Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts


Abstract
:1. Introduction
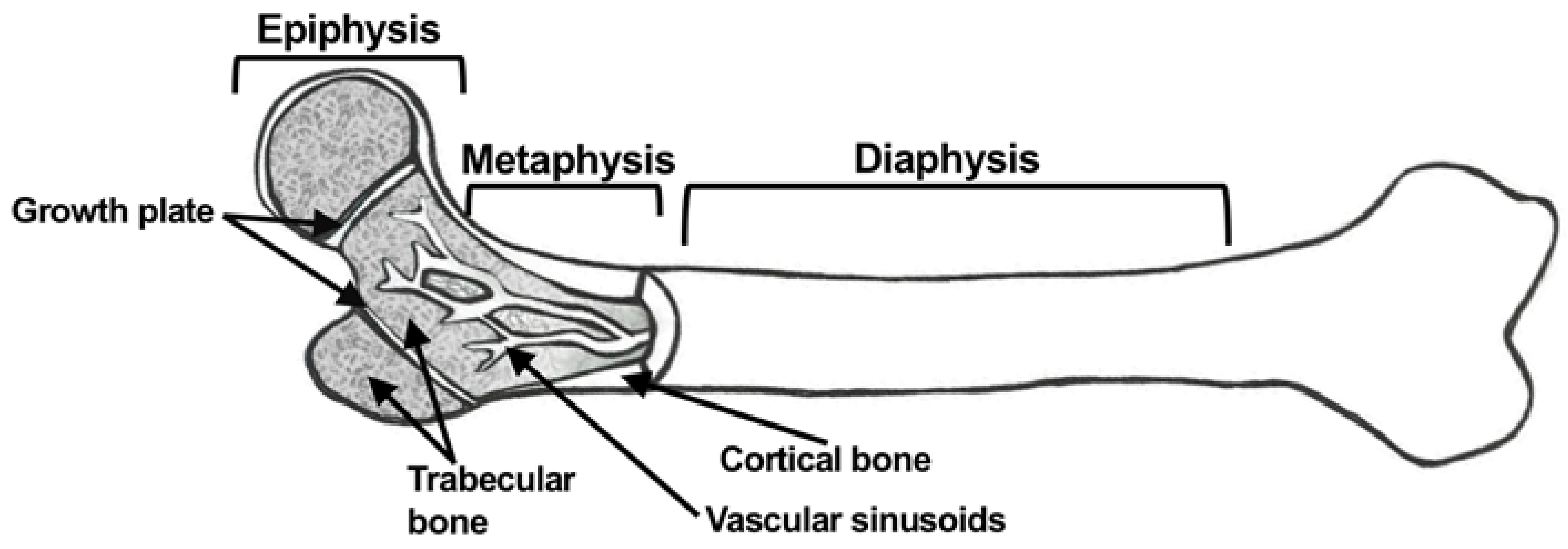
1.1. Gross Anatomy of Bone
1.2. Bone Physiology, Remodeling, and Metabolism
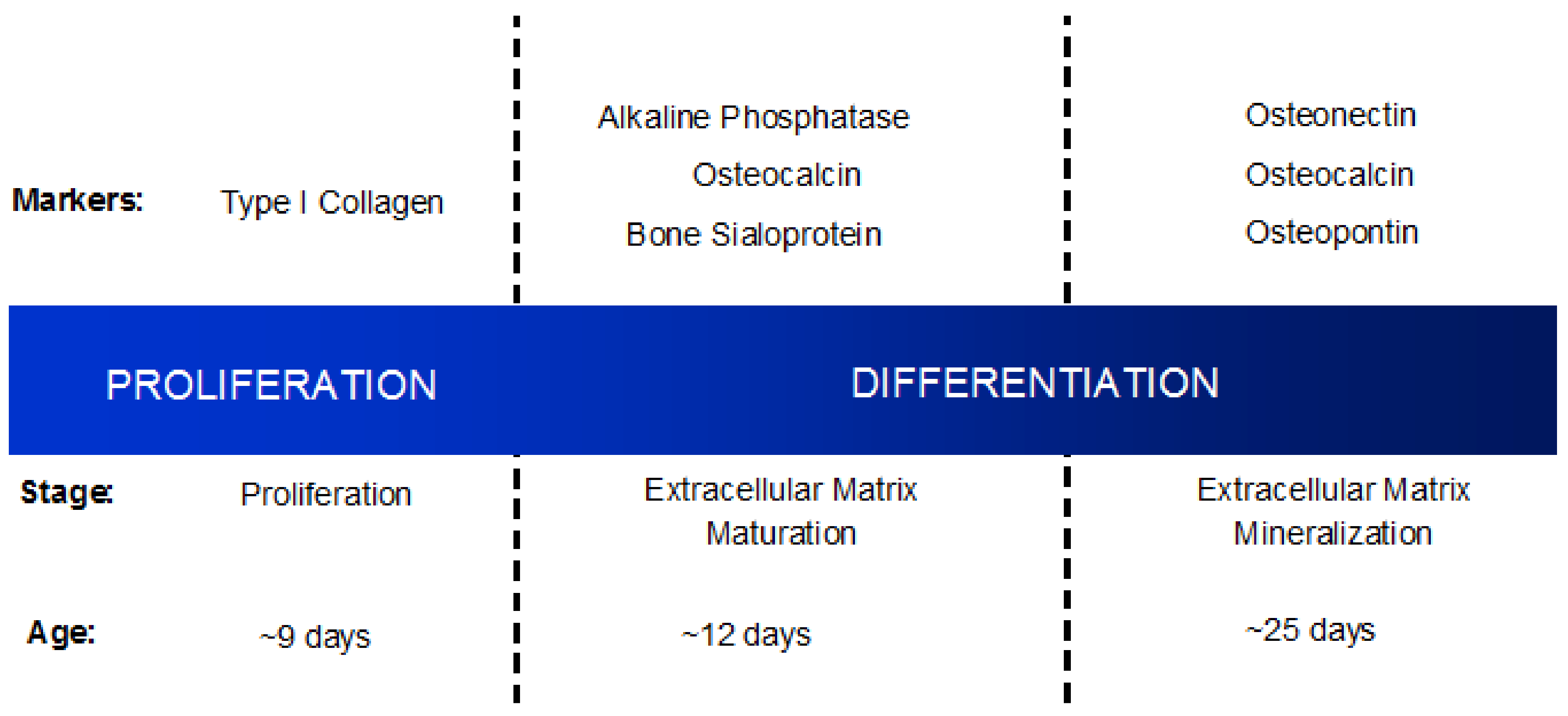
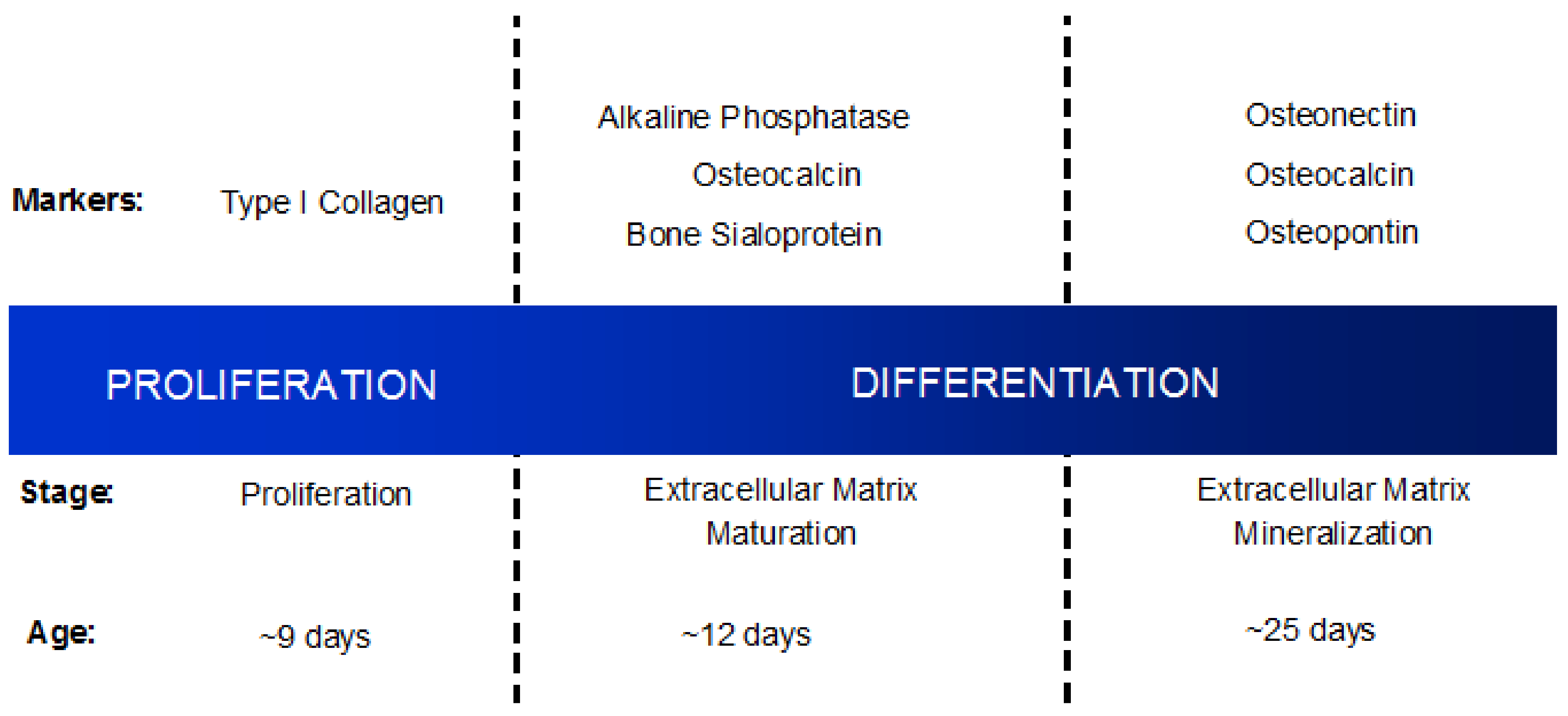
1.3. Osteoblast Differentiation
1.4. Osteoblasts Work in Concert with Osteoclasts to Regulate Bone Remodeling
2. Osteoblasts in the Bone Microenvironment as Contributors to Bone Disease and Degradation
3. Bone Is a Favored Site for Cancer Cell Metastasis
3.1. Breast Cancer Metastases to Bone
3.2. Multiple Myeloma Colonization of the Skeleton
3.3. Prostate Cancer Metastases to Bone
4. Osteoblasts as Mediators in Cancer Cell Dormancy in Bone
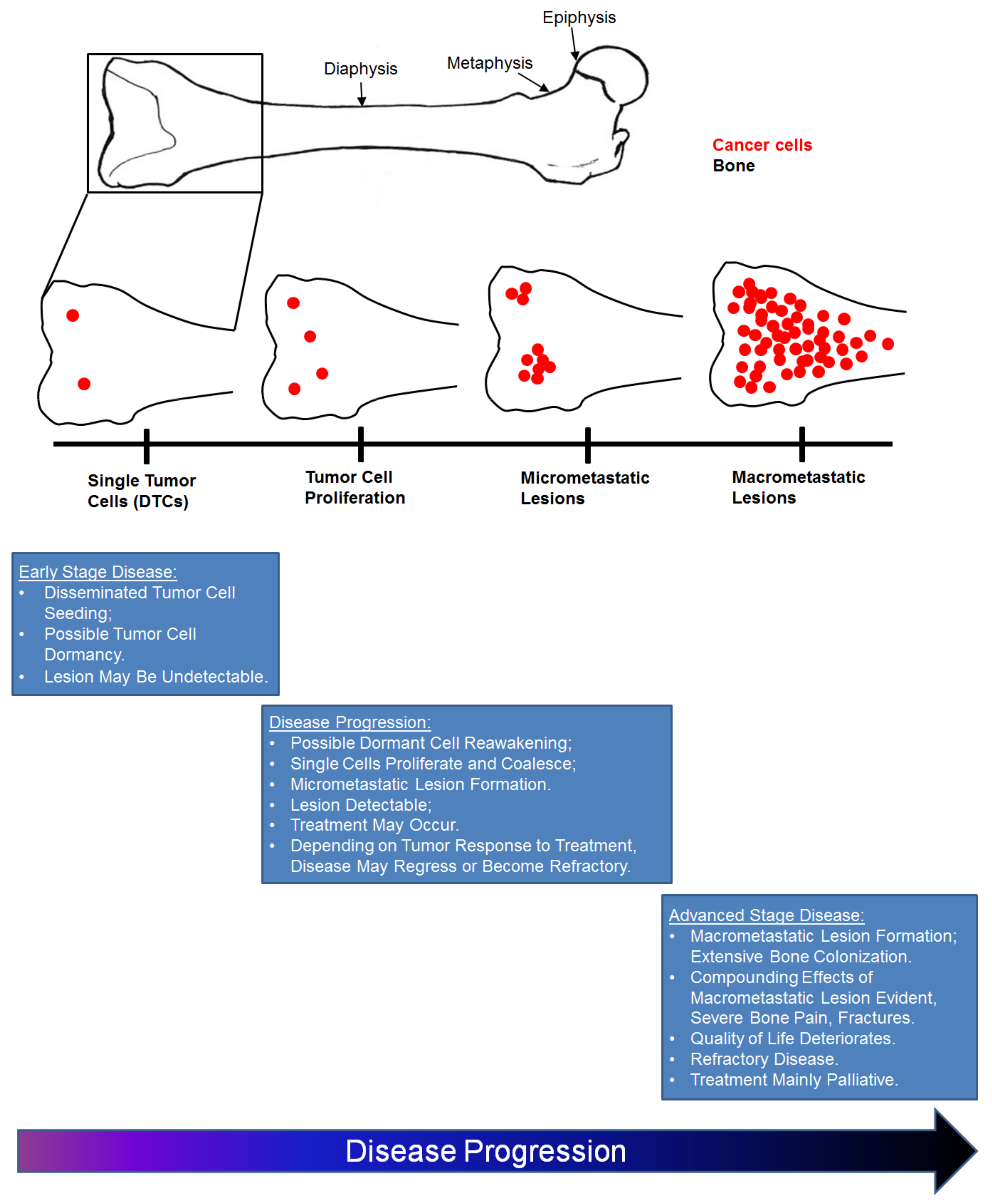
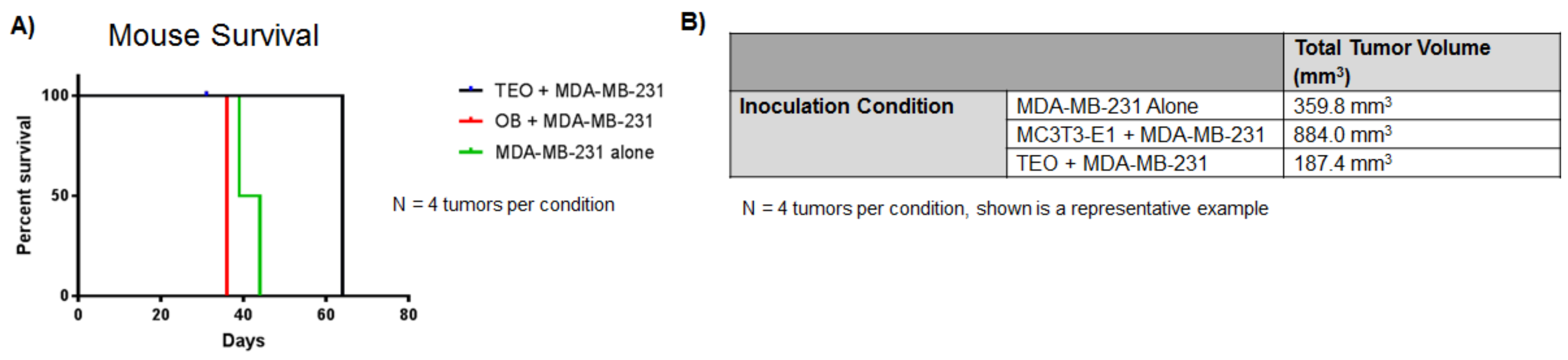
5. Models to Study the Effect of the Bone Niche on Cancer Cell Dormancy
6. Dormant Cancer Cell Re-Activation in Bone
7. Osteoblasts in the Early Stages of Cancer Metastasis to Bone
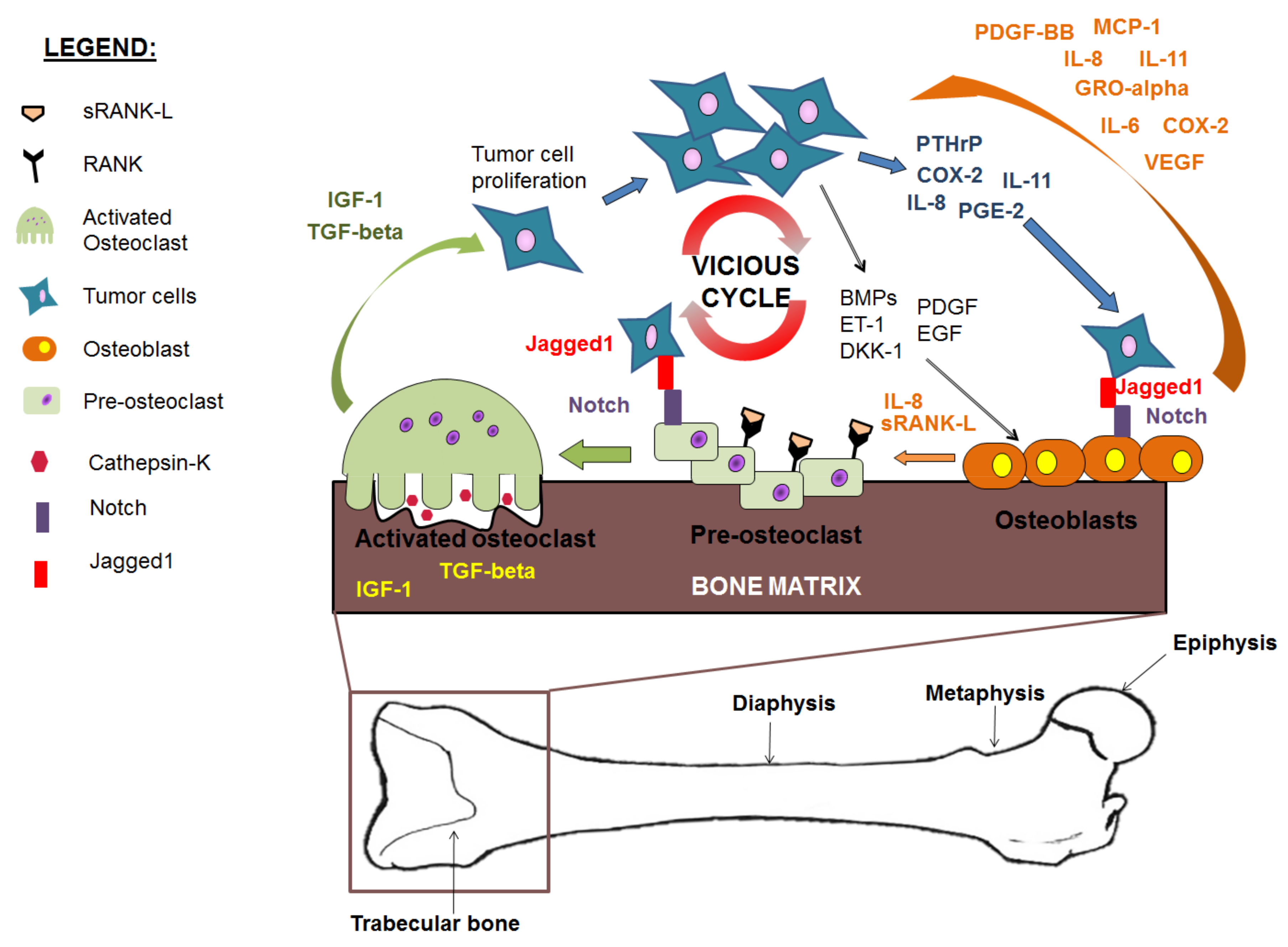
8. The ‘Vicious Cycle’ of Cancer Metastasis to Bone
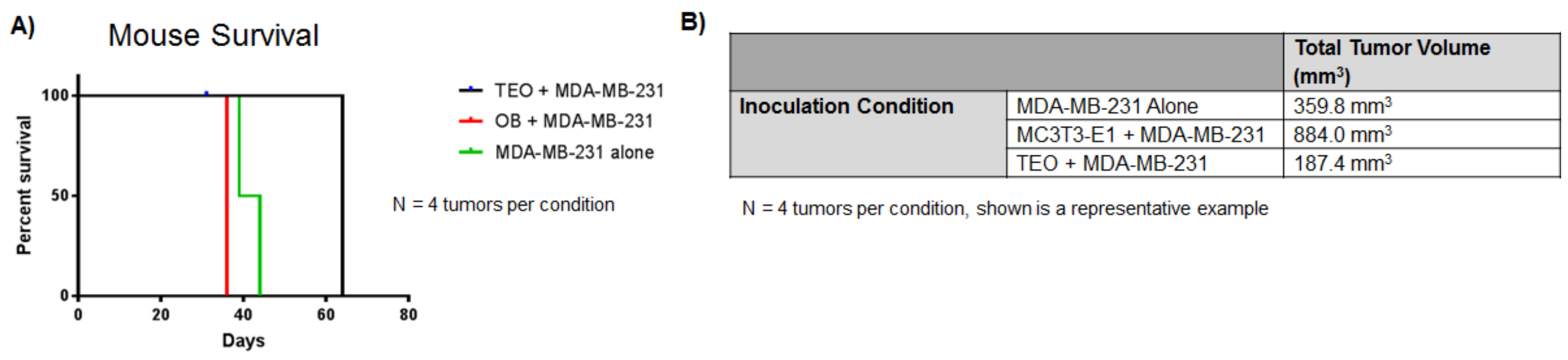
9. Osteoblasts in Advanced Stage Metastatic Disease
10. Osteoblast Alignment and Bone Matrix Organization Are Altered by Direct Interaction with Bone Metastatic Cancer Cells
11. Cancer Cells Are Capable of Mimicking Osteoblasts in the Tumor Microenvironment
12. Future Questions to Be Considered
13. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Marks, S.C., Jr.; Odgren, P.R. Structure and development of the skeleton. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: San Diego, CA, USA, 2002; Volume 1, pp. 3–16. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Augat, P.; Schorlemmer, S. The role of cortical bone and its microstructure in bone strength. Age Ageing 2006, 35 (Suppl. 2), ii27–ii31. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Kaibara, K.; Tabata, Y.; Nagata, N.; Enomoto, S.; Marukawa, E.; Umakoshi, Y. Unique alignment and texture of biological apatite crystallites in typical calcified tissues analyzed by microbeam x-ray diffractometer system. Bone 2002, 31, 479–487. [Google Scholar] [CrossRef]
- Keaveny, T.M.; Morgan, E.F.; Niebur, G.L.; Yeh, O.C. Biomechanics of trabecular bone. Annu. Rev. Biomed. Eng. 2001, 3, 307–333. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, C.J.; Lin, S.H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer 2005, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Institutes of Health, N.C.I. Seer Training Modules, Cancer Registration & Surveillance Modules: Classificatoin of Bones. Available online: https://training.seer.cancer.gov (accessed on 5 May 2018).
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of bone tissue: Structure, function, and factors that influence bone cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Oftadeh, R.; Perez-Viloria, M.; Villa-Camacho, J.C.; Vaziri, A.; Nazarian, A. Biomechanics and mechanobiology of trabecular bone: A review. J. Biomech. Eng. 2015, 137. [Google Scholar] [CrossRef] [PubMed]
- Price, J.S.; Oyajobi, B.O.; Russell, R.G. The cell biology of bone growth. Eur. J. Clin. Nutr. 1994, 48 (Suppl. 1), S131–S149. [Google Scholar] [PubMed]
- Mazo, I.B.; von Andrian, U.H. Adhesion and homing of blood-borne cells in bone marrow microvessels. J. Leukoc. Biol. 1999, 66, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullender, M.G.; van der Meer, D.D.; Huiskes, R.; Lips, P. Osteocyte density changes in aging and osteoporosis. Bone 1996, 18, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Udagawa, N.; Takami, M.; Suda, T. Cells of bone: Osteoclast generation. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: San Diego, CA, USA, 2002; Volume 1, pp. 109–126. [Google Scholar]
- Stenbeck, G. Formation and function of the ruffled border in osteoclasts. Semin. Cell Dev. Biol. 2002, 13, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Hauschka, P.V.; Mavrakos, A.E.; Iafrati, M.D.; Doleman, S.E.; Klagsburn, M. Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-sepharose. J. Biol. Chem. 1986, 261, 12665–12674. [Google Scholar] [PubMed]
- Minguell, J.J.; Erices, A.; Conget, P. Mesenchymal stem cells. Exp. Biol. Med. 2001, 226, 507–520. [Google Scholar] [CrossRef]
- Lian, J.B.; Stein, G.S. Concepts of osteoblast growth and differentiation: Basis for modulation of bone cell development and tissue formation. Crit. Rev. Oral Biol. Med. 1992, 3, 269–305. [Google Scholar] [CrossRef] [PubMed]
- Renema, N.; Navet, B.; Heymann, M.F.; Lezot, F.; Heymann, D. Rank-rankl signalling in cancer. Biosci. Rep. 2016, 36, e00366. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of osteoclast differentiation by cytokine networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Ren, H.; Li, X.; Yu, D.; Mu, S.; Chen, Z.; Fu, Q. Effects of intermedin on proliferation, apoptosis and the expression of opg/rankl/m-csf in the mc3t3-e1 osteoblast cell line. Mol. Med. Rep. 2015, 12, 6711–6717. [Google Scholar] [CrossRef] [PubMed]
- Kanis, J.A.; McCloskey, E.V. Bone turnover and biochemical markers in malignancy. Cancer 1997, 80, 1538–1545. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.R.; Peters, C.; Saftig, P.; Bromme, D. Cathepsin k activity-dependent regulation of osteoclast actin ring. J. Biol. Chem. 2009, 284, 2584–2592. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C. How the osteoclast degrades bone. Bioessays 1998, 20, 837–846. [Google Scholar] [CrossRef]
- Mastro, A.M.; Gay, C.V.; Welch, D.R.; Donahue, H.J.; Jewell, J.; Mercer, R.; DiGirolamo, D.; Chislock, E.M.; Guttridge, K. Breast cancer cells induce osteoblast apoptosis: A possible contributor to bone degradation. J. Cell. Biochem. 2004, 91, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Josse, J.; Velard, F.; Gangloff, S.C. Staphylococcus aureus vs. Osteoblast: Relationship and consequences in osteomyelitis. Front. Cell. Infect. Microbiol. 2015, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Deberg, M.A.; Bellahcene, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y.E. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheum. 2008, 58, 442–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, C.; Mazzucchelli, G.; Lambert, C.; Comblain, F.; DePauw, E.; Henrotin, Y. Comparison of secretome from osteoblasts derived from sclerotic versus non-sclerotic subchondral bone in oa: A pilot study. PLoS ONE 2018, 13, e0194591. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T. Mechanisms of preferential metastasis of breast cancer to bone. Int. J. Oncol. 1996, 9, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Hamza, T.; Li, B. Differential responses of osteoblasts and macrophages upon Staphylococcus aureus infection. BMC Microbiol. 2014, 14, 207. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Chan, P.M.B.; Wen, C. Do immune cells lead the way in subchondral bone disturbance in osteoarthritis? Prog. Biophys. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tofino-Vian, M.; Guillen, M.I.; Perez Del Caz, M.D.; Castejon, M.A.; Alcaraz, M.J. Extracellular vesicles from adipose-derived mesenchymal stem cells downregulate senescence features in osteoarthritic osteoblasts. Oxid. Med. Cell. Longev. 2017, 2017, 7197598. [Google Scholar] [CrossRef] [PubMed]
