The Dynamic Roles of TGF-β Signalling in EBV-Associated Cancers


 ,
,
Abstract
1. Introduction
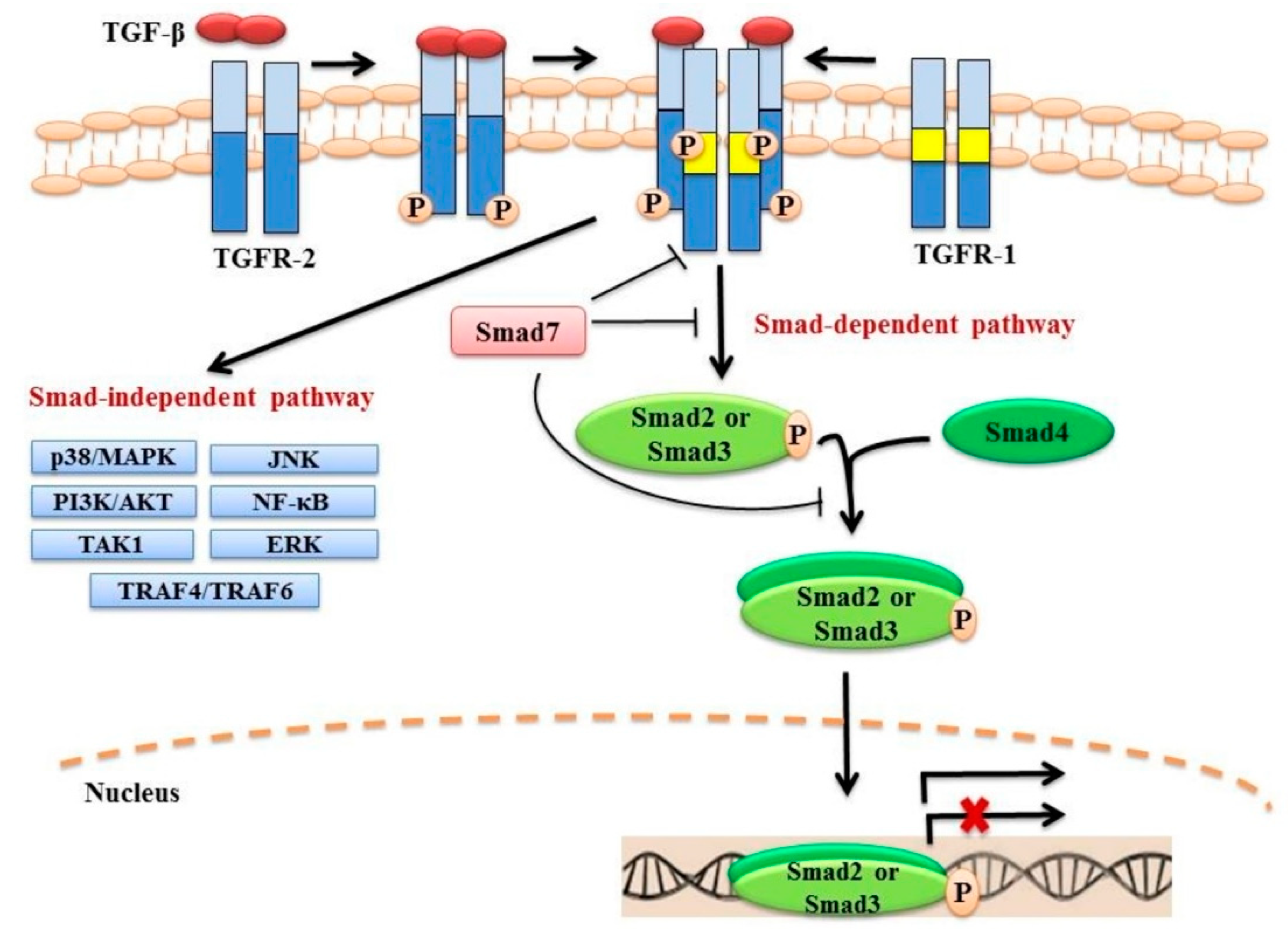
2. TGF-β Signalling
2.1. Canonical Smad-Dependent Signalling
2.2. Non-Canonical Smad-Independent Signalling
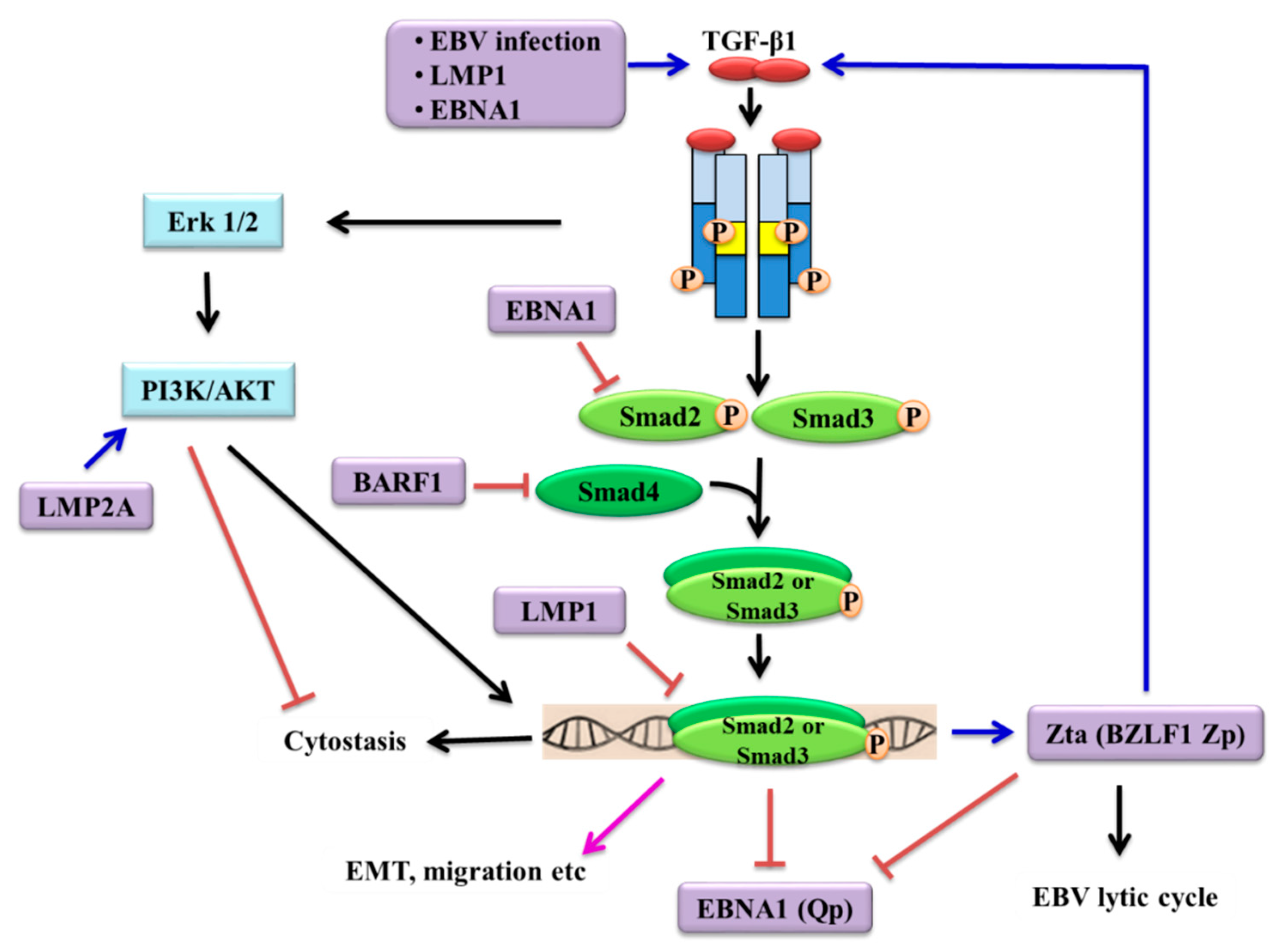
3. Resistance of EBV-Positive Cells to TGF-β-Mediated Cytostasis
3.1. Contribution of EBV Latent Genes
3.2. Dysregulation of TGF-β Receptors
4. Tumour Promoting Roles of TGF-β
4.1. High Levels of TGF-β in EBV-Associated Cancers
4.2. Contribution of TGF-β Signalling to the Aggressive Phenotypes of EBV-Associated Cancers
5. Induction of EBV Lytic Reactivation by TGF-β
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Takehara, K.; LeRoy, E.C.; Grotendorst, G.R. TGF-β inhibition of endothelial cell proliferation: Alteration of EGF binding and EGF-induced growth-regulatory (competence) gene expression. Cell 1987, 49, 415–422. [Google Scholar] [CrossRef]
- Moses, H.L.; Coffey, R.J., Jr.; Leof, E.B.; Lyons, R.M.; Keski-Oja, J. Transforming growth factor beta regulation of cell proliferation. J. Cell. Physiol. Suppl. 1987, (Suppl. 5), 1–7. [Google Scholar] [CrossRef]
- Stoeck, M.; Howe, R.C.; Miescher, S.; von Fliedner, V.; MacDonald, H.R. Effect of transforming growth factor beta on the EL4 thymoma variant EL4/6.1: Dissociation of inhibition of proliferation from expression of IL-1 and IL-2 receptors. Immunobiology 1990, 181, 13–21. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGF-β in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, K.; Tsumuraya, M. Transforming growth factor beta 1 induces apoptotic cell death in cultured human gastric carcinoma cells. Cancer Res. 1992, 52, 4042–4045. [Google Scholar] [PubMed]
- Lebrun, J.J. The Dual Role of TGF-β in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 381–428. [Google Scholar] [CrossRef] [PubMed]
- Oft, M.; Heider, K.H.; Beug, H. TGF-β signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 1998, 8, 1243–1252. [Google Scholar] [CrossRef]
- Ueki, N.; Nakazato, M.; Ohkawa, T.; Ikeda, T.; Amuro, Y.; Hada, T.; Higashino, K. Excessive production of transforming growth-factor beta 1 can play an important role in the development of tumorigenesis by its action for angiogenesis: Validity of neutralizing antibodies to block tumor growth. Biochim. Biophys. Acta 1992, 1137, 189–196. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Beauchamp, R.D.; Koeppen, H.; Park, B.H.; Schreiber, H.; Moses, H.L.; Rowley, D.A. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc. Natl. Acad. Sci. USA 1990, 87, 1486–1490. [Google Scholar] [CrossRef] [PubMed]
- McEarchern, J.A.; Kobie, J.J.; Mack, V.; Wu, R.S.; Meade-Tollin, L.; Arteaga, C.L.; Dumont, N.; Besselsen, D.; Seftor, E.; Hendrix, M.J.; et al. Invasion and metastasis of a mammary tumor involves TGF-β signaling. Int. J. Cancer 2001, 91, 76–82. [Google Scholar] [CrossRef]
- Lehmann, K.; Janda, E.; Pierreux, C.E.; Rytomaa, M.; Schulze, A.; McMahon, M.; Hill, C.S.; Beug, H.; Downward, J. Raf induces TGF-β production while blocking its apoptotic but not invasive responses: A mechanism leading to increased malignancy in epithelial cells. Genes Dev. 2000, 14, 2610–2622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Li, X.; Sui, L.H.; Wang, Q.; Li, P.; Fu, S.B. Functional study on TGF-β/Smads signaling pathway in human ovarian cancer cells. Yi Chuan Xue Bao 2004, 31, 759–765. [Google Scholar] [PubMed]
