Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment

Abstract
:1. Introduction
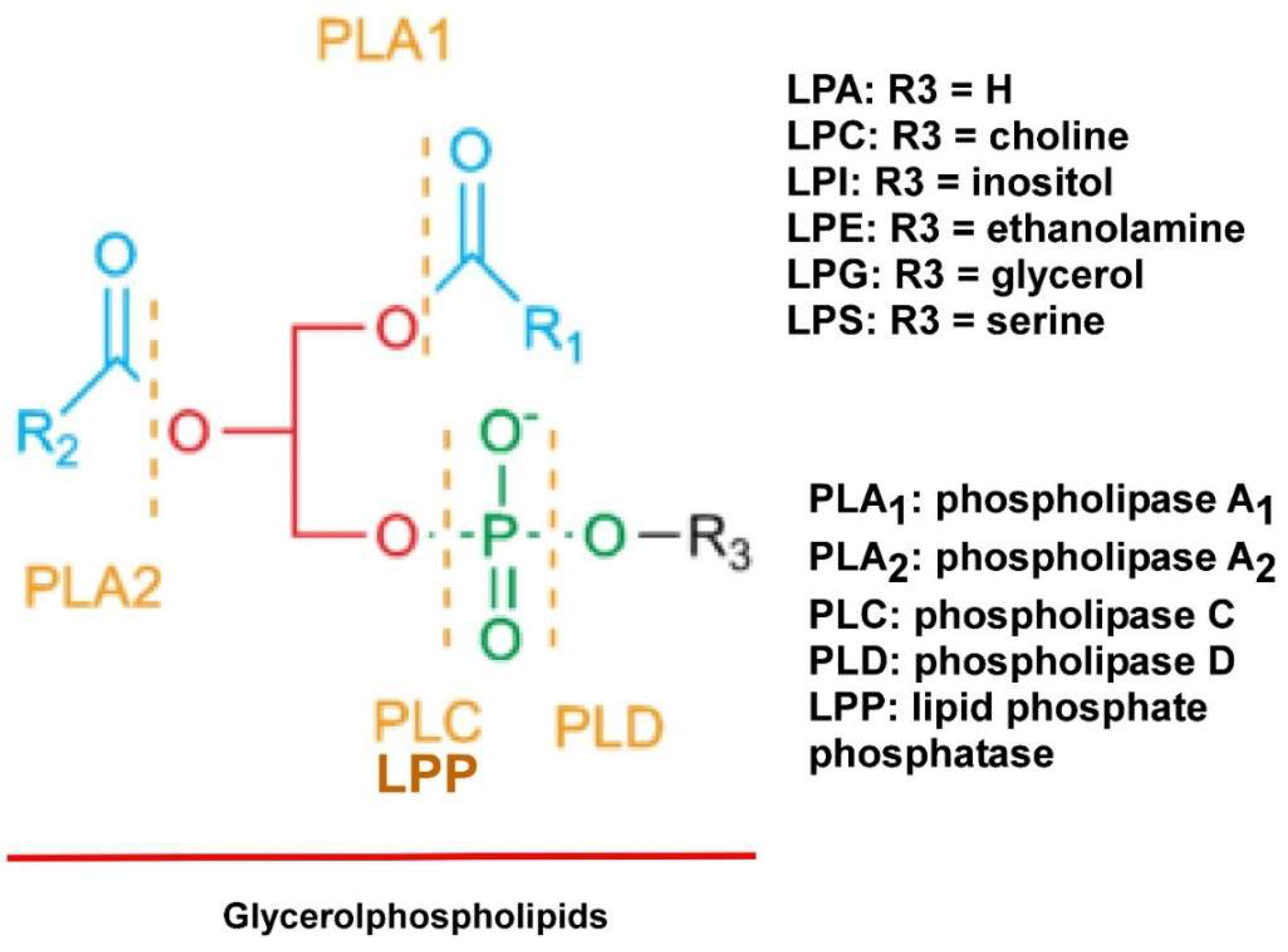
2. LPLs
2.1. LPA
2.1.1. Increased LPA Levels in EOC
2.1.2. LPA Production and Regulation
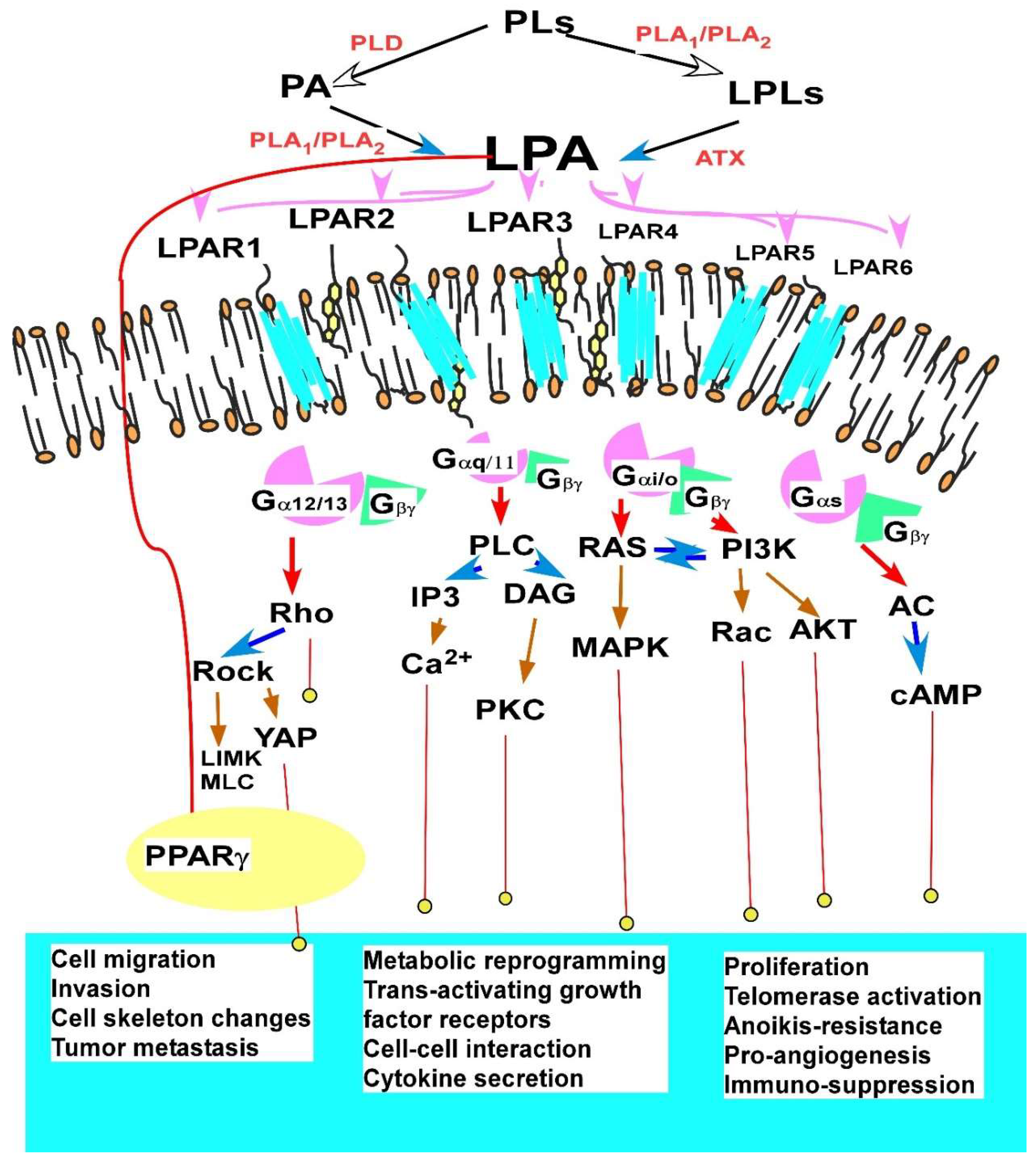
2.1.3. Major Cellular Functions and Signaling Mechanisms of LPA in EOC
2.1.4. LPA in the Immune System
2.2. LPC
2.3. Lysophosphatidylinositol (LPI) and Other LPLs
2.4. Sphingosine-1-Phosphate (S1P)
2.4.1. S1P Levels and Production
2.4.2. S1P Functions and Signaling Mechanisms in EOC
2.5. Sphingosylphosphorylcholine (SPC)
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Westermann, A.M.; Beijnen, J.H.; Moolenaar, W.H.; Rodenhuis, S. Growth factors in human ovarian cancer. Cancer Treat. Rev. 1997, 23, 113–131. [Google Scholar] [CrossRef]
- Amsterdam, A. Novel role of growth factors in ovary function. Harefuah 2010, 149, 789–793. [Google Scholar] [PubMed]
- Thibault, B.; Castells, M.; Delord, J.P.; Couderc, B. Ovarian cancer microenvironment: Implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 2014, 33, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Wang, Q.; Lau, W.B.; Lau, B.; Xu, L.; Zhao, L.; Yang, H.; Feng, M.; Xuan, Y.; Yang, Y.; et al. Tumor microenvironment: The culprit for ovarian cancer metastasis? Cancer Lett. 2016, 377, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Hodeib, M.; Serna-Gallegos, T.; Tewari, K.S. A review of HER2-targeted therapy in breast and ovarian cancer: Lessons from antiquity—Cleopatra and Penelope. Future Oncol. 2015, 11, 3113–3131. [Google Scholar] [CrossRef] [PubMed]
- Ntanasis-Stathopoulos, I.; Fotopoulos, G.; Tzanninis, I.G.; Kotteas, E.A. The emerging role of tyrosine kinase inhibitors in ovarian cancer treatment: A systematic review. Cancer Investig. 2016, 34, 313–339. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chiorean, E.G.; Chiorean, M.V.; Rex, D.K.; Robb, B.W.; Hahn, N.M.; Liu, Z.; Loehrer, P.J.; Harrison, M.L.; Xu, Y. Elevated phospholipase A2 activities in plasma samples from multiple cancers. PLoS ONE 2013, 8, e57081. [Google Scholar] [CrossRef] [PubMed]
- Pap, E.; Pallinger, E.; Pasztoi, M.; Falus, A. Highlights of a new type of intercellular communication: Microvesicle-based information transfer. Inflamm. Res. 2009, 58, 1–8. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zheng, S.; Luo, Y.; Wang, B. Exosome theranostics: Biology and translational medicine. Theranostics 2018, 8, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Beach, A.; Zhang, H.G.; Ratajczak, M.Z.; Kakar, S.S. Exosomes: An overview of biogenesis, composition and role in ovarian cancer. J. Ovarian Res. 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S.N.; Abdel-Mageed, A.B. Tumor-derived exosomes in oncogenic reprogramming and cancer progression. Cell. Mol. Life Sci. 2015, 72, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, S.; Zhang, K.; Qing, Y.; Xu, T. A comprehensive overview of exosomes in ovarian cancer: Emerging biomarkers and therapeutic strategies. J. Ovarian Res. 2017, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Maddalena, F.; Condelli, V.; Landriscina, M.; Esposito, F. Stress-adaptive response in ovarian cancer drug resistance: Role of trap1 in oxidative metabolism-driven inflammation. Adv. Protein Chem. Struct. Biol. 2017, 108, 163–198. [Google Scholar] [PubMed]
- Ke, C.; Li, A.; Hou, Y.; Sun, M.; Yang, K.; Cheng, J.; Wang, J.; Ge, T.; Zhang, F.; Li, Q.; et al. Metabolic phenotyping for monitoring ovarian cancer patients. Sci. Rep. 2016, 6, 23334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the lipid maps comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Tania, M.; Khan, M.A.; Song, Y. Association of lipid metabolism with ovarian cancer. Curr. Oncol. 2010, 17, 6–11. [Google Scholar] [PubMed]
- Tsujiuchi, T.; Araki, M.; Hirane, M.; Dong, Y.; Fukushima, N. Lysophosphatidic acid receptors in cancer pathobiology. Histol. Histopathol. 2014, 29, 313–321. [Google Scholar] [PubMed]
- Pua, T.L.; Wang, F.Q.; Fishman, D.A. Roles of LPA in ovarian cancer development and progression. Future Oncol. 2009, 5, 1659–1673. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A. Physiological and pathophysiological roles of lysophosphatidic acids produced by secretory lysophospholipase d in body fluids. Biochim. Biophys. Acta 2002, 1582, 18–25. [Google Scholar] [CrossRef]
- Turkoglu, O.; Zeb, A.; Graham, S.; Szyperski, T.; Szender, J.B.; Odunsi, K.; Bahado-Singh, R. Metabolomics of biomarker discovery in ovarian cancer: A systematic review of the current literature. Metabolomics 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Varas-Godoy, M.; Rice, G.; Illanes, S.E. The crosstalk between ovarian cancer stem cell niche and the tumor microenvironment. Stem Cells Int. 2017, 2017, 5263974. [Google Scholar] [CrossRef] [PubMed]