- Lentino, J.R. Prosthetic joint infections: Bane of orthopedists, challenge for infectious disease specialists. Clin. Infect. Dis. 2003, 36, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Berendt, T.; Byren, I. Bone and joint infection. Clin. Med. 2004, 4, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Tillander, J.; Hagberg, K.; Berlin, O.; Hagberg, L.; Branemark, R. Osteomyelitis risk in patients with transfemoral amputations treated with osseointegration prostheses. Clin. Orthop. Relat. Res. 2017, 475, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Kellesarian, S.V.; Javed, F.; Romanos, G.E. Osteomyelitis arising around osseointegrated dental implants: A systematic review. Implant Dent. 2018. [Google Scholar] [CrossRef] [PubMed]
- Semel, G.; Wolff, A.; Shilo, D.; Akrish, S.; Emodi, O.; Rachmiel, A. Mandibular osteomyelitis associated with dental implants. A case series. Eur. J. Oral Implantol. 2016, 9, 435–442. [Google Scholar] [PubMed]
- Heilmann, C. Adhesion mechanisms of staphylococci. Adv. Exp. Med. Biol. 2011, 715, 105–123. [Google Scholar] [PubMed]
- Bosse, M.J.; Gruber, H.E.; Ramp, W.K. Internalization of bacteria by osteoblasts in a patient with recurrent, long-term osteomyelitis. A case report. J. Bone Jt. Surg. Am. 2005, 87, 1343–1347. [Google Scholar] [CrossRef]
- Dapunt, U.; Maurer, S.; Giese, T.; Gaida, M.M.; Hansch, G.M. The macrophage inflammatory proteins mip1alpha (ccl3) and mip2alpha (cxcl2) in implant-associated osteomyelitis: Linking inflammation to bone degradation. Mediat. Inflamm. 2014, 2014, 728619. [Google Scholar] [CrossRef] [PubMed]
- Bost, K.L.; Bento, J.L.; Ellington, J.K.; Marriott, I.; Hudson, M.C. Induction of colony-stimulating factor expression following staphylococcus or salmonella interaction with mouse or human osteoblasts. Infect. Immun. 2000, 68, 5075–5083. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Friedland, J.S. Regulation of chemokine gene expression and secretion in staphylococcus aureus-infected osteoblasts. Microbes Infect. 2004, 6, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Ning, R.; Zhang, X.; Guo, X.; Li, Q. Staphylococcus aureus regulates secretion of interleukin-6 and monocyte chemoattractant protein-1 through activation of nuclear factor kappab signaling pathway in human osteoblasts. Braz. J. Infect. Dis. 2011, 15, 189–194. [Google Scholar] [PubMed]
- Gasper, N.A.; Petty, C.C.; Schrum, L.W.; Marriott, I.; Bost, K.L. Bacterium-induced cxcl10 secretion by osteoblasts can be mediated in part through toll-like receptor 4. Infect. Immun. 2002, 70, 4075–4082. [Google Scholar] [CrossRef] [PubMed]
- Somayaji, S.N.; Ritchie, S.; Sahraei, M.; Marriott, I.; Hudson, M.C. Staphylococcus aureus induces expression of receptor activator of nf-kappab ligand and prostaglandin e2 in infected murine osteoblasts. Infect. Immun. 2008, 76, 5120–5126. [Google Scholar] [CrossRef] [PubMed]
- Widaa, A.; Claro, T.; Foster, T.J.; O'Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein a plays a critical role in mediating bone destruction and bone loss in osteomyelitis. PLoS ONE 2012, 7, e40586. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell. Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinder, M.; Chislock, E.M.; Bussard, K.M.; Shuman, L.A.; Mastro, A.M. Metastatic breast cancer induces an osteoblast inflammatory response. Exp. Cell Res. 2008, 314, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Sosnoski, D.; Krishnan, V.; Kraemer, W.J.; Dunn-Lewis, C.; Mastro, A.M. Changes in cytokines of the bone microenvironment during breast cancer metastasis. Int. J. Breast Cancer 2012, 2012, 160265. [Google Scholar] [CrossRef] [PubMed]
- Claro, T.; Widaa, A.; O’Seaghdha, M.; Miajlovic, H.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein a binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PLoS ONE 2011, 6, e18748. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.J., Jr.; Ward, C.L.; Romano, D.R.; Hurtgen, B.J.; Hardy, S.K.; Woodbury, R.L.; Trevino, A.V.; Rathbone, C.R.; Wenke, J.C. Staphylococcus aureus biofilms decrease osteoblast viability, inhibits osteogenic differentiation, and increases bone resorption in vitro. BMC Musculoskelet. Disord. 2013, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Rasigade, J.P.; Trouillet-Assant, S.; Ferry, T.; Diep, B.A.; Sapin, A.; Lhoste, Y.; Ranfaing, J.; Badiou, C.; Benito, Y.; Bes, M.; et al. Psms of hypervirulent staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS ONE 2013, 8, e63176. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; O’Neill, L.A.J.; Gearing, A.J.H.; Callard, R.E. The Cytokine Facts Book, 2nd ed.; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Sanchez, C.; Pesesse, L.; Gabay, O.; Delcour, J.P.; Msika, P.; Baudouin, C.; Henrotin, Y.E. Regulation of subchondral bone osteoblast metabolism by cyclic compression. Arthritis Rheum. 2012, 64, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, D.; Todorov, A.; Cengic, T.; Pagenstert, G.; Scharen, S.; Netzer, C.; Hugle, T.; Geurts, J. Alterations of subchondral bone progenitor cells in human knee and hip osteoarthritis lead to a bone sclerosis phenotype. Int. J. Mol. Sci. 2018, 19, 475. [Google Scholar] [CrossRef] [PubMed]
- Martineau, X.; Abed, E.; Martel-Pelletier, J.; Pelletier, J.P.; Lajeunesse, D. Alteration of wnt5a expression and of the non-canonical wnt/pcp and wnt/pkc-ca2+ pathways in human osteoarthritis osteoblasts. PLoS ONE 2017, 12, e0180711. [Google Scholar] [CrossRef] [PubMed]
- Tat, S.K.; Padrines, M.; Theoleyre, S.; Couillaud-Battaglia, S.; Heymann, D.; Redini, F.; Fortun, Y. Opg/membranous—Rankl complex is internalized via the clathrin pathway before a lysosomal and a proteasomal degradation. Bone 2006, 39, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Phadke, P.A.; Mercer, R.R.; Harms, J.F.; Yujiang, J.; Frost, A.R.; Jewell, J.L.; Bussard, K.M.; Nelson, S.; Moore, C.; Kappes, J.C.; et al. Kinetics of metastatic breast cancer cell trafficking in bone. Clin. Cancer Res. 2006, 12, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.-J.; Liang, Q.; Zhong, J.-H.; Zhu, M.; Meng, F.-Y.; Wu, N.; Liang, R.; Yuan, B.-Y. Ibandronate to treat skeletal-related events and bone pain in metastatic bone disease or multiple myeloma: A meta-analysis of randomised clinical trials. BMJ Open 2015, 5, e007258. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: Evidence from in vivo models. J. Bone Miner. Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Belic, J.; Graf, R.; Bauernhofer, T.; Cherkas, Y.; Ulz, P.; Waldispuehl-Geigl, J.; Perakis, S.; Gormley, M.; Patel, J.; Li, W.; et al. Genomic alterations in plasma DNA from patients with metastasized prostate cancer receiving abiraterone or enzalutamide. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Pulido, C.; Vendrell, I.; Ferreira, A.R.; Casimiro, S.; Mansinho, A.; Alho, I.; Costa, L. Bone metastasis risk factors in breast cancer. Ecancermedicalscience 2017, 11, 715. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.J.; Rajkumar, S.V.; Therneau, T.M.; Singh, P.P.; Dispenzieri, A.; Kumar, S.K. Relationship between initial clinical presentation and the molecular cytogenetic classification of myeloma. Leukemia 2014, 28, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Santini, D.; Barni, S.; Intagliata, S.; Falcone, A.; Ferraù, F.; Galetta, D.; Moscetti, L.; La Verde, N.; Ibrahim, T.; Petrelli, F.; et al. Corrigendum: Natural history of non-small-cell lung cancer with bone metastases. Sci. Rep. 2016, 6, 22205. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Sandro, B.; Salvatore, I.; Alfredo, F.; Francesco, F.; Domenico, G.; Luca, M.; Nicla, L.V.; Toni, I.; Fausto, P.; et al. Natural history of non-small-cell lung cancer with bone metastases. Sci. Rep. 2015, 5, 18670. [Google Scholar]