- Chen, C.R.; Kang, Y.; Massague, J. Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proc. Natl. Acad. Sci. USA. 2001, 98, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Faghihloo, E. Viruses as key modulators of the TGF-β pathway; a double-edged sword involved in cancer. Rev. Med. Virol. 2018, 28. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt's Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [PubMed]
- Childs, C.B.; Proper, J.A.; Tucker, R.F.; Moses, H.L. Serum contains a platelet-derived transforming growth factor. Proc. Natl. Acad. Sci. USA 1982, 79, 5312–5316. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Lindquist, P.B.; Lee, A.; Wen, D.; Tamm, J.; Graycar, J.L.; Rhee, L.; Mason, A.J.; Miller, D.A.; Coffey, R.J.; et al. A new type of transforming growth factor-beta, TGF-β 3. EMBO J. 1988, 7, 3737–3743. [Google Scholar] [PubMed]
- Segarini, P.R.; Roberts, A.B.; Rosen, D.M.; Seyedin, S.M. Membrane binding characteristics of two forms of transforming growth factor-beta. J. Biol. Chem. 1987, 262, 14655–14662. [Google Scholar] [PubMed]
- Sporn, M.B.; Roberts, A.B.; Wakefield, L.M.; Assoian, R.K. Transforming growth factor-beta: Biological function and chemical structure. Science 1986, 233, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Jarrett, J.A.; Chen, E.Y.; Eaton, D.H.; Bell, J.R.; Assoian, R.K.; Roberts, A.B.; Sporn, M.B.; Goeddel, D.V. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Gentry, L.E.; Lioubin, M.N.; Purchio, A.F.; Marquardt, H. Molecular events in the processing of recombinant type 1 pre-pro-transforming growth factor beta to the mature polypeptide. Mol. Cell. Biol. 1988, 8, 4162–4168. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.M.; Laprise, M.H.; Blanchette, F.; Gentry, L.E.; Leduc, R. Processing of transforming growth factor beta 1 precursor by human furin convertase. J. Biol. Chem. 1995, 270, 10618–10624. [Google Scholar] [CrossRef] [PubMed]
- Gentry, L.E.; Nash, B.W. The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry 1990, 29, 6851–6857. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Derynck, R.; Tsang, M.L.; Weatherbee, J.A. Transforming growth factor-beta activation in irradiated murine mammary gland. J. Clin. Investig. 1994, 93, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.; Berg, T.M.; Lawrence, D.A. Acidic cellular environments: Activation of latent TGF-β and sensitization of cellular responses to TGF-beta and EGF. Int. J. Cancer 1989, 43, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N. TGF-β: From latent to active. Microbes Infect 1999, 1, 1255–1263. [Google Scholar] [CrossRef]
- Lin, H.Y.; Wang, X.F.; Ng-Eaton, E.; Weinberg, R.A.; Lodish, H.F. Expression cloning of the TGF-β type II receptor, a functional transmembrane serine/threonine kinase. Cell 1992, 68, 775–785. [Google Scholar] [CrossRef]
- Ebner, R.; Chen, R.H.; Shum, L.; Lawler, S.; Zioncheck, T.F.; Lee, A.; Lopez, A.R.; Derynck, R. Cloning of a type I TGF-β receptor and its effect on TGF-β binding to the type II receptor. Science 1993, 260, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.; We, R.; Derynck, R. Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 1996, 383, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Sekelsky, J.J.; Newfeld, S.J.; Raftery, L.A.; Chartoff, E.H.; Gelbart, W.M. Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics 1995, 139, 1347–1358. [Google Scholar] [PubMed]
- Raftery, L.A.; Twombly, V.; Wharton, K.; Gelbart, W.M. Genetic screens to identify elements of the decapentaplegic signaling pathway in Drosophila. Genetics 1995, 139, 241–254. [Google Scholar] [PubMed]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-β signal transduction. J. Cell Sci. 2001, 114, 4359–4369. [Google Scholar] [PubMed]