- Ray, U.; Roy Chowdhury, S.; Vasudevan, M.; Bankar, K.; Roychoudhury, S.; Roy, S.S. Gene regulatory networking reveals the molecular cue to lysophosphatidic acid-induced metabolic adaptations in ovarian cancer cells. Mol. Oncol. 2017, 11, 491–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Fang, X.J.; Casey, G.; Mills, G.B. Lysophospholipids activate ovarian and breast cancer cells. Biochem. J. 1995, 309, 933–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Gaudette, D.C.; Boynton, J.D.; Frankel, A.; Fang, X.J.; Sharma, A.; Hurteau, J.; Casey, G.; Goodbody, A.; Mellors, A.; et al. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin. Cancer Res. 1995, 1, 1223–1232. [Google Scholar] [PubMed]
- Benesch, M.G.K.; MacIntyre, I.T.K.; McMullen, T.P.W.; Brindley, D.N. Coming of age for autotaxin and lysophosphatidate signaling: Clinical applications for preventing, detecting and targeting tumor-promoting inflammation. Cancers 2018, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.J.; Schwartz, B.; Washington, M.; Kennedy, A.; Webster, K.; Belinson, J.; Xu, Y. Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: Comparison of the lysophospholipid contents in malignant vs. nonmalignant ascitic fluids. Anal. Biochem. 2001, 290, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Casey, G.; Mills, G.B. Effect of lysophospholipids on signaling in the human jurkat T cell line. J. Cell. Physiol. 1995, 163, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Wang, Z.; Tipps, R.; Xu, Y. Biology of lpa in health and disease. Semin. Cell Dev. Biol. 2004, 15, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Murph, M.; Mills, G.B. Targeting the lipids LPA and S1p and their signalling pathways to inhibit tumour progression. Expert. Rev. Mol. Med. 2007, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Sutphen, R.; Xu, Y.; Wilbanks, G.D.; Fiorica, J.; Grendys, E.C., Jr.; LaPolla, J.P.; Arango, H.; Hoffman, M.S.; Martino, M.; Wakeley, K.; et al. Lysophospholipids are potential biomarkers of ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 1185–1191. [Google Scholar] [PubMed]
- Xiao, Y.; Chen, Y.; Kennedy, A.W.; Belinson, J.; Xu, Y. Evaluation of plasma lysophospholipids for diagnostic significance using electrospray ionization mass spectrometry (ESI-MS) analyses. Ann. N. Y. Acad. Sci. 2000, 905, 242–259. [Google Scholar] [CrossRef] [PubMed]
- Sedlakova, I.; Vavrova, J.; Tosner, J.; Hanousek, L. Lysophosphatidic acid in ovarian cancer patients. Ceska Gynekol. 2006, 71, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Meleh, M.; Pozlep, B.; Mlakar, A.; Meden-Vrtovec, H.; Zupancic-Kralj, L. Determination of serum lysophosphatidic acid as a potential biomarker for ovarian cancer. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 858, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Sedlakova, I.; Vavrova, J.; Tosner, J.; Hanousek, L. Lysophosphatidic acid: An ovarian cancer marker. Eur. J. Gynaecol. Oncol. 2008, 29, 511–514. [Google Scholar] [PubMed]
- Nakamura, K.; Igarashi, K.; Ohkawa, R.; Yokota, H.; Masuda, A.; Nakagawa, S.; Yano, T.; Ikeda, H.; Aoki, J.; Yatomi, Y. Serum autotaxin is not a useful biomarker for ovarian cancer. Lipids 2012, 47, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Chen, Y.; Hu, Z.; Hu, C. Diagnostic value of total plasma lysophosphatidic acid in ovarian cancer: A meta-analysis. Int. J. Gynecol. Cancer 2015, 25, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Cao, L.Y.; Fu, Z.Z.; Wang, Y.J.; Wang, G.X.; Gu, T. Clinical significance of plasma lysophosphatidic acid levels in the differential diagnosis of ovarian cancer. J. Cancer Res. Ther. 2015, 11, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Zhang, W.C.; Zhang, J.L.; Zheng, C.J.; Zhu, H.; Yu, H.M.; Fan, L.M. Plasma levels of lysophosphatidic acid in ovarian cancer versus controls: A meta-analysis. Lipids Health Dis. 2015, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.M.; Havik, E.; Postma, F.R.; Beijnen, J.H.; Dalesio, O.; Moolenaar, W.H.; Rodenhuis, S. Malignant effusions contain lysophosphatidic acid (LPA)-like activity. Ann. Oncol. 1998, 9, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Zhao, Z.; Antalis, C.; Yan, L.; Del Priore, G.; Hamed, A.H.; Stehman, F.B.; Schilder, J.M.; Xu, Y. Elevated and secreted phospholipase A(2) activities as new potential therapeutic targets in human epithelial ovarian cancer. FASEB J. 2012, 26, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Murph, M.; Tanaka, T.; Pang, J.; Felix, E.; Liu, S.; Trost, R.; Godwin, A.K.; Newman, R.; Mills, G. Liquid chromatography mass spectrometry for quantifying plasma lysophospholipids: Potential biomarkers for cancer diagnosis. Methods Enzymol. 2007, 433, 1–25. [Google Scholar] [PubMed]
