood 2005, 106, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Rousseau, R.F.; Middleton, S.A.; Nichols, G.L.; Newell, D.R.; Lunec, J.; Tweddle, D.A. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 2015, 6, 10207–10221. [Google Scholar] [CrossRef] [PubMed]
- Zanjirband, M.; Edmondson, R.J.; Lunec, J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget 2016, 7, 40115–40134. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Friess, T.; Birzele, F.; Kiialainen, A.; Dangl, M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J. Hematol. Oncol. 2016, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.M.; Simeone, E.; Ascierto, P.A. The role of MEK inhibitors in the treatment of metastatic melanoma. Curr. Opin. Oncol. 2014, 26, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.R.; Hwang, E.; Firestone, A.J.; Huang, T.; Xu, J.; Zuber, J.; Bohin, N.; Wen, T.; Kogan, S.C.; Haigis, K.M.; et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood 2014, 124, 3947–3955. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. Out of the jaws of death: PRMT5 steers p53. Nat. Cell Biol. 2008, 10, 1389–1390. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.; Durant, S.T.; Cho, E.-C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Parkin, B.; Ouillette, P.; Bixby, D.; Shedden, K.; Erba, H.; Wang, S.; Malek, S.N. Multiple distinct molecular mechanisms influence sensitivity and resistance to MDM2 inhibitors in adult acute myelogenous leukemia. Blood 2010, 116, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Reis, B.; Jukofsky, L.; Chen, G.; Martinelli, G.; Zhong, H.; So, W.V.; Dickinson, M.J.; Drummond, M.; Assouline, S.; Hashemyan, M.; et al. Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica 2016, 101, e185–e188. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef] [PubMed]
- Dry, J.R.; Pavey, S.; Pratilas, C.A.; Harbron, C.; Runswick, S.; Hodgson, D.; Chresta, C.; McCormack, R.; Byrne, N.; Cockerill, M.; et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res. 2010, 70, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Martinelli, P.; Zamponi, R.; Shing, D.C.; Bonetti, P.; Luzi, L.; Volorio, S.; Bernard, L.; Pruneri, G.; Alcalay, M.; et al. Delocalization and destabilization of the Arf tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 2006, 66, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.; Biederer, C.; Woods, D.; Shifman, O.; Shirasawa, S.; Sasazuki, T.; McMahon, M.; Oren, M.; McCormick, F. Opposing effects of Ras on p53: Transcriptional activation of mdm2 and induction of p19ARF. Cell 2000, 103, 321–330. [Google Scholar] [CrossRef]
- Phillips, A.; Blaydes, J.P. MNK1 and EIF4E are downstream effectors of MEKs in the regulation of the nuclear export of HDM2 mRNA. Oncogene 2007, 27, 1645–1649. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | FLT3 | TP53 | Mutated Genes |
|---|---|---|---|
| OCI-AML2 | wt/A680V | wt | DNMT3A |
| FLT3wt-S (5) | wt | wt | NPM1 (3) NRAS (1) |
| MOLM-13 | ITD | wt | MLL-AF9 mTOR |
| FLT3-ITD-S (7) | ITD | wt | NPM1 (4) |
| MV4-11 | ITD | wt | MLL-AF4 |
| FLT3-ITD-R (6) | ITD | wt | NPM1 (2) RUNX1 (2) |
| PL-21 | wt/P336L | wt/P36fs | KRAS |
| HL-60 | wt | del | NRAS |
| FLT3wt-R (5) | wt | wt | NPM1 (2) DNMT3A (1) |
| MOLM-16 | wt | mut | MLL V1368L |
| TP53mut (3) | wt | mut | PTPN11 (1) NRAS (1) |
| Clinical Characteristic | Absolute Number | Relative Number (%) | |
|---|---|---|---|
| AML | 27 | 100 | |
| FAB classification | M0 | 0 | 0 |
| M1 | 6 | 22 | |
| M2 | 7 | 26 | |
| M3 | 0 | 0 | |
| M4 | 6 | 22 | |
| M5 | 8 | 30 | |
| M6 | 0 | 0 | |
| M7 | 0 | 0 | |
| Pathogenesis | de novo | 26 | 96 |
| relapse | 1 | 4 | |
| Molecular Analysis | FLT3wt/NPM1wt | 15 | 56 |
| FLT3-ITD | 12 | 44 | |
| NPM1mut | 12 | 44 | |
| FLT3-ITD and NPM1mut | 6 | 22 | |
| Cytogenetic Analysis | normal karyotype (NK) | 20 | 74 |
| −5, −5q | 0 | 0 | |
| −7, −7q | 3 | 11 | |
| inv(16) | 1 | 4 | |
| t(8;21) | 0 | 0 | |
| t(15;17) | 0 | 0 | |
| complex karyotype (CK) | 3 | 11 | |
| Risk Category * | favorable | 6 | 22 |
| intermediate | 6 | 22 | |
| adverse | 15 | 56 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seipel, K.; Marques, M.A.T.; Sidler, C.; Mueller, B.U.; Pabst, T. The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia. Cancers 2018, 10, 170. https://doi.org/10.3390/cancers10060170
Seipel K, Marques MAT, Sidler C, Mueller BU, Pabst T. The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia. Cancers. 2018; 10(6):170. https://doi.org/10.3390/cancers10060170
Chicago/Turabian StyleSeipel, Katja, Miguel A. T. Marques, Corinne Sidler, Beatrice U. Mueller, and Thomas Pabst. 2018. "The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia" Cancers 10, no. 6: 170. https://doi.org/10.3390/cancers10060170
APA StyleSeipel, K., Marques, M. A. T., Sidler, C., Mueller, B. U., & Pabst, T. (2018). The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia. Cancers, 10(6), 170. https://doi.org/10.3390/cancers10060170