- Savage, C.; Das, P.; Finelli, A.L.; Townsend, S.R.; Sun, C.Y.; Baird, S.E.; Padgett, R.W. Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor beta pathway components. Proc. Natl. Acad. Sci. USA 1996, 93, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Chen, Y.G.; Ozdamar, B.; Gyuricza, C.A.; Chong, P.A.; Wrana, J.L.; Massague, J.; Shi, Y. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000, 287, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGF-β receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef]
- Wu, J.W.; Hu, M.; Chai, J.; Seoane, J.; Huse, M.; Li, C.; Rigotti, D.J.; Kyin, S.; Muir, T.W.; Fairman, R.; et al. Crystal structure of a phosphorylated Smad2. Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-β signaling. Mol. Cell 2001, 8, 1277–1289. [Google Scholar] [CrossRef]
- Chacko, B.M.; Qin, B.Y.; Tiwari, A.; Shi, G.; Lam, S.; Hayward, L.J.; De Caestecker, M.; Lin, K. Structural basis of heteromeric smad protein assembly in TGF-β signaling. Mol. Cell 2004, 15, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Topper, J.N.; DiChiara, M.R.; Brown, J.D.; Williams, A.J.; Falb, D.; Collins, T.; Gimbrone, M.A., Jr. CREB binding protein is a required coactivator for Smad-dependent, transforming growth factor beta transcriptional responses in endothelial cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9506–9511. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.V.; Shen, L.; Anderson, S.A.; Massague, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Yamamura, Y.; Hua, X.; Bergelson, S.; Lodish, H.F. Critical role of Smads and AP-1 complex in transforming growth factor-beta -dependent apoptosis. J. Biol. Chem. 2000, 275, 36295–36302. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, A.; Ishidou, Y.; Ebisawa, T.; Shimanuki, T.; Imamura, T.; Miyazono, K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 2001, 155, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-β signaling by Smad7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF -β receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGF-β receptor and functions as an antagonist of TGF-β signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Zhang, S.; Fei, T.; Zhang, L.; Zhang, R.; Chen, F.; Ning, Y.; Han, Y.; Feng, X.H.; Meng, A.; Chen, Y.G. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell. Biol. 2007, 27, 4488–4499. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liao, H.; Cheng, M.; Shi, X.; Lin, X.; Feng, X.H.; Chen, Y.G. Smad7 Protein Interacts with Receptor-regulated Smads (R-Smads) to Inhibit Transforming Growth Factor-beta (TGF-β)/Smad Signaling. J. Biol. Chem. 2016, 291, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007, 67, 3752–3758. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Shimotohno, K. Transforming growth factor-beta-mediated signaling via the p38 MAP kinase pathway activates Smad-dependent transcription through SUMO-1 modification of Smad4. J. Biol. Chem. 2003, 278, 50833–50842. [Google Scholar] [CrossRef] [PubMed]
- Kamaraju, A.K.; Roberts, A.B. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 2005, 280, 1024–1036. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, I.; Polyak, K.; Iavarone, A.; Massague, J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev. 1995, 9, 1831–1845. [Google Scholar] [CrossRef] [PubMed]