- Zhao, Z.; Xu, Y. An extremely simple method for extraction of lysophospholipids and phospholipids from blood samples. J. Lipid Res. 2010, 51, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Michels, M.; Japtok, L.; Alisjahbana, B.; Wisaksana, R.; Sumardi, U.; Puspita, M.; Kleuser, B.; de Mast, Q.; van der Ven, A.J. Decreased plasma levels of the endothelial protective sphingosine-1-phosphate are associated with dengue-induced plasma leakage. J. Infect. 2015, 71, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Raza, A.; Sturgill, J.; Lyon, D.; Young, J.; Hait, N.C.; Takabe, K. Paradoxical association of postoperative plasma sphingosine-1-phosphate with breast cancer aggressiveness and chemotherapy. Mediators Inflamm. 2017, 2017, 5984819. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Uchida, H.; Nagai, J.; Inoue, M.; Aoki, J.; Ueda, H. Evidence for de novo synthesis of lysophosphatidic acid in the spinal cord through phospholipase A2 and autotaxin in nerve injury-induced neuropathic pain. J. Pharmacol. Exp. Ther. 2010, 333, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Eder, A.M.; Sasagawa, T.; Mao, M.; Aoki, J.; Mills, G.B. Constitutive and lysophosphatidic acid (LPA)-induced LPA production: Role of phospholipase D and phospholipase a2. Clin. Cancer Res. 2000, 6, 2482–2491. [Google Scholar] [PubMed]
- Sengupta, S.; Xiao, Y.J.; Xu, Y. A novel laminin-induced LPA autocrine loop in the migration of ovarian cancer cells. FASEB J. 2003, 17, 1570–1572. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Belinson, J.; Morton, R.E.; Xu, Y.; Xu, Y. Phorbol 12-myristate 13-acetate stimulates lysophosphatidic acid secretion from ovarian and cervical cancer cells but not from breast or leukemia cells. Gynecol. Oncol. 1998, 71, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell. Dev. Biol. 2004, 15, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Tang, X.; Venkatraman, G.; Bekele, R.T.; Brindley, D.N. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J. Biomed. Res. 2016, 30, 272–284. [Google Scholar] [PubMed]
- Nikolaou, A.; Kokotou, M.G.; Limnios, D.; Psarra, A.; Kokotos, G. Autotaxin inhibitors: A patent review (2012–2016). Expert. Opin. Ther. Pat. 2017, 27, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Gaits, F.; Fourcade, O.; Le Balle, F.; Gueguen, G.; Gaige, B.; Gassama-Diagne, A.; Fauvel, J.; Salles, J.P.; Mauco, G.; Simon, M.F.; et al. Lysophosphatidic acid as a phospholipid mediator: Pathways of synthesis. FEBS Lett. 1997, 410, 54–58. [Google Scholar] [CrossRef]
- Pages, C.; Simon, M.F.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat. 2001, 64, 1–10. [Google Scholar] [CrossRef]
- Roszkowski, I.; Niewiarowska, M.; Czerwinska, J.; Bar-Pratkowska, J.; Obrebski, T. Problems of surgical treatment of a patient with blood platelet disorders. Ginekol. Pol. 1971, 42, 1499–1501. [Google Scholar] [PubMed]
- Hisada, Y.; Geddings, J.E.; Ay, C.; Mackman, N. Venous thrombosis and cancer: From mouse models to clinical trials. J. Thromb. Haemost. 2015, 13, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Menczer, J. Preoperative elevated platelet count and thrombocytosis in gynecologic malignancies. Arch. Gynecol. Obstet. 2017, 295, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Swier, N.; Versteeg, H.H. Reciprocal links between venous thromboembolism, coagulation factors and ovarian cancer progression. Thromb. Res. 2017, 150, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Huang, F.; He, Z.; Zuo, M.Z. Clinicopathological and prognostic significance of platelet count in patients with ovarian cancer. Climacteric 2018, 21, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Afshar-Kharghan, V.; Schafer, A.I. Paraneoplastic thrombocytosis: The secrets of tumor self-promotion. Blood 2014, 124, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Nugent, D.; Belinson, J.L.; Xu, Y. The synergistic interactions of oleoyl-lysophosphatidic acid in platelet aggregation. Med. Sci. Res. 1999, 27, 435–441. [Google Scholar]
- Nugent, D.; Xu, Y. Sphingosine-1-phosphate: Characterization of its inhibition of platelet aggregation. Platelets 2000, 11, 226–232. [Google Scholar] [PubMed]