- Kim, S.; Chun, M.; Wang, H.; Cho, S.; Oh, Y.-T.; Kang, S.-H.; Yang, J. Bone metastasis from primary hepatocellular carcinoma: Characteristics of soft tissue formation. Cancer Res. Treat. 2007, 39, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-C.; Kuo, P.-L. Bone metastasis from renal cell carcinoma. Int. J. Mol. Sci. 2016, 17, 987. [Google Scholar] [CrossRef] [PubMed]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: Benefits and limits of radioiodine therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Liede, A.; Jerzak, K.J.; Hernandez, R.K.; Wade, S.W.; Sun, P.; Narod, S.A. The incidence of bone metastasis after early-stage breast cancer in canada. Breast Cancer Res. Treat. 2016, 156, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Uzzo, R.; Amato, R.J.; Ellis, G.K.; Hakimian, B.; Roodman, G.D.; Smith, M.R. The science and practice of bone health in oncology: Managing bone loss and metastasis in patients with solid tumors. J. Natl. Compr. Cancer Netw. 2009, 7 (Suppl. 7), S1–S29. [Google Scholar] [CrossRef]
- Manders, K.; van de Poll-Franse, L.V.; Creemers, G.J.; Vreugdenhil, G.; van der Sangen, M.J.; Nieuwenhuijzen, G.A.; Roumen, R.M.; Voogd, A.C. Clinical management of women with metastatic breast cancer: A descriptive study according to age group. BMC Cancer 2006, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Bone Remodeling and Its Disorders; Martin Dunitz Ltd.: London, UK, 1999. [Google Scholar]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Yi, B.; Williams, P.J.; Niewolna, M.; Wang, Y.; Yoneda, T. Tumor-derived platelet-derived growth factor-bb plays a critical role in osteosclerotic bone metastasis in an animal model of human breast cancer. Cancer Res. 2002, 62, 917–923. [Google Scholar] [PubMed]
- Yin, J.J.; Mohammad, K.S.; Kakonen, S.M.; Harris, S.; Wu-Wong, J.R.; Wessale, J.L.; Padley, R.J.; Garrett, I.R.; Chirgwin, J.M.; Guise, T.A. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 10954–10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guise, T.A.; Mundy, G.R. Cancer and bone. Endocr. Rev. 1998, 19, 18–54. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Marathe, D.D.; Marathe, A.; Mager, D.E. Integrated model for denosumab and ibandronate pharmacodynamics in postmenopausal women. Biopharm. Drug Dispos. 2011, 32, 471–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagnostis, P.; Vakalopoulou, S.; Christoulas, D.; Paschou, S.A.; Papatheodorou, A.; Garipidou, V.; Kokkoris, P.; Terpos, E. The role of sclerostin/dickkopf-1 and receptor activator of nuclear factor kb ligand/osteoprotegerin signalling pathways in the development of osteoporosis in patients with haemophilia a and b: A cross-sectional study. Haemoph. Off. J. World Fed. Hemoph. 2018, 24, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.N.; Lie, J.D.; Wan, C.K.V.; Cameron, M.; Austel, A.G.; Nguyen, J.K.; Van, K.; Hyun, D. Osteoporosis: A review of treatment options. Pharm. Ther. 2018, 43, 92–104. [Google Scholar]
- Guise, T.A. Molecular mechanisms of osteolytic bone metastases. Cancer 2000, 88, 2892–2898. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Yasuda, H.; Mizuno, A.; Itoh, K.; Ueno, Y.; Shinki, T.; Gillespie, M.T.; Martin, T.J.; Higashio, K.; et al. Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 2000, 141, 3478–3484. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of rank, rankl, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Williams, P.J.; Mundy, G.R. The bisphosphonate ibandronate promotes apoptosis in mda-mb-231 human breast cancer cells in bone metastases. Cancer Res. 2001, 61, 4418–4424. [Google Scholar] [PubMed]
- Taube, T.; Elomaa, I.; Blomqvist, C.; Benton, N.C.; Kanis, J.A. Histomorphometric evidence for osteoclast-mediated bone resorption in metastatic breast cancer. Bone 1994, 15, 161–166. [Google Scholar] [CrossRef]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Roodman, G.D. Multiple myeloma and bone: The fatal interaction. Cold Spring Harbor Perspect. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bataille, R.; Chappard, D.; Marcelli, C.; Dessauw, P.; Sany, J.; Baldet, P.; Alexandre, C. Mechanisms of bone destruction in multiple myeloma: The importance of an unbalanced process in determining the severity of lytic bone disease. J. Clin. Oncol. 1989, 7, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.A.; Chung, H.Y.; Ghobrial, I.; Choi, S.J.; Morandi, F.; Colla, S.; Rizzoli, V.; Roodman, G.D.; Giuliani, N. Il-3 is a potential inhibitor of osteoblast differentiation in multiple myeloma. Blood 2005, 106, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Silbermann, R.; Bolzoni, M.; Storti, P.; Guasco, D.; Bonomini, S.; Zhou, D.; Wu, J.; Anderson, J.L.; Windle, J.J.; Aversa, F.; et al. Bone marrow monocyte-/macrophage-derived activin a mediates the osteoclastogenic effect of il-3 in multiple myeloma. Leukemia 2014, 28, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess tgf-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Nyman, J.S.; Merkel, A.R.; Uppuganti, S.; Nayak, B.; Rowland, B.; Makowski, A.J.; Oyajobi, B.O.; Sterling, J.A. Combined treatment with a transforming growth factor beta inhibitor (1d11) and bortezomib improves bone architecture in a mouse model of myeloma-induced bone disease. Bone 2016, 91, 81–91. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, S.; del Prete, D.; Jin, S.; Sun, Q.; Huston, A.J.; Kostov, F.E.; Sammut, B.; Hong, C.S.; Anderson, J.L.; Patrene, K.D.; et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 2011, 118, 6871–6880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, S.; Fulciniti, M.; Yan, H.; Vallet, S.; Eda, H.; Patel, K.; Santo, L.; Cirstea, D.; Hideshima, T.; Schirtzinge, L.; et al. In vivo and in vitro effects of a novel anti-dkk1 neutralizing antibody in multiple myeloma. Bone 2013, 53, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. Ezh2 or hdac1 inhibition reverses multiple myeloma-induced epigenetic suppression of osteoblast differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Silbermann, R.; Roodman, G.D. Bone effects of cancer therapies: Pros and cons. Curr. Opin. Support. Palliat. Care 2011, 5, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Munshi, N.C. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015, 125, 3049–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.P.; Beck, J.T.; Stewart, A.K.; Shah, J.; Kelly, K.R.; Isaacs, R.; Bilic, S.; Sen, S.; Munshi, N.C. A phase ib multicentre dose-determination study of bhq880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br. J. Haematol. 2014, 167, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.-T.; Chauhan, D.; et al. Anti-dkk1 mab (bhq880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371. [Google Scholar] [CrossRef] [PubMed]