- Laiho, M.; DeCaprio, J.A.; Ludlow, J.W.; Livingston, D.M.; Massague, J. Growth inhibition by TGF-β linked to suppression of retinoblastoma protein phosphorylation. Cell 1990, 62, 175–185. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Yang, W.; Shen, J.; Wu, M.; Arsura, M.; FitzGerald, M.; Suldan, Z.; Kim, D.W.; Hofmann, C.S.; Pianetti, S.; Romieu-Mourez, R.; et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 2001, 20, 1688–1702. [Google Scholar] [CrossRef] [PubMed]
- Claassen, G.F.; Hann, S.R. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor beta -induced cell-cycle arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 9498–9503. [Google Scholar] [CrossRef] [PubMed]
- Warner, B.J.; Blain, S.W.; Seoane, J.; Massague, J. Myc downregulation by transforming growth factor beta required for activation of the p15(Ink4b) G(1) arrest pathway. Mol. Cell. Biol. 1999, 19, 5913–5922. [Google Scholar] [CrossRef] [PubMed]
- Bakhshayesh, M.; Zaker, F.; Hashemi, M.; Katebi, M.; Solaimani, M. TGF- β1-mediated apoptosis associated with SMAD-dependent mitochondrial Bcl-2 expression. Clin. Lymphoma Myeloma Leuk. 2012, 12, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Hagimoto, N.; Kuwano, K.; Inoshima, I.; Yoshimi, M.; Nakamura, N.; Fujita, M.; Maeyama, T.; Hara, N. TGF-β 1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J. Immunol. 2002, 168, 6470–6478. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, I.; Wang, H.; Grand, R.; Armitage, R.J.; Fanslow, W.C.; Gregory, C.D.; Gordon, J. Transforming growth factor-beta 1 cooperates with anti-immunoglobulin for the induction of apoptosis in group I (biopsy-like) Burkitt lymphoma cell lines. Blood 1996, 87, 1147–1154. [Google Scholar] [PubMed]
- Saltzman, A.; Munro, R.; Searfoss, G.; Franks, C.; Jaye, M.; Ivashchenko, Y. Transforming growth factor-beta-mediated apoptosis in the Ramos B-lymphoma cell line is accompanied by caspase activation and Bcl-XL downregulation. Exp. Cell Res. 1998, 242, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Allday, M.J. Apoptosis induced by TGF-β 1 in Burkitt's lymphoma cells is caspase 8 dependent but is death receptor independent. J. Immunol. 2000, 165, 2500–2510. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, H.K.; Smeland, E.; Mustafa, A.S.; Godal, T.; Ohlsson, R. Epstein-Barr virus mediates a switch in responsiveness to transforming growth factor, type beta, in cells of the B cell lineage. Eur. J. Immunol. 1987, 17, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, L.; Yaseen, N.; Sharma, S. Latent membrane protein-1 induces cyclin D2 expression, pRb hyperphosphorylation, and loss of TGF-β1-mediated growth inhibition in EBV-positive B cells. J. Immunol. 1995, 155, 1047–1056. [Google Scholar] [PubMed]
- Kumar, A.; Rogers, T.; Maizel, A.; Sharma, S. Loss of transforming growth factor beta 1 receptors and its effects on the growth of EBV-transformed human B cells. J. Immunol. 1991, 147, 998–1006. [Google Scholar] [PubMed]
- Fukuda, M.; Ikuta, K.; Yanagihara, K.; Tajima, M.; Kuratsune, H.; Kurata, T.; Sairenji, T. Effect of transforming growth factor-β1 on the cell growth and Epstein-Barr virus reactivation in EBV-infected epithelial cell lines. Virology 2001, 288, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Xiang, Q.; Xiao, Y.C.; Su, Z.J.; Huang, Z.F.; Zhang, Q.H.; Tan, Y.; Li, X.K.; Huang, Y.D. The effect of transforming growth factor-β1 on nasopharyngeal carcinoma cells: Insensitive to cell growth but functional to TGF-β/Smad pathway. J. Exp. Clin. Cancer Res. 2010, 29, 35. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Tann, B.; Zeng, Y.; Zhou, W.; Li, K.; Zhao, M. Establishment of an epithelial cell line (CNE-2) from an NPC patient with poorly differentiated squamous cell carcinoma. Chin. J. Cancer 1983, 2, 70. [Google Scholar]