- Leblanc, R.; Houssin, A.; Peyruchaud, O. Platelets, autotaxin and lysophosphatidic acid signaling: Win-win factors for cancer metastasis. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Clark, R.; Chekmareva, M.; Johnson, A.; George, S.; Shaw, P.; Seewaldt, V.; Rinker-Schaeffer, C. In vivo and ex vivo approaches to study ovarian cancer metastatic colonization of milky spot structures in peritoneal adipose. J. Vis. Exp. 2015, e52721. [Google Scholar] [CrossRef] [PubMed]
- Feist, P.E.; Loughran, E.A.; Stack, M.S.; Hummon, A.B. Quantitative proteomic analysis of murine white adipose tissue for peritoneal cancer metastasis. Anal. Bioanal. Chem. 2018, 410, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yan, L.; Xu, Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene 2015, 34, 3315–3324. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase d activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell. Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Volden, P.A.; Skor, M.N.; Johnson, M.B.; Singh, P.; Patel, F.N.; McClintock, M.K.; Brady, M.J.; Conzen, S.D. Mammary adipose tissue-derived lysophospholipids promote estrogen receptor-negative mammary epithelial cell proliferation. Cancer Prev. Res. 2016, 9, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.G.K.; Yang, Z.; Tang, X.; Meng, G.; Brindley, D.N. Lysophosphatidate signaling: The tumor microenvironment’s new nemesis. Trends Cancer 2017, 3, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xiao, Y.J.; Singh, L.S.; Zhao, X.; Zhao, Z.; Feng, L.; Rose, T.M.; Prestwich, G.D.; Xu, Y. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res. 2006, 66, 3006–3014. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, S.; Georas, S.N. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Guerrouahen, B.S.; Pasquier, J.; Satheesh, N.J.; Suhre, K.; Rafii, A. Nesting of colon and ovarian cancer cells in the endothelial niche is associated with alterations in glycan and lipid metabolism. Sci. Rep. 2017, 7, 39999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 induction by IFNgamma and TNFalpha self-limits type-1 immunity in the human tumor microenvironment. Cancer Immunol. Res. 2016, 4, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Reinartz, S.; Finkernagel, F.; Adhikary, T.; Rohnalter, V.; Schumann, T.; Schober, Y.; Nockher, W.A.; Nist, A.; Stiewe, T.; Jansen, J.M.; et al. A transcriptome-based global map of signaling pathways in the ovarian cancer microenvironment associated with clinical outcome. Genome Biol. 2016, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Raines, T.A.; Lynch, K.R.; Slack-Davis, J.K. Decreased peritoneal ovarian cancer growth in mice lacking expression of lipid phosphate phosphohydrolase 1. PLoS ONE 2015, 10, e0120071. [Google Scholar] [CrossRef] [PubMed]
- Baudhuin, L.M.; Cristina, K.L.; Lu, J.; Xu, Y. AKT activation induced by lysophosphatidic acid and sphingosine-1-phosphate requires both mitogen-activated protein kinase kinase and p38 mitogen-activated protein kinase and is cell-line specific. Mol. Pharmacol. 2002, 62, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xu, Y. The role of lpa and yap signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 2013, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cai, Q.; Li, P.; Wang, W.; Wang, J.; Gerry, E.; Wang, T.L.; Shih, I.M.; Nephew, K.P.; Xu, Y. The novel zip4 regulation and its role in ovarian cancer. Oncotarget 2017, 8, 90090–90107. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cai, Q.; Xu, Y. LPA Regulates sox9 in Ovarian Cancer Cells; Gavin Publishers: Lisle, IL, USA, 2017. [Google Scholar]
- Fang, X.; Yu, S.; Bast, R.C.; Liu, S.; Xu, H.J.; Hu, S.X.; LaPushin, R.; Claret, F.X.; Aggarwal, B.B.; Lu, Y.; et al. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. J. Biol. Chem. 2004, 279, 9653–9661. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Ward, J.D.; Radhakrishnan, R.; Jayaraman, M.; Song, Y.S.; Dhanasekaran, D.N. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker slug/snail2 in ovarian cancer cells via galphai2, src, and hif1alpha signaling nexus. Oncotarget 2016, 7, 37664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Berk, M.; Singh, L.S.; Tan, H.; Yin, L.; Powell, C.T.; Xu, Y. Kiss1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase c alpha. Clin. Exp. Metastasis 2005, 22, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Sengupta, S.; Berk, M.; Kwak, Y.G.; Escobar, P.F.; Belinson, J.; Mok, S.C.; Xu, Y. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 2006, 66, 7983–7990. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, D.; Zhang, H.; Kirmani, K.; Zhao, Z.; Steinmetz, R.; Xu, Y. Lysophosphatidic acid stimulates cell migration, invasion, and colony formation as well as tumorigenesis/metastasis of mouse ovarian cancer in immunocompetent mice. Mol. Cancer Ther. 2009, 8, 1692–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhao, Z.; Wei, G.; Yan, L.; Wang, D.; Zhang, H.; Sandusky, G.E.; Turk, J.; Xu, Y. Group via phospholipase a2 in both host and tumor cells is involved in ovarian cancer development. FASEB J. 2010, 24, 4103–4116. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xiao Yj, Y.J.; Baudhuin, L.M.; Hong, G.; Xu, Y. Role of ether-linked lysophosphatidic acids in ovarian cancer cells. J. Lipid Res. 2002, 43, 463–476. [Google Scholar] [PubMed]