- Kocemba, K.A.; Groen, R.W.; van Andel, H.; Kersten, M.J.; Mahtouk, K.; Spaargaren, M.; Pals, S.T. Transcriptional silencing of the wnt-antagonist dkk1 by promoter methylation is associated with enhanced wnt signaling in advanced multiple myeloma. PLoS ONE 2012, 7, e30359. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Dou, L.; Song, J.; Luo, J. Cbfa2t2 is required for bmp-2-induced osteogenic differentiation of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2018, 496, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Charhon, S.A.; Chapuy, M.C.; Delvin, E.E.; Valentin-Opran, A.; Edouard, C.M.; Meunier, P.J. Histomorphometric analysis of sclerotic bone metastases from prostatic carcinoma special reference to osteomalacia. Cancer 1983, 51, 918–924. [Google Scholar] [CrossRef]
- Clarke, N.W.; McClure, J.; George, N.J. Morphometric evidence for bone resorption and replacement in prostate cancer. Br. J. Urol. 1991, 68, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Scher, H.I. Clinical approaches to osseous metastases in prostate cancer. Oncologist 2003, 8, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, C.; Morris, M.J.; Den, R.; Coleman, R.E. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastasis Rev. 2018, 37, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekita, A.; Matsugaki, A.; Nakano, T. Disruption of collagen/apatite alignment impairs bone mechanical function in osteoblastic metastasis induced by prostate cancer. Bone 2017, 97, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Matsugaki, A.; Aramoto, G.; Ninomiya, T.; Sawada, H.; Hata, S.; Nakano, T. Abnormal arrangement of a collagen/apatite extracellular matrix orthogonal to osteoblast alignment is constructed by a nanoscale periodic surface structure. Biomaterials 2015, 37, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Corn, P.G.; Yang, J.; Palanisamy, N.; Starbuck, M.W.; Efstathiou, E.; Li Ning Tapia, E.M.; Zurita, A.J.; Aparicio, A.; Ravoori, M.K.; et al. Prostate cancer cell-stromal cell crosstalk via fgfr1 mediates antitumor activity of dovitinib in bone metastases. Sci. Transl. Med. 2014, 6, 252ra122. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Yang, J.; Peleg, S.; Sikes, C.R.; Kreimann, E.L.; Daliani, D.; Olive, M.; Raymond, K.A.; Janus, T.J.; Logothetis, C.J.; et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clin. Cancer Res. 2003, 9, 2587–2597. [Google Scholar] [PubMed]
- Oberneder, R.; Riesenberg, R.; Kriegmair, M.; Bitzer, U.; Klammert, R.; Schneede, P.; Hofstetter, A.; Riethmuller, G.; Pantel, K. Immunocytochemical detection and phenotypic characterization of micrometastatic tumour cells in bone marrow of patients with prostate cancer. Urol. Res. 1994, 22, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Li, X.; Koh, A.J.; Berry, J.E.; Thudi, N.; Rosol, T.J.; Pienta, K.J.; McCauley, L.K. Tumor expressed pthrp facilitates prostate cancer-induced osteoblastic lesions. Int. J. Cancer 2008, 123, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Clines, G.A.; Mohammad, K.S.; Bao, Y.; Stephens, O.W.; Suva, L.J.; Shaughnessy, J.D., Jr.; Fox, J.W.; Chirgwin, J.M.; Guise, T.A. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol. Endocrinol. 2007, 21, 486–498. [Google Scholar] [CrossRef] [PubMed]
- David Roodman, G.; Silbermann, R. Mechanisms of osteolytic and osteoblastic skeletal lesions. Bonekey Rep. 2015, 4, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carducci, M.A.; Saad, F.; Abrahamsson, P.A.; Dearnaley, D.P.; Schulman, C.C.; North, S.A.; Sleep, D.J.; Isaacson, J.D.; Nelson, J.B. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer 2007, 110, 1959–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carducci, M.A.; Nelson, J.B.; Bowling, M.K.; Rogers, T.; Eisenberger, M.A.; Sinibaldi, V.; Donehower, R.; Leahy, T.L.; Carr, R.A.; Isaacson, J.D.; et al. Atrasentan, an endothelin-receptor antagonist for refractory adenocarcinomas: Safety and pharmacokinetics. J. Clin. Oncol. 2002, 20, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Carducci, M.A.; Padley, R.J.; Breul, J.; Vogelzang, N.J.; Zonnenberg, B.A.; Dallani, D.D.; Schulman, C.C.; Nabulsi, A.A.; Humerickhouse, R.A.; Weinberg, M.A.; et al. Effect of endothelin-a receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer; a randomized, phase ii, placebo-controlled trial. J. Clin. Oncol. 2003, 21, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.I.; Tangen, C.M.; Hussain, M.; Lara, P.N., Jr.; Goldkorn, A.; Moinpour, C.M.; Garzotto, M.G.; Mack, P.C.; Carducci, M.A.; Monk, J.P.; et al. Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (swog s0421): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 893–900. [Google Scholar] [CrossRef]
- Suominen, M.I.; Fagerlund, K.M.; Rissanen, J.P.; Konkol, Y.M.; Morko, J.P.; Peng, Z.; Alhoniemi, E.J.; Laine, S.K.; Corey, E.; Mumberg, D.; et al. Radium-223 inhibits osseous prostate cancer growth by dual targeting of cancer cells and bone microenvironment in mouse models. Clin. Cancer Res. 2017, 23, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, J.; et al. Skeletal localization and neutralization of the sdf-1 (cxcl12)/cxcr4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2004, 20, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T.; Sundar, R.; Widmark, A.; Landstrom, M.; Persson, E. Osteoblast-derived factors promote metastatic potential in human prostate cancer cells, in part via non-canonical transforming growth factor beta (tgfbeta) signaling. Prostate 2018, 78, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Okita, N.; Sharkey, N.; Neuberger, T.; Webb, A.; Mastro, A.M. Localization of mcp-1, vegf, and il-6 in the bone microenvironment of mice bearing metastatic breast cancer. Clin. Exp. Metas 2010, 27, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Whang, Y.M.; Campbell, P.; Mulcrone, P.L.; Elefteriou, F.; Cho, S.W.; Park, S.I. Dual targeting c-met and vegfr2 in osteoblasts suppresses growth and osteolysis of prostate cancer bone metastasis. Cancer Lett. 2018, 414, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Bussard, K.M. Thomas Jefferson University, Philadelphia, PA, USA. Unpublished data. 2018. [Google Scholar]
- Shulby, S.A.; Dolloff, N.G.; Stearns, M.E.; Meucci, O.; Fatatis, A. Cx3cr1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004, 64, 4693–4698. [Google Scholar] [CrossRef] [PubMed]
- Jamieson-Gladney, W.L.; Zhang, Y.; Fong, A.M.; Meucci, O.; Fatatis, A. The chemokine receptor cx(3)cr1 is directly involved in the arrest of breast cancer cells to the skeleton. Breast Cancer Res. 2011, 13, R91. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zhang, Y.; Jernigan, D.L.; Feng, X.; Yan, J.; Garcia, F.U.; Meucci, O.; Salvino, J.M.; Fatatis, A. Novel small-molecule cx3cr1 antagonist impairs metastatic seeding and colonization of breast cancer cells. Mol. Cancer Res. 2016, 14, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for tgf-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Leconet, W.; Chentouf, M.; du Manoir, S.; Chevalier, C.; Sirvent, A.; Ait-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic activity of anti-axl antibody against triple-negative breast cancer patient-derived xenografts and metastasis. Clin. Cancer Res. 2017, 23, 2806–2816. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. Gas6/axl axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Decker, A.M.; Wang, J.; Lee, E.; Kana, L.A.; Yumoto, K.; Cackowski, F.C.; Rhee, J.; Carmeliet, P.; Buttitta, L.; et al. Endogenous gas6 and mer receptor signaling regulate prostate cancer stem cells in bone marrow. Oncotarget 2016, 7, 25698–25711. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Nie, D.; Li, J.; Du, X.; Lu, Y.; Li, Y.; Liu, C.; Zhou, J.; Pan, J. Gas6/axl signaling regulates self-renewal of chronic myelogenous leukemia stem cells by stabilizing beta-catenin. Clin. Cancer Res. 2017, 23, 2842–2855. [Google Scholar] [CrossRef] [PubMed]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the tgfbetariii-p38mapk-ps249/t252rb pathway. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Ghajar, C.M.; Bissell, M.J. The need for complex 3d culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv. Drug Deliv. Rev. 2014, 69, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse models of metastasis: Progress and prospects. Dis. Models Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Christofori, G. Rebuilding cancer metastasis in the mouse. Mol. Oncol. 2013, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Wikman, H.; Vessella, R.; Pantel, K. Cancer micrometastasis and tumour dormancy. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2008, 116, 754–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Naumov, G.N.; MacDonald, I.C.; Weinmeister, P.M.; Kerkvliet, N.; Nadkarni, K.V.; Wilson, S.M.; Morris, V.L.; Groom, A.C.; Chambers, A.F. Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res. 2002, 62, 2162–2168. [Google Scholar] [PubMed]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.-Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. Aml-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jacamo, R.; Shi, Y.X.; Wang, R.Y.; Battula, V.L.; Konoplev, S.; Strunk, D.; Hofmann, N.A.; Reinisch, A.; Konopleva, M.; et al. Human extramedullary bone marrow in mice: A novel in vivo model of genetically controlled hematopoietic microenvironment. Blood 2012, 119, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Li, M.; Milwid, J.; Dunham, J.; Vinegoni, C.; Gorbatov, R.; Iwamoto, Y.; Wang, F.; Shen, K.; Hatfield, K.; et al. Implantable microenvironments to attract hematopoietic stem/cancer cells. Proc. Natl Acad. Sci. USA 2012, 109, 19638–19643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, B.P.; Naved, B.A.; Nyberg, E.L.; Dias, M.; Holmes, C.A.; Elisseeff, J.H.; Dorafshar, A.H.; Grayson, W.L. Three-dimensional printing of bone extracellular matrix for craniofacial regeneration. ACS Biomater. Sci. Eng. 2016, 2, 1806–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Heckl, D.; Parekkadan, B. Multiple genetically engineered humanized microenvironments in a single mouse. Biomater. Res. 2016, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Bersani, F.; Lee, J.; Yu, M.; Morris, R.; Desai, R.; Ramaswamy, S.; Toner, M.; Haber, D.A.; Parekkadan, B. Bioengineered implantable scaffolds as a tool to study stromal-derived factors in metastatic cancer models. Cancer Res. 2014, 74, 7229–7238. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, A.; Izadmehr, S.; Yao, S.; O'Connor-Chapman, K.L.; Huang, A.; Gregoriades, E.M.; Yakar, S.; Levine, A.C. Prostatic acid phosphatase alters the rankl/opg system and induces osteoblastic prostate cancer bone metastases. Endocrinology 2016, 157, 4526–4533. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The bmp inhibitor coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Kokabu, S.; Rosen, V. Bmp3 expression by osteoblast lineage cells is regulated by canonical wnt signaling. FEBS Open Bio 2018, 8, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, T.; Kawaguchi, H.; Jinno, S.; Hoshi, K.; Itaka, K.; Takato, T.; Nakamura, K.; Okayama, H. Bone morphogenetic protein 2-induced osteoblast differentiation requires smad-mediated down-regulation of cdk6. Mol. Cell. Biol. 2004, 24, 6560–6568. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Deng, C.; Li, Y.P. Tgf-β and bmp signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Quayle, L.; Ottewell, P.D.; Holen, I. Bone metastasis: Molecular mechanisms implicated in tumour cell dormancy in breast and prostate cancer. Curr. Cancer Drug Targets 2015, 15, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Bodenstine, T.M.; Beck, B.H.; Cao, X.; Cook, L.M.; Ismail, A.; Powers, S.J.; Powers, J.K.; Mastro, A.M.; Welch, D.R. Pre-osteoblastic mc3t3-e1 cells promote breast cancer growth in bone in a murine xenograft model. Chin. J. Cancer 2011, 30, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Antonacci, C.; Toschi, N.; Giannini, E.; Bonfiglio, R.; Buonomo, C.O.; Pistolese, C.A.; Tarantino, U.; Bonanno, E. Breast osteoblast-like cells: A reliable early marker for bone metastases from breast cancer. Clin. Breast Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pienta, K.J.; Taichamn, R.S. Hematopoietic stem cell niche is a potential therapeutic target for bone metastatic tumors. Clin. Cancer Res. 2011, 17, 5553–5558. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, E.A.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. The prostate cancer bone marrow niche: More than just ‘fertile soil’. Asian J. Androl. 2012, 14, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast-cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.G.; Cusano, N.E.; Silva, B.C.; Cremers, S.; Bilezikian, J.P. Cathepsin k: Its skeletal actions and role as a therapeutic target in osteoporosis. Nat. Rev. Rheumatol. 2011, 7, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Walia, B.; Lingenheld, E.; Duong, L.; Sanjay, A.; Drissi, H. A novel role for cathepsin k in periosteal osteoclast precursors during fracture repair. Ann. N. Y. Acad. Sci. 2018, 1415, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.A.; Fournier, P.G.J.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609. [Google Scholar] [CrossRef] [PubMed]
- Bendre, M.; Montague, D.C.; Peery, T.; Akel, N.S.; Gaddy, D.; Suva, L.J. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone 2003, 33, 28–37. [Google Scholar] [CrossRef]
- Bendre, M.S.; Margulies, A.G.; Walser, B.; Akel, N.S.; Bhattacharrya, S.; Skinner, R.A.; Swain, F.; Ramani, V.; Mohammad, K.S.; Wessner, L.L.; et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kb ligand pathway. Cancer Res. 2005, 65, 11001–11009. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.F.; Vignery, A.; Silverglate, A.; Ravin, N.D.; Livolsi, V.; Broadus, A.E.; Baron, R. Quantitative bone histomorphology in humoral hypercalcemia of malignancy: Uncoupling of bone cell activity. J. Clin. Endocrinol. Metab. 1982, 55, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.; Miyasaka, C.; Mastro, A.M. Metastatic breast cancer cells suppress osteoblast adhesion and differentiation. Clin. Exp. Metas 2004, 21, 427–435. [Google Scholar] [CrossRef]