- Takanashi, M.; Li, J.; Shirakata, M.; Mori, S.; Hirai, K. Tumorigenicity of mouse BALB/c 3T3 fibroblast cells which express Epstein-Barr virus-encoded LMP1 and show normal growth phenotypes in vitro is correlated with loss of transforming growth factor-β1-mediated growth inhibition. Arch. Virol. 1999, 144, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Countryman, J.; Miller, G. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 4085–4089. [Google Scholar] [CrossRef] [PubMed]
- Countryman, J.; Gradoville, L.; Bhaduri-McIntosh, S.; Ye, J.; Heston, L.; Himmelfarb, S.; Shedd, D.; Miller, G. Stimulus duration and response time independently influence the kinetics of lytic cycle reactivation of Epstein-Barr virus. J. Virol. 2009, 83, 10694–10709. [Google Scholar] [CrossRef] [PubMed]
- Bhende, P.M.; Dickerson, S.J.; Sun, X.; Feng, W.H.; Kenney, S.C. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J. Virol. 2007, 81, 7363–7370. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Thorley-Lawson, D.A. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 2007, 81, 13566–13577. [Google Scholar] [CrossRef] [PubMed]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Rickinson, A.B. Co-infections, inflammation and oncogenesis: future directions for EBV research. Semin. Cancer Biol. 2014, 26, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Yap, L.F.; Lo, K.W. Epstein-Barr virus and epithelial carcinogenesis. In DNA Tumour Viruses: Virology, Pathogenesis and Vaccines; Roberts, I.S., Ed.; Caister Academic Press: Poole, UK, 2018; pp. 139–161. ISBN 978-1-910190-80-7. [Google Scholar]
- Kenney, J.L.; Guinness, M.E.; Reiss, M.; Lacy, J. Antisense to the Epstein-Barr virus (EBV)-encoded latent membrane protein 1 (LMP-1) sensitizes EBV-immortalized B cells to transforming growth factor-beta and chemotherapeutic agents. Int. J. Cancer 2001, 91, 89–98. [Google Scholar] [CrossRef]
- Inman, G.J.; Allday, M.J. Resistance to TGF-β1 correlates with a reduction of TGF-β type II receptor expression in Burkitt’s lymphoma and Epstein-Barr virus-transformed B lymphoblastoid cell lines. J. Gen. Virol. 2000, 81, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Prokova, V.; Mosialos, G.; Kardassis, D. Inhibition of transforming growth factor beta signaling and Smad-dependent activation of transcription by the Latent Membrane Protein 1 of Epstein-Barr virus. J. Biol. Chem. 2002, 277, 9342–9350. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Morishita, M.; Tsukazaki, T.; Yamamoto, N. Repression of Smad-dependent transforming growth factor-beta signaling by Epstein-Barr virus latent membrane protein 1 through nuclear factor-κB. Int. J. Cancer 2003, 105, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Dawson, C.W.; Lo, K.W.; Yu, Y.; Young, L.S. Upregulation of Id1 by Epstein-Barr Virus-encoded LMP1 confers resistance to TGFβ-mediated growth inhibition. Mol. Cancer 2010, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.A.; Dawson, C.W.; Laverick, L.; Davis, A.M.; Dudman, J.P.; Raveenthiraraj, S.; Ahmad, Z.; Yap, L.-F.; Young, L.S. The Epstein-Barr virus encoded LMP1 oncoprotein modulates cell adhesion via regulation of activin A/TGFβ and β1 integrin signalling. Sci. Rep. 2016, 6, 19533. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guan, X.; Lv, J.; Li, X.; Wang, Y.; Li, L. Limb-bud and Heart (LBH) functions as a tumor suppressor of nasopharyngeal carcinoma by inducing G1/S cell cycle arrest. Sci. Rep. 2015, 5, 7626. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-β1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Wood, V.H.; O’Neil, J.D.; Wei, W.; Stewart, S.E.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGF-β signaling pathways. Oncogene 2007, 26, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.L.; Tsai, C.N.; Chung, P.J.; Chen, J.L.; Sun, C.M.; Chen, R.H.; Hong, J.H.; Chang, Y.S. Transcription of Epstein-Barr virus-encoded nuclear antigen 1 promoter Qp is repressed by transforming growth factor-beta via Smad4 binding element in human BL cells. Virology 2000, 277, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Foote, L.C.; Marshak-Rothstein, A.; Rothstein, T.L. Tolerant B lymphocytes acquire resistance to Fas-mediated apoptosis after treatment with interleukin 4 but not after treatment with specific antigen unless a surface immunoglobulin threshold is exceeded. J. Exp. Med. 1998, 187, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Holder, M.J.; Knox, K.; Gordon, J. Factors modifying survival pathways of germinal center B cells. Glucocorticoids and transforming growth factor-beta, but not cyclosporin A or anti-CD19, block surface immunoglobulin-mediated rescue from apoptosis. Eur. J. Immunol. 1992, 22, 2725–2728. [Google Scholar] [CrossRef] [PubMed]
- Sater, R.A.; Sandel, P.C.; Monroe, J.G. B cell receptor-induced apoptosis in primary transitional murine B cells: signaling requirements and modulation by T cell help. Int. Immunol. 1998, 10, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Horndasch, M.; Raschke, E.E.; Bommer, G.; Schuhmacher, M.; Dumont, E.; Kuklik-Roos, C.; Eick, D.; Kempkes, B. Epstein-Barr virus antagonizes the antiproliferative activity of transforming growth factor-beta but does not abolish its signaling. Int. J. Cancer 2002, 101, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Campion, E.M.; Hakimjavadi, R.; Loughran, S.T.; Phelan, S.; Smith, S.M.; D'Souza, B.N.; Tierney, R.J.; Bell, A.I.; Cahill, P.A.; Walls, D. Repression of the proapoptotic cellular BIK/NBK gene by Epstein-Barr virus antagonizes transforming growth factor beta1-induced B-cell apoptosis. J. Virol. 2014, 88, 5001–5013. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Chang, M.S.; Yoon, C.J.; Middeldorp, J.M.; Martinez, O.M.; Byeon, S.J.; Rha, S.Y.; Kim, S.H.; Kim, Y.S.; Woo, J.H. Epstein-Barr virus BARF1-induced NFkappaB/miR-146a/SMAD4 alterations in stomach cancer cells. Oncotarget 2016, 7, 82213–82227. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Kurosaki, H.; Sairenji, T. Loss of functional transforming growth factor (TGF)-beta type II receptor results in insensitivity to TGF-beta1-mediated apoptosis and Epstein-Barr virus reactivation. J. Med. Virol. 2006, 78, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Yang, W.; Ai, L.; Li, Z.; Jin, J. Transforming growth factor beta type II receptor as a marker in diffuse large B cell lymphoma. Tumour Biol. 2015, 36, 9903–9908. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Li, X.; Jiang, Q.; Liu, Z.; Yang, H.; Wang, S.; Xie, S.; Liu, Q.; Liu, T.; Huang, J.; et al. Transcriptional patterns, biomarkers and pathways characterizing nasopharyngeal carcinoma of Southern China. J. Transl. Med. 2008, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zeng, Z.; Fan, S.; Wang, J.; Yang, J.; Zhou, Y.; Li, X.; Huang, D.; Liang, F.; Wu, M.; et al. Evaluation of the prognostic value of TGF-beta superfamily type I receptor and TGF-beta type II receptor expression in nasopharyngeal carcinoma using high-throughput tissue microarrays. J. Mol. Histol. 