- Ren, J.; Li, Y.; Zhang, Y.L.; Zhou, X.H.; Zhang, L.; Yang, Y.; Li, Y. Effect of inhibitors of phospholipase A(2); on the metastasis potentials of human ovarian cancer cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2010, 26, 992–995. (In Chinese) [Google Scholar] [PubMed]
- Schwartz, B.M.; Hong, G.; Morrison, B.H.; Wu, W.; Baudhuin, L.M.; Xiao, Y.J.; Mok, S.C.; Xu, Y. Lysophospholipids increase interleukin-8 expression in ovarian cancer cells. Gynecol. Oncol. 2001, 81, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Kim, K.S.; Berk, M.P.; Oates, R.; Escobar, P.; Belinson, J.; Li, W.; Lindner, D.J.; Williams, B.; Xu, Y. Lysophosphatidic acid downregulates tissue inhibitor of metalloproteinases, which are negatively involved in lysophosphatidic acid-induced cell invasion. Oncogene 2007, 26, 2894–2901. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Gaudette, D.; Furui, T.; Mao, M.; Estrella, V.; Eder, A.; Pustilnik, T.; Sasagawa, T.; Lapushin, R.; Yu, S.; et al. Lysophospholipid growth factors in the initiation, progression, metastases, and management of ovarian cancer. Ann. N. Y. Acad. Sci. 2000, 905, 188–208. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Schummer, M.; Mao, M.; Yu, S.; Tabassam, F.H.; Swaby, R.; Hasegawa, Y.; Tanyi, J.L.; LaPushin, R.; Eder, A.; et al. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim. Biophys. Acta 2002, 1582, 257–264. [Google Scholar] [CrossRef]
- Mills, G.B.; Eder, A.; Fang, X.; Hasegawa, Y.; Mao, M.; Lu, Y.; Tanyi, J.; Tabassam, F.H.; Wiener, J.; Lapushin, R.; et al. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat. Res. 2002, 107, 259–283. [Google Scholar] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. Lpa receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Jesionowska, A.; Cecerska-Heryc, E.; Matoszka, N.; Dolegowska, B. Lysophosphatidic acid signaling in ovarian cancer. J. Recept Signal. Transduct. Res. 2015, 35, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Wen, Z.Q.; Xu, W.P.; Wang, Z.Y.; Du, X.L.; Wang, F. Inhibition of lysophosphatidic acid receptor-2 expression by rna interference decreases lysophosphatidic acid-induced urokinase plasminogen activator activation, cell invasion, and migration in ovarian cancer SKOV-3 cells. Croat Med. J. 2008, 49, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zheng, D.; Deng, X.; Bai, L.; Xu, Y.; Cong, Y.S. Lysophosphatidic acid activates telomerase in ovarian cancer cells through hypoxia-inducible factor-1alpha and the PI3K pathway. J. Cell Biochem. 2008, 105, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.Q.; Yao, Y.W.; Liu, C.H.; Zhang, H.; Xu, X.B.; Zeng, J.L.; Liang, W.J.; Yang, W.; Song, Y. Diagnostic and prognostic significance of lysophosphatidic acid in malignant pleural effusions. J. Thorac. Dis. 2014, 6, 483–490. [Google Scholar] [PubMed]
- Fan, Q.; Cai, Q.; Xu, Y. Foxm1 is a downstream target of lpa and yap oncogenic signaling pathways in high grade serous ovarian cancer. Oncotarget 2015, 6, 27688–27699. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.A.; Liu, Y.; Ellerbroek, S.M.; Stack, M.S. Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and mmp-dependent invasion in ovarian cancer cells. Cancer Res. 2001, 61, 3194–3199. [Google Scholar] [PubMed]
- Burkhalter, R.J.; Westfall, S.D.; Liu, Y.; Stack, M.S. Lysophosphatidic acid initiates epithelial to mesenchymal transition and induces beta-catenin-mediated transcription in epithelial ovarian carcinoma. J. Biol. Chem. 2015, 290, 22143–22154. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, Y.; Wang, X. The roles of glycodelin in cancer development and progression. Front. Immunol. 2017, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xiao, Y.J.; Zhu, K.; Baudhuin, L.M.; Lu, J.; Hong, G.; Kim, K.S.; Cristina, K.L.; Song, L.; F, S.W.; et al. Unfolding the pathophysiological role of bioactive lysophospholipids. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2003, 3, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, Y.; Indiana University School of Medicine, Indianapolis, IN, USA. Unpublished observation. 2007.
- Seo, E.J.; Kwon, Y.W.; Jang, I.H.; Kim, D.K.; Lee, S.I.; Choi, E.J.; Kim, K.H.; Suh, D.S.; Lee, J.H.; Choi, K.U.; et al. Autotaxin regulates maintenance of ovarian cancer stem cells through lysophosphatidic acid-mediated autocrine mechanism. Stem Cells 2016, 34, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M.; Tanaka, Y.; Kato, H.E.; Nakada-Nakura, Y.; et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Hope, J.M.; Wang, F.Q.; Whyte, J.S.; Ariztia, E.V.; Abdalla, W.; Long, K.; Fishman, D.A. LPA receptor 2 mediates LPA-induced endometrial cancer invasion. Gynecol. Oncol. 2009, 112, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, D.; Iyer, S.; Ghaleb, A.M.; Shim, H.; Yang, V.W.; Chun, J.; Yun, C.C. The absence of lpa2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology 2009, 136, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, N.C.; Chun, J. Promising pharmacological directions in the world of lysophosphatidic acid signaling. Biomol. Ther. 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Fukushima, K.; Onishi, Y.; Inui, K.; Node, Y.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Lysophosphatidic acid (LPA) signaling via LPA4 and LPA6 negatively regulates cell motile activities of colon cancer cells. Biochem. Biophys. Res. Commun. 2017, 483, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Hirane, M.; Fukushima, K.; Tomimatsu, A.; Fukushima, N.; Tsujiuchi, T. Diverse effects of LPA4, LPA5 and LPA6 on the activation of tumor progression in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Fukushima, K.; Otagaki, S.; Ishimoto, K.; Minami, K.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Effects of LPA1 and LPA6 on the regulation of colony formation activity in colon cancer cells treated with anticancer drugs. J. Recept. Signal. Transduct. Res. 2018, 38, 71–75. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular ppargamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Tsukahara, R.; Yasuda, S.; Makarova, N.; Valentine, W.J.; Allison, P.; Yuan, H.; Baker, D.L.; Li, Z.; Bittman, R.; et al. Different residues mediate recognition of 1-O-oleyllysophosphatidic acid and rosiglitazone in the ligand binding domain of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2006, 281, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T. Ppar gamma networks in cell signaling: Update and impact of cyclic phosphatidic acid. J. Lipids 2013, 2013, 246597. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Haniu, H.; Matsuda, Y. Effect of alkyl glycerophosphate on the activation of peroxisome proliferator-activated receptor gamma and glucose uptake in C2C12 cells. Biochem. Biophys. Res. Commun. 2013, 433, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Finkernagel, F.; Reinartz, S.; Konzer, A.; Adhikary, T.; Nist, A.; Stiewe, T.; Wagner, U.; Looso, M.; Graumann, J.; et al. Proteotranscriptomics reveal signaling networks in the ovarian cancer microenvironment. Mol. Cell. Proteom. 2018, 17, 270–289. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, S.A.; Capece, T.; Popovic, M.; Chapman, T.J.; Rezaee, F.; Kim, M.; Georas, S.N. Regulation of T cell motility in vitro and in vivo by LPA and LPA2. PLoS ONE 2014, 9, e101655. [Google Scholar] [CrossRef] [PubMed]
- Okita, M.; Gaudette, D.C.; Mills, G.B.; Holub, B.J. Elevated levels and altered fatty acid composition of plasma lysophosphatidylcholine(lysopc) in ovarian cancer patients. Int. J. Cancer 1997, 71, 31–34. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Li, L.; Wei, J.; Xiong, S.; Zhao, Z. High resolution mass spectrometry coupled with multivariate data analysis revealing plasma lipidomic alteration in ovarian cancer in asian women. Talanta 2016, 150, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xiao, Y.; Elson, P.; Tan, H.; Plummer, S.J.; Berk, M.; Aung, P.P.; Lavery, I.C.; Achkar, J.P.; Li, L.; et al. Plasma lysophosphatidylcholine levels: Potential biomarkers for colorectal cancer. J. Clin. Oncol. 2007, 25, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xu, Y. Measurement of endogenous lysophosphatidic acid by ESI-MS/MS in plasma samples requires pre-separation of lysophosphatidylcholine. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 3739–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, T.; Jakubzig, B.; Grundmann, M.; Massing, U.; Kostenis, E.; Schlesinger, M.; Bendas, G. The molecular mechanism by which saturated lysophosphatidylcholine attenuates the metastatic capacity of melanoma cells. FEBS Open Bio 2016, 6, 1297–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, T.; Floegel, A.; Sookthai, D.; Johnson, T.; Rolle-Kampczyk, U.; Otto, W.; von Bergen, M.; Boeing, H.; Kaaks, R. Higher plasma levels of lysophosphatidylcholine 18:0 are related to a lower risk of common cancers in a prospective metabolomics study. BMC Med. 2016, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Terada, N.; Inoue, T.; Kobayashi, T.; Nakayama, K.; Okada, Y.; Yoshikawa, T.; Miyazaki, Y.; Uegaki, M.; Utsunomiya, N.; et al. Decreased expression of lysophosphatidylcholine (16:0/OH) in high resolution imaging mass spectrometry independently predicts biochemical recurrence after surgical treatment for prostate cancer. Prostate 2015, 75, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wilkins, P.; Hu, W.; Murthy, K.S.; Chen, J.; Lee, Z.; Oyesanya, R.; Wu, J.; Barbour, S.E.; Fang, X. Inhibition of calcium-independent phospholipase A2 suppresses proliferation and tumorigenicity of ovarian carcinoma cells. Biochem. J. 2007, 406, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, Z.; Antalis, C.; Zhao, Z.; Emerson, R.; Wei, G.; Zhang, S.; Zhang, Z.Y.; Xu, Y. Combination therapy of an inhibitor of group via phospholipase A2 with paclitaxel is highly effective in blocking ovarian cancer development. Am. J. Pathol. 2011, 179, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.B.; Iaciura, B.M.; Nohara, L.L.; Lopes, C.D.; Veas, E.M.; Mariano, V.S.; Bozza, P.T.; Lopes, U.G.; Atella, G.C.; Almeida, I.C.; et al. Lysophosphatidylcholine triggers TLR2- and TLR4-mediated signaling pathways but counteracts LPS-induced no synthesis in peritoneal macrophages by inhibiting NF-kappab translocation and MAPK/ERK phosphorylation. PLoS ONE 2013, 8, e76233. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Li, Y.; Kuo, Y.M.; Andrews, A.J.; Nanayakkara, G.; Johnson, C.; Fu, H.; Shan, H.; Du, F.; et al. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- de Bony, J.; Dufourcq, J.; Clin, B. Lipid-protein interactions: NMR study of melittin and its binding to lysophosphatidylcholine. Biochim. Biophys. Acta 1979, 552, 531–534. [Google Scholar] [CrossRef]
- Kim, Y.L.; Im, Y.J.; Ha, N.C.; Im, D.S. Albumin inhibits cytotoxic activity of lysophosphatidylcholine by direct binding. Prostaglandins Other Lipid Mediat. 2007, 83, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Fang, P.; Sun, Y.; Jiang, X.; Wang, H.; Yang, X.F. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C.; et al. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017, 27, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Cai, Q.; Xu, Y. The lipidomic analyses in low and highly aggressive ovarian cancer cell lines. Lipids 2016, 51, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine kinases and sphingosine 1-phosphate receptors: Signaling and actions in the cardiovascular system. Front. Pharmacol. 2017, 8, 556. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, D.; Haddadi, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphingosine kinase (SPHK) isoenzymes and isoform expression: Challenges for sphk as an oncotarget. Oncotarget 2017, 8, 36898–36929. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.I.; Campos, L.E.; Castro, M.G.; Aladhami, A.; Oskeritzian, C.A.; Alvarez, S.E. Sphingosine-1 phosphate: A new modulator of immune plasticity in the tumor microenvironment. Front. Oncol. 2016, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Takabe, K.; Terracina, K.P.; Soma, D.; Hirose, Y.; Kobayashi, T.; Matsuda, Y.; Wakai, T. Sphingosine-1-phosphate transporters as targets for cancer therapy. Biomed. Res. Int. 2014, 2014, 651727. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostenis, E. Novel clusters of receptors for sphingosine-1-phosphate, sphingosylphosphorylcholine, and (lyso)-phosphatidic acid: New receptors for “old” ligands. J. Cell. Biochem. 2004, 92, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.C.; Kim, K.M.; Lee, K.S.; Namkoong, S.; Lee, S.J.; Han, J.A.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Serum bioactive lysophospholipids prevent trail-induced apoptosis via PI3K/AKT-dependent cflip expression and bad phosphorylation. Cell Death Differ. 2004, 11, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Kim, M.K.; Lee, H.Y.; Kim, S.D.; Lee, S.Y.; Kim, J.M.; Ryu, S.H.; Bae, Y.S. S1p stimulates chemotactic migration and invasion in ovcar3 ovarian cancer cells. Biochem. Biophys Res. Commun. 2007, 356, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Xia, P.; Di, W. Sphingosine 1-phosphate: A potential molecular target for ovarian cancer therapy? Cancer Investig. 2014, 32, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Liu, W.R.; Tian, M.X.; Fan, J.; Shi, Y.H. The SPHKS/S1P/S1PR1 axis in immunity and cancer: More ore to be mined. World J. Surg. Oncol. 2016, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; Baudhuin, L.M.; Xu, Y. Sphingosine-1-phosphate modulates growth and adhesion of ovarian cancer cells. FEBS Lett. 1999, 460, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Baudhuin, L.M.; Jiang, Y.; Zaslavsky, A.; Ishii, I.; Chun, J.; Xu, Y. S1p3-mediated AKT activation and cross-talk with platelet-derived growth factor receptor (PDGFR). FASEB J. 2004, 18, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhao, Z.; Caperell-Grant, A.; Yang, G.; Mok, S.C.; Liu, J.; Bigsby, R.M.; Xu, Y. S1p differentially regulates migration of human ovarian cancer and human ovarian surface epithelial cells. Mol. Cancer Ther. 2008, 7, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Devine, K.M.; Smicun, Y.; Hope, J.M.; Fishman, D.A. S1p induced changes in epithelial ovarian cancer proteolysis, invasion, and attachment are mediated by GI and RAC. Gynecol. Oncol. 2008, 110, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Smicun, Y.; Gil, O.; Devine, K.; Fishman, D.A. S1p and LPA have an attachment-dependent regulatory effect on invasion of epithelial ovarian cancer cells. Gynecol. Oncol. 2007, 107, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Smicun, Y.; Reierstad, S.; Wang, F.Q.; Lee, C.; Fishman, D.A. S1p regulation of ovarian carcinoma invasiveness. Gynecol. Oncol. 2006, 103, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Troupiotis-Tsailaki, A.; Zachmann, J.; Gonzalez-Gil, I.; Gonzalez, A.; Ortega-Gutierrez, S.; Lopez-Rodriguez, M.L.; Pardo, L.; Govaerts, C. Ligand chain length drives activation of lipid g protein-coupled receptors. Sci. Rep. 2017, 7, 2020. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Liu, Y.; Xie, L.; Wu, X.; Qiu, L.; Di, W. Sphingosine kinase 1/sphingosine-1-phosphate (S1p)/S1p receptor axis is involved in ovarian cancer angiogenesis. Oncotarget 2017, 8, 74947–74961. [Google Scholar] [CrossRef] [PubMed]
- Fyrst, H.; Saba, J.D. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 2010, 6, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Illuzzi, G.; Bernacchioni, C.; Aureli, M.; Prioni, S.; Frera, G.; Donati, C.; Valsecchi, M.; Chigorno, V.; Bruni, P.; Sonnino, S.; et al. Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl)retinamide in human ovarian cancer cells. J. Biol. Chem. 2010, 285, 18594–18602. [Google Scholar] [CrossRef] [PubMed]
- Snider, A.J.; Orr Gandy, K.A.; Obeid, L.M. Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 2010, 92, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Patmanathan, S.N.; Wang, W.; Yap, L.F.; Herr, D.R.; Paterson, I.C. Mechanisms of sphingosine 1-phosphate receptor signalling in cancer. Cell Signal. 2017, 34, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cheng, Y.; Chang, H.M.; Deguchi, M.; Hsueh, A.J.; Leung, P.C.K. Sphingosine-1-phosphate promotes ovarian cancer cell proliferation by disrupting hippo signaling. Oncotarget 2017, 8, 27166–27176. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhao, S.; Liu, S.; Liu, Y.; Li, X.; Li, S. MicroRNA-148a inhibits migration and invasion of ovarian cancer cells via targeting sphingosine-1-phosphate receptor 1. Mol. Med. Rep. 2015, 12, 3775–3780. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Takuwa, N.; Yoshioka, K.; Okamoto, Y.; Gonda, K.; Sugihara, K.; Fukamizu, A.; Asano, M.; Takuwa, Y. S1p(2), the G protein-coupled receptor for sphingosine-1-phosphate, negatively regulates tumor angiogenesis and tumor growth in vivo in mice. Cancer Res. 2010, 70, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Liu, Y.X.; Xie, L.; Di, W. Effect of S1PR2 inhibition on epithelial ovarian cancer SKOV3 cell proliferation in vitro and in vivo. Zhonghua Fu Chan Ke Za Zhi 2018, 53, 106–110. (In Chinese) [Google Scholar] [PubMed]
- Michaud, J.; Im, D.S.; Hla, T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J. Immunol. 2010, 184, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Dolezalova, H.; Kong, Y.; Hu, Y.L.; Jaffe, R.B.; Kalli, K.R.; Conover, C.A. Distinctive expression and functions of the type 4 endothelial differentiation gene-encoded G protein-coupled receptor for lysophosphatidic acid in ovarian cancer. Cancer Res. 1999, 59, 5370–5375. [Google Scholar] [PubMed]
- Argraves, K.M.; Wilkerson, B.A.; Argraves, W.S. Sphingosine-1-phosphate signaling in vasculogenesis and angiogenesis. World J. Biol. Chem. 2010, 1, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Lucke, S.; Levkau, B. Endothelial functions of sphingosine-1-phosphate. Cell. Physiol. Biochem. 2010, 26, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Ren, J.; Jiang, Y.; Ebrahem, Q.; Tipps, R.; Cristina, K.; Xiao, Y.J.; Qiao, J.; Taylor, K.L.; Lum, H.; et al. GPR4 plays a critical role in endothelial cell function and mediates the effects of sphingosylphosphorylcholine. FASEB J. 2005, 19, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Beach, J.A.; Aspuria, P.J.; Cheon, D.J.; Lawrenson, K.; Agadjanian, H.; Walsh, C.S.; Karlan, B.Y.; Orsulic, S. Sphingosine kinase 1 is required for TGF-beta mediated fibroblastto- myofibroblast differentiation in ovarian cancer. Oncotarget 2016, 7, 4167–4182. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SPHK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van, I.W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.Y.; Shin, E.H.; Bae, Y.S. Sphingosylphosphorylcholine stimulates human monocyte-derived dendritic cell chemotaxis. Acta Pharmacol. Sin. 2006, 27, 1359–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, A.; Sabatte, J.; Nahmod, K.; Martinez, D.; Salamone, G.; Vermeulen, M.; Maggini, J.; Salomon, H.; Geffner, J. Sphingosylphosphorylcholine activates dendritic cells, stimulating the production of interleukin-12. Immunology 2007, 121, 328–336. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Lipid | EOC or BC Plasma | Healthy Control Plasma | EOC Ascites | Benign Ascites |
|---|---|---|---|---|
| Acyl-LPA | 2–22 [33,34] | 0.6–0.9 [33,34] | 19–95 [28,43] | 2.9 [28,43] |
| Alkyl-, and alkenyl-LPA | 3.7 ± 1.7 [28] | 0.9 ± 0.7 [28] | ||
| LPI | to 3.0 [28,35] | 0–1.5 [28,35] | 14.7 ± 9.7 [28,35] | 2.9 ± 2.0 [28,35] |
| LPC | 120 ± 0.30 [45] 117–153 [34] | 128 ± 46 [45] 122 [34] | ||
| S1P | 0.52 ± 0.12 [137] | 0.58 ± 0.18 [45,46,47,48] | sub-µM to low µM [45,46,47,48] | sub-µM to low µM [45,46,47,48] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y. Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment. Cancers 2018, 10, 227. https://doi.org/10.3390/cancers10070227
Xu Y. Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment. Cancers. 2018; 10(7):227. https://doi.org/10.3390/cancers10070227
Chicago/Turabian StyleXu, Yan. 2018. "Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment" Cancers 10, no. 7: 227. https://doi.org/10.3390/cancers10070227
APA StyleXu, Y. (2018). Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment. Cancers, 10(7), 227. https://doi.org/10.3390/cancers10070227