- Lee, J.W.; Chung, H.Y.; Ehrlich, L.A.; Jelinek, D.F.; Callander, N.S.; Roodman, G.D.; Choi, S.J. Il-3 expression by myeloma cells increases both osteoclast formation and growth of myeloma cells. Blood 2004, 103, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Otsuka, F.; Otani, H.; Yamashita, M.; Takasugi, K.; Inagaki, K.; Yamamura, M.; Makino, H. Tnf-alpha inhibits bmp-induced osteoblast differentiation through activating sapk/jnk signaling. Biochem. Biophys. Res. Commun. 2007, 356, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Ballester, O.F.; Moscinski, L.C.; Lyman, G.H.; Chaney, J.V.; Saba, H.I.; Spiers, A.S.; Klein, C. High levels of interleukin-6 are associated with low tumor burden and low growth fraction in multiple myeloma. Blood 1994, 83, 1903–1908. [Google Scholar] [PubMed]
- Heider, U.; Zavrski, I.; Jakob, C.; Bangeroth, K.; Fleissner, C.; Langelotz, C.; Possinger, K.; Hofbauer, L.C.; Viereck, V.; Sezer, O. Expression of receptor activator of nf-kappab ligand (rankl) mrna in human multiple myeloma cells. J. Cancer Res. Clin. Oncol. 2004, 130, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Sati, H.I.; Greaves, M.; Apperley, J.F.; Russell, R.G.; Croucher, P.I. Expression of interleukin-1beta and tumour necrosis factor-alpha in plasma cells from patients with multiple myeloma. Br. J. Haematol. 1999, 104, 350–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.J.; Cruz, J.C.; Craig, F.; Chung, H.; Devlin, R.D.; Roodman, G.D.; Alsina, M. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood 2000, 96, 671–675. [Google Scholar] [PubMed]
- Park, B.M.; Kim, E.J.; Nam, H.J.; Zhang, D.; Bae, C.H.; Kang, M.; Kim, H.; Lee, W.; Bogen, B.; Lim, S.K. Cyclized oligopeptide targeting lrp5/6-dkk1 interaction reduces the growth of tumor burden in a multiple myeloma mouse model. Yonsei Med. J. 2017, 58, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, C.; Hojfeldt, G.; Hojman, P. The role of intratumoral and systemic il-6 in breast cancer. Breast Cancer Res. Treat. 2013, 138, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-derived jagged 1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; Song, L.; Shimada, H.; Keshelava, N.; Russell, H.V.; Metelitsa, L.S.; Groshen, S.G.; Seeger, R.C.; DeClerck, Y.A. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009, 69, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (xl184), a novel met and vegfr2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Basel, D.; Chow, S.-O.; Fong-Yee, C.; Kim, S.; Buttgereit, F.; Dunstan, C.R.; Zhou, H.; Seibel, M.J. Targeting il-6 and rankl signaling inhibits prostate cancer growth in bone. Clin. Exp. Metastasis 2014, 31, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Castro, N.J.; Cui, H.; Zhou, X.; Boualam, B.; McGrane, R.; Glazer, R.I.; Zhang, L.G. A 3d printed nano bone matrix for characterization of breast cancer cell and osteoblast interactions. Nanotechnology 2016, 27, 315103. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, Y.; Fujita, N.; Ohishi, K.; Tsuruo, T. Stimulation of interleukin-11 production from osteoblast-like cells by transforming growth factor-beta and tumor cell factors. Int. J. Cancer 1997, 71, 422–428. [Google Scholar] [CrossRef]
- Gonzalez, A.; Garcia de Durango, C.; Alonso, V.; Bravo, B.; Rodriguez de Gortazar, A.; Wells, A.; Forteza, J.; Vidal-Vanaclocha, F. Distinct osteomimetic response of androgen-dependent and independent human prostate cancer cells to mechanical action of fluid flow: Prometastatic implications. Prostate 2017, 77, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Bendre, M.S.; Gaddy-Kurten, D.; Mon-Foote, T.; Akel, N.S.; Skinner, R.A.; Nicholas, R.W.; Suva, L.J. Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Res. 2002, 62, 5571–5579. [Google Scholar] [PubMed]
- Singh, B.; Berry, J.A.; Vincent, L.E.; Lucci, A. Involvement of il-8 in cox-2-mediated bone metastases from breast cancer. J. Surg. Res. 2006, 134, 44–51. [Google Scholar] [CrossRef] [PubMed]
- McCoy, E.M.; Hong, H.; Pruitt, H.C.; Feng, X. Il-11 produced by breast cancer cells augments osteoclastogenesis by sustaining the pool of osteoclast progenitor cells. BMC Cancer 2013, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, Q.; Corey, E.; Xie, W.; Fan, J.; Mizokami, A.; Zhang, J. Activation of mcp-1/ccr2 axis promotes prostate cancer growth in bone. Clin. Exp. Metastasis 2009, 26, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Berry, J.A.; Shoher, A.; Ayers, G.D.; Wei, C.; Lucci, A. Cox-2 involvement in breast cancer metastasis to bone. Oncogene 2007, 26, 3789. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, S.E.; Lennard, T.W.; Williams, J.R.; Birch, M.A. Vascular endothelial growth factor acts as an osteolytic factor in breast cancer metastases to bone. Br. J. Cancer 2005, 92, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; You, S.; Kumar, V.; Zhang, C.; Cao, Y. In vitro the behaviors of metastasis with suppression of vegf in human bone metastatic lncap-derivative c4-2b prostate cancer cell line. J. Exp. Clin. Cancer Res. CR 2012, 31, 40. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Sosnoski, D.M.; Gandhi, U.H.; Novinger, L.J.; Prabhu, K.S.; Mastro, A.M. Selenium modifies the osteoblast inflammatory stress response to bone metastatic breast cancer. Carcinogenesis 2009, 30, 1941–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiirevnyamba, A.; Takahashi, T.; Shan, H.; Ogawa, H.; Yano, S.; Kanayama, H.; Izumi, K.; Uehara, H. Enhancement of osteoclastogenic activity in osteolytic prostate cancer cells by physical contact with osteoblasts. Br. J. Cancer 2011, 104, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Wang, R.; Chen, D.; Mao, J.; Shi, R.; Wu, Z.; Kang, J.; Tian, W.; Zhang, C. Cox2 is involved in hypoxia-induced tnf-alpha expression in osteoblast. Sci. Rep. 2015, 5, 10020. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumimoto, T.; Williams, P.; Yoneda, T. The sk-n-as human neuroblastoma cell line develops osteolytic bone metastases with increased angiogenesis and cox-2 expression. J. Bone Oncol. 2014, 3, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Berry, J.A.; Shoher, A.; Lucci, A. Cox-2 induces il-11 production in human breast cancer cells. J. Surg. Res. 2006, 131, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. Tgf-beta signaling blockade inhibits pthrp secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Yan, J.; Lu, X.; Xu, S.; Lerit, D.A.; Kang, Y. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat. Med. 2009, 15, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Myoui, A.; Hashimoto, N.; Sasaki, A.; Hata, K.; Morita, Y.; Yoshikawa, H.; Rosen, C.J.; Mundy, G.R.; Yoneda, T. Bone-derived igf mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res. 2012, 72, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; Lee, C.; Sadler, W.D.; Koh, A.J.; Jones, J.; Seo, J.W.; Soki, F.N.; Cho, S.W.; Daignault, S.D.; McCauley, L.K. Parathyroid hormone-related protein drives a cd11b+gr1+ cell-mediated positive feedback loop to support prostate cancer growth. Cancer Res. 2013, 73, 6574–6583. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hanai, J.I.; Le, P.T.; Bi, R.; Maridas, D.; DeMambro, V.; Figueroa, C.A.; Kir, S.; Zhou, X.; Mannstadt, M.; et al. Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab. 2017, 25, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.L.; Huang, W.D.; Li, B.; Chen, T.R.; Li, Z.X.; Zhao, C.L.; Li, H.Y.; Wu, Y.M.; Yan, W.J.; Xiao, J.R. Microrna-124 inhibits bone metastasis of breast cancer by repressing interleukin-11. Mol. Cancer 2018, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Ohshiba, T.; Miyaura, C.; Ito, A. Role of prostaglandin e produced by osteoblasts in osteolysis due to bone metastasis. Biochem. Biophys. Res. Commun. 2003, 300, 957–964. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, B.; Jin, W.J.; Kim, J.W.; Kim, H.H.; Ha, H.; Lee, Z.H. Trolox inhibits osteolytic bone metastasis of breast cancer through both pge2-dependent and independent mechanisms. Biochem. Pharmacol. 2014, 91, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Verbovsek, U.; Van Noorden, C.J.; Lah, T.T. Complexity of cancer protease biology: Cathepsin k expression and function in cancer progression. Semin. Cancer Biol. 2015, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Guise, T.; Kang, Y. The biology of bone metastasis. Cold Spring Harbor Perspect. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dayyani, F.; Gallick, G.E.; Logothetis, C.J.; Corn, P.G. Novel therapies for metastatic castrate-resistant prostate cancer. J. Natl. Cancer Inst. 2011, 103, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Gobel, A.; Browne, A.J.; Thiele, S.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Potentiated suppression of dickkopf-1 in breast cancer by combined administration of the mevalonate pathway inhibitors zoledronic acid and statins. Breast Cancer Res. Treat. 2015, 154, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.W.; Barlogie, B.; Rudikoff, S.; Shaughnessy, J.D., Jr. Dkk1-induced inhibition of wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 2008, 42, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.-Q.; Wu, J.-J.; Troiano, N.; Insogna, K. Targeted over-expression of dkk1 in osteoblasts reduces bone mass but does not impair the anabolic response to intermittent pth treatment in mice. J. Bone Miner. Metab. 2011, 29, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a rankl-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Kwan Tat, S.; Padrines, M.; Theoleyre, S.; Heymann, D.; Fortun, Y. Il-6, rankl, tnf-alpha/il-1: Interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004, 15, 49–60. [Google Scholar] [PubMed]
- Zhu, Z.; Huang, P.; Chong, Y.; George, S.K.; Wen, B.; Han, N.; Liu, Z.; Kang, L.; Lin, N. Nucleus pulposus cells derived igf-1 and mcp-1 enhance osteoclastogenesis and vertebrae disruption in lumbar disc herniation. Int. J. Clin. Exp. Pathol. 2014, 7, 8520–8531. [Google Scholar] [PubMed]
- Ohba, T.; Cole, H.A.; Cates, J.M.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Bisphosphonates inhibit osteosarcoma-mediated osteolysis via attenuation of tumor expression of mcp-1 and rankl. J. Bone Miner. Res. 2014, 29, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Jiang, Y.; Valente, A.J. The expression of monocyte chemoattractant protein-1 and other chemokines by osteoblasts. Front. Biosci. 1999, 4, D571–D580. [Google Scholar] [CrossRef] [PubMed]
- Koide, N.; Nishio, A.; Sato, T.; Sugiyama, A.; Miyagawa, S. Significance of macrophage chemoattractant protein-1 expression and macrophage infiltration in squamous cell carcinoma of the esophagus. Am. J. Gastroenterol. 2004, 99, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cai, Z.; Galson, D.L.; Xiao, G.; Liu, Y.; George, D.E.; Melhem, M.F.; Yao, Z.; Zhang, J. Monocyte chemotactic protein-1 (mcp-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate 2006, 66, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Mestdagt, M.; Polette, M.; Buttice, G.; Noel, A.; Ueda, A.; Foidart, J.-M.; Gilles, C. Transactivation of mcp-1/ccl2 by β-catenin/tcf-4 in human breast cancer cells. Int. J. Cancer 2006, 118, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Neumark, E.; Sagi-Assif, O.; Shalmon, B.; Ben-Baruch, A.; Witz, I.P. Progression of mouse mammary tumors: Mcp-1-tnf-alpha cross regulatory pathway and clonal expression of promalignancy and antimalignancy factors. Int J. Cancer 2003, 106, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.; Mastro, A.M. Cytokines secreted by bone-metastatic breast cancer cells alter the expression pattern of f-actin and reduce focal adhesion plaques in osteoblasts through PI3K. Exp. Cell Res. 2005, 310, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Wang, J.; Schneider, A.; Sun, Y.X.; Koh-Paige, A.J.; Osman, N.I.; McCauley, L.K.; Taichman, R.S. Regulation of sdf-1 (cxcl12) production by osteoblasts; a possible mechanism for stem cell homing. Bone 2006, 38, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Fang, M.; Wang, J.; Cooper, C.R.; Pienta, K.J.; Taichman, R.S. Expression and activation of alpha v beta 3 integrins by sdf-1/cxc12 increases the aggressiveness of prostate cancer cells. Prostate 2007, 67, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.J.; Li, J.K.; Lee, Y.C.; Yu, G.; Lin, S.C.; Pan, T.; Satcher, R.L.; Titus, M.A.; Yu-Lee, L.Y.; Weng, W.H.; et al. Cabozantinib-induced osteoblast secretome promotes survival and migration of metastatic prostate cancer cells in bone. Oncotarget 2017, 8, 74987–75006. [Google Scholar] [CrossRef] [PubMed]
- Kiyama, S.; Morrison, K.; Zellweger, T.; Akbari, M.; Cox, M.; Yu, D.; Miyake, H.; Gleave, M.E. Castration-induced increases in insulin-like growth factor-binding protein 2 promotes proliferation of androgen-independent human prostate lncap tumors. Cancer Res. 2003, 63, 3575–3584. [Google Scholar] [PubMed]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through wnt16b. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- van Andel, H.; Ren, Z.; Koopmans, I.; Joosten, S.P.; Kocemba, K.A.; de Lau, W.; Kersten, M.J.; de Bruin, A.M.; Guikema, J.E.; Clevers, H.; et al. Aberrantly expressed lgr4 empowers wnt signaling in multiple myeloma by hijacking osteoblast-derived r-spondins. Proc. Natl. Acad. Sci. USA 2017, 114, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Sukhdeo, K.; Mani, M.; Zhang, Y.; Dutta, J.; Yasui, H.; Rooney, M.D.; Carrasco, D.E.; Zheng, M.; He, H.; Tai, Y.T.; et al. Targeting the beta-catenin/tcf transcriptional complex in the treatment of multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 7516–7521. [Google Scholar] [CrossRef] [PubMed]
- Derksen, P.W.; Tjin, E.; Meijer, H.P.; Klok, M.D.; MacGillavry, H.D.; van Oers, M.H.; Lokhorst, H.M.; Bloem, A.C.; Clevers, H.; Nusse, R.; et al. Illegitimate wnt signaling promotes proliferation of multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6122–6127. [Google Scholar] [CrossRef] [PubMed]
- Nemani, N.; Santo, L.; Eda, H.; Cirstea, D.; Mishima, Y.; Patel, C.; O’Donnell, E.; Yee, A.; Raje, N. Role of decorin in multiple myeloma (mm) bone marrow microenvironment. J. Bone Miner. Res. 2015, 30, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, S.L.; Harris, S.T.; Bone, H.; Miller, P.D.; Orwoll, E.S.; Watts, N.B.; Rosen, C.J. Bisphosphonates: Safety and efficacy in the treatment and prevention of osteoporosis. Am. Fam. Physician 2000, 61, 2731–2736. [Google Scholar] [PubMed]
- Hillner, B.E.; Ingle, J.N.; Berenson, J.R.; Janjan, N.A.; Albain, K.S.; Lipton, A.; Yee, G.; Biermann, J.S.; Chlebowski, R.T.; Pfister, D.G. American society of clinical oncology guideline on the role of bisphosphonates in breast cancer. J. Clin. Oncol. 2000, 18, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P.D.; Demiaux, B.; Malaval, L.; Chapuy, M.C.; Edouard, C.; Meunier, P.J. Serum bone gamma carboxyglutamic acid-containing protein in primary hyperthyroidism and in malignant hypercalcemia. J. Clin. Investig. 1986, 77, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, S.C.; Rosol, T.J.; Shevrin, D.H.; York, P.A. Quantitative bone histomorphometry in nude mice bearing a human squamous cell lung cancer. J. Bone Min. Res. 1998, 3, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Galasko, C.S. Mechanisms of lytic and blastic metastatic disease of bone. Clin. Orthop. 1982, 169, 20–27. [Google Scholar] [CrossRef]
- Martin, T.J.; Moseley, J.M. Mechanisms in the skeletal complications of breast cancer. Endocr. Relat. Cancer 2000, 7, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fioramonti, M.; Santini, D.; Iuliani, M.; Ribelli, G.; Manca, P.; Papapietro, N.; Spiezia, F.; Vincenzi, B.; Denaro, V.; Russo, A.; et al. Cabozantinib targets bone microenvironment modulating human osteoclast and osteoblast functions. Oncotarget 2017, 8, 20113–20121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, M.T.; Hunter, K.D.; Robinson, S.P.; Graham, T.J.; Corey, E.; Dear, T.N.; Hughes, R.; Brown, N.J.; Holen, I. Rapid modification of the bone microenvironment following short-term treatment with cabozantinib in vivo. Bone 2015, 81, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zhang, H.; Karatsinides, A.; Keller, J.M.; Kozloff, K.M.; Aftab, D.T.; Schimmoller, F.; Keller, E.T. Cabozantinib inhibits prostate cancer growth and prevents tumor-induced bone lesions. Clin. Cancer Res. 2014, 20, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Pond, G.R.; Sonpavde, G.; Fizazi, K.; De Bono, J.S.; Basch, E.M.; Scher, H.I.; Smith, M.R. Cabozantinib for metastatic castration-resistant prostate cancer (mcrpc) following docetaxel: Combined analysis of two phase iii trials. J. Clin. Oncol. 2018, 36, 225–225. [Google Scholar] [CrossRef]
- Smith, M.; De Bono, J.; Sternberg, C.; Le Moulec, S.; Oudard, S.; De Giorgi, U.; Krainer, M.; Bergman, A.; Hoelzer, W.; De Wit, R.; et al. Phase iii study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: Comet-1. J. Clin. Oncol. 2016, 34, 3005–3013. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Matsugaki, A.; Sekita, A.; Nakano, T. Alteration of osteoblast arrangement via direct attack by cancer cells: New insights into bone metastasis. Sci. Rep. 2017, 7, 44824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Row, S.; Liu, Y.; Alimperti, S.; Agarwal, S.K.; Andreadis, S.T. Cadherin-11 is a novel regulator of extracellular matrix synthesis and tissue mechanics. J. Cell Sci. 2016, 129, 2950–2961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhe, R.P.; Gudzenko, T.; Bachmann, M.; Becker, S.F.; Gonnermann, C.; Winter, C.; Abbruzzese, G.; Alfandari, D.; Kratzer, M.-C.; Franz, C.M.; et al. Cadherin-11 localizes to focal adhesions and promotes cell–substrate adhesion. Nat. Commun. 2016, 7, 10909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjenseth, A.; Fykerud, T.A.; Sirnes, S.; Bruun, J.; Yohannes, Z.; Kolberg, M.; Omori, Y.; Rivedal, E.; Leithe, E. The gap junction channel protein connexin 43 is covalently modified and regulated by sumoylation. J. Biol. Chem. 2012, 287, 15851–15861. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Nakano, T.; Umakoshi, Y.; Yamamoto, M.; Tabata, Y. Degree of biological apatite c-axis orientation rather than bone mineral density controls mechanical function in bone regenerated using recombinant bone morphogenetic protein-2. J. Bone Miner. Res. 2013, 28, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Skedros, J.G.; Dayton, M.R.; Sybrowsky, C.L.; Bloebaum, R.D.; Bachus, K.N. The influence of collagen fiber orientation and other histocompositional characteristics on the mechanical properties of equine cortical bone. J. Exp. Biol. 2006, 209, 3025. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, A.; Miyabe, S.; Nakano, T.; Umakoshi, Y.; Ito, M.; Mihara, M. The combination therapy with alfacalcidol and risedronate improves the mechanical property in lumbar spine by affecting the material properties in an ovariectomized rat model of osteoporosis. BMC Musculoskelet. Disord. 2009, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.Q.; Maeda, Y.; Taipaleenmaki, H.; Zhang, W.; Jafferji, M.; Gordon, J.A.; Li, Z.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; et al. Mir-218 directs a wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J. Biol. Chem. 2012, 287, 42084–42092. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Li, G.X.; Tan, L.D.; Du, X.; Li, X.Q.; He, R.; Wang, Q.S.; Feng, Y.M. Breast cancer cells obtain an osteomimetic feature via epithelial-mesenchymal transition that have undergone bmp2/runx2 signaling pathway induction. Oncotarget 2016, 7, 79688–79705. [Google Scholar] [CrossRef] [PubMed]
- Hagberg Thulin, M.; Jennbacken, K.; Damber, J.E.; Welen, K. Osteoblasts stimulate the osteogenic and metastatic progression of castration-resistant prostate cancer in a novel model for in vitro and in vivo studies. Clin. Exp. Metastasis 2014, 31, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rucci, N.; Teti, A. Osteomimicry: How the seed grows in the soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Agrawal, K.C.; Abdel-Mageed, A.B. Independent and cooperative roles of tumor necrosis factor-alpha, nuclear factor-kappab, and bone morphogenetic protein-2 in regulation of metastasis and osteomimicry of prostate cancer cells and differentiation and mineralization of mc3t3-e1 osteoblast-like cells. Cancer Sci. 2010, 101, 103–111. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Osteoblast | Osteoclast | Cancer Cells |
|---|---|---|---|
| IL-6 | [50,60,189,190] | -- | [50,191] |
| IL-8 | [50,176,192,193] | [176] | [176,177,194,195,196,197] |
| MCP-1 | [50,198,199] | -- | -- |
| VEGF | [50,130,131] | -- | [50,200,201] |
| GRO-alpha | [50] | -- | -- |
| COX-2 | [202,203,204] | -- | [196,199,205,206] |
| TGF-beta | [129,175] | [207,208] | -- |
| IGF-1 | [175] | [19,209] | -- |
| PTHrP | -- | -- | [28,84,175,210,211] |
| IL-11 | [193] | -- | [177,197,206,212] |
| PGE-2 | [213] | -- | [196,214] |
| Cathepsin-K | -- | [173,174,215] | -- |
| Jagged-1 | -- | -- | [188,216] |
| Notch-1 | [216] | [188] | -- |
| RANK-L | [84,172] | -- | -- |
| RANK | -- | [84,172] | -- |
| Endothelin-1 | [217] | -- | [121,217] |
| DKK-1 | -- | -- | [121,218,219,220] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shupp, A.B.; Kolb, A.D.; Mukhopadhyay, D.; Bussard, K.M. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers 2018, 10, 182. https://doi.org/10.3390/cancers10060182
Shupp AB, Kolb AD, Mukhopadhyay D, Bussard KM. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers. 2018; 10(6):182. https://doi.org/10.3390/cancers10060182
Chicago/Turabian StyleShupp, Alison B., Alexus D. Kolb, Dimpi Mukhopadhyay, and Karen M. Bussard. 2018. "Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts" Cancers 10, no. 6: 182. https://doi.org/10.3390/cancers10060182
APA StyleShupp, A. B., Kolb, A. D., Mukhopadhyay, D., & Bussard, K. M. (2018). Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers, 10(6), 182. https://doi.org/10.3390/cancers10060182