2012, 43, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Fang, W.; Cai, L.; Zheng, H.; Ye, Y.; Zhang, L.; Li, J.; Peng, H.; Cho, W.C.; Wang, E.; et al. TGFbetaR2 is a major target of miR-93 in nasopharyngeal carcinoma aggressiveness. Mol. Cancer 2014, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Wang, Z.; Wang, J.; Liu, X.; Hu, C. MicroRNA-19a promotes nasopharyngeal carcinoma by targeting transforming growth factor beta receptor 2. Exp. Ther. Med. 2017, 14, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xuan, S.H.; Li, Y.; Zhang, Z.P.; Li, X.H. Role of the TGFbeta/PDCD4/AP-1 Signaling Pathway in Nasopharyngeal Carcinoma and Its Relationship to Prognosis. Cell. Physiol. Biochem. 2017, 43, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.C.; Matthews, J.B.; Huntley, S.; Robinson, C.M.; Fahey, M.; Parkinson, E.K.; Prime, S.S. Decreased expression of TGF-beta cell surface receptors during progression of human oral squamous cell carcinoma. J. Pathol. 2001, 193, 458–467. [Google Scholar] [CrossRef]
- Chan, A.S.; To, K.F.; Lo, K.W.; Mak, K.F.; Pak, W.; Chiu, B.; Tse, G.M.; Ding, M.; Li, X.; Lee, J.C.; et al. High frequency of chromosome 3p deletion in histologically normal nasopharyngeal epithelia from southern Chinese. Cancer Res. 2000, 60, 5365–5370. [Google Scholar] [PubMed]
- Lo, K.W.; Teo, P.M.; Hui, A.B.; To, K.F.; Tsang, Y.S.; Chan, S.Y.; Mak, K.F.; Lee, J.C.; Huang, D.P. High resolution allelotype of microdissected primary nasopharyngeal carcinoma. Cancer Res. 2000, 60, 3348–3353. [Google Scholar] [PubMed]
- Chen, H.C.; Chen, G.H.; Chen, Y.H.; Liao, W.L.; Liu, C.Y.; Chang, K.P.; Chang, Y.S.; Chen, S.J. MicroRNA deregulation and pathway alterations in nasopharyngeal carcinoma. Br. J. Cancer 2009, 100, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.X.; Yi, H.; Qu, J.Q.; He, Q.Y.; Xiao, Z.Q. Integrated analysis of the differential cellular and EBV miRNA expression profiles in microdissected nasopharyngeal carcinoma and non-cancerous nasopharyngeal tissues. Oncol. Rep. 2015, 34, 2585–2601. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Li, Y.Q.; Guo, R.; He, Q.M.; Ren, X.Y.; Tang, X.R.; Jia, W.H.; Kang, T.B.; Zeng, M.S.; Sun, Y.; et al. Identification of miR-143 as a tumour suppressor in nasopharyngeal carcinoma based on microRNA expression profiling. Int. J. Biochem. Cell Biol. 2015, 61, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Lin, Y.X.; Qi, X.K.; He, G.P.; Zhang, Y.; Zhang, H.J.; Xu, M.; Feng, Q.S.; Bei, J.X.; Zeng, Y.X.; et al. TNFRSF19 inhibits TGFbeta signaling through interaction with TGFbeta receptor type I to promote tumorigenesis. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Dai, W.; Cheung, A.K.; Ko, J.M.; Kan, R.; Wong, B.W.; Leong, M.M.; Deng, M.; Kwok, T.C.; Chan, J.Y.; et al. Whole-exome sequencing identifies multiple loss-of-function mutations of NF-kappaB pathway regulators in nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 11283–11288. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Chung, G.T.; Lui, V.W.; To, K.F.; Ma, B.B.; Chow, C.; Woo, J.K.; Yip, K.Y.; Seo, J.; Hui, E.P.; et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat. Commun. 2017, 8, 14121. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Yao, X.; Tang, S.; Zhang, J.; Yau, T.O.; Li, X.; Tang, C.M.; Kang, W.; Lung, R.W.; Li, J.W.; et al. Integrative identification of Epstein-Barr virus-associated mutations and epigenetic alterations in gastric cancer. Gastroenterology 2014, 147, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Boger, C.; Kruger, S.; Behrens, H.M.; Bock, S.; Haag, J.; Kalthoff, H.; Rocken, C. Epstein-Barr virus-associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann. Oncol. 2017, 28, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Abdul Razak, F.R.; Terpstra, M.; Chan, F.C.; Saber, A.; Nijland, M.; van Imhoff, G.; Visser, L.; Gascoyne, R.; Steidl, C.; et al. The mutational landscape of Hodgkin lymphoma cell lines determined by whole-exome sequencing. Leukemia 2014, 28, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Hudnall, S.D.; Meng, H.; Lozovatsky, L.; Li, P.; Strout, M.; Kleinstein, S.H. Recurrent genetic defects in classical Hodgkin lymphoma cell lines. Leuk. Lymphoma 2016, 57, 2890–2900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.J.; Asmann, Y.W.; Maurer, M.J.; Wang, C.; Slager, S.L.; Hodge, L.S.; Manske, M.; Price-Troska, T.; Yang, Z.Z.; Zimmermann, M.T.; et al. Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated diffuse large B-cell lymphoma. Blood Cancer J. 2015, 5, e346. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Lee, S.B.; Yoo, H.Y.; Kim, S.J.; Kim, W.S.; Kim, J.I.; Ko, Y.H. Whole-exome and transcriptome sequencing of refractory diffuse large B-cell lymphoma. Oncotarget 2016, 7, 86433–86445. [Google Scholar] [CrossRef] [PubMed]
- Greenawalt, D.M.; Liang, W.S.; Saif, S.; Johnson, J.; Todorov, P.; Dulak, A.; Enriquez, D.; Halperin, R.; Ahmed, A.; Saveliev, V.; et al. Comparative analysis of primary versus relapse/refractory DLBCL identifies shifts in mutation spectrum. Oncotarget 2017, 8, 99237–99244. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Dalal, B.I.; Keown, P.A.; Greenberg, A.H. Immunocytochemical localization of secreted transforming growth factor-beta 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am. J. Pathol. 1993, 143, 381–389. [Google Scholar] [PubMed]
- Friedman, E.; Gold, L.I.; Klimstra, D.; Zeng, Z.S.; Winawer, S.; Cohen, A. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol. Biomark. Prev. 1995, 4, 549–554. [Google Scholar]
- Gorsch, S.M.; Memoli, V.A.; Stukel, T.A.; Gold, L.I.; Arrick, B.A. Immunohistochemical staining for transforming growth factor beta 1 associates with disease progression in human breast cancer. Cancer Res. 1992, 52, 6949–6952. [Google Scholar] [PubMed]
- Shim, K.S.; Kim, K.H.; Han, W.S.; Park, E.B. Elevated serum levels of transforming growth factor-beta1 in patients with colorectal carcinoma: Its association with tumor progression and its significant decrease after curative surgical resection. Cancer 1999, 85, 554–561. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Verspaget, H.W.; van Duijn, W.; van der Zon, J.M.; Zuidwijk, K.; Kubben, F.J.; Verheijen, J.H.; Hommes, D.W.; Lamers, C.B.; Sier, C.F. Tissue level, activation and cellular localisation of TGF-beta1 and association with survival in gastric cancer patients. Br. J. Cancer 2007, 97, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.; Bakin, A.V.; Arteaga, C.L. Autocrine transforming growth factor-beta signaling mediates Smad-independent motility in human cancer cells. J. Biol. Chem. 2003, 278, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Lu, S.L.; Li, A.G.; He, W.; Corless, C.L.; Kulesz-Martin, M.; Wang, X.J. Distinct mechanisms of TGF-beta1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J. Clin. Investig. 2005, 115, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Vinals, F.; Pouyssegur, J. Transforming growth factor beta1 (TGF-beta1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-α signaling. Mol. Cell. Biol. 2001, 21, 7218–7230. [Google Scholar] [CrossRef] [PubMed]
- Newcom, S.R.; Kadin, M.E.; Ansari, A.A. Production of transforming growth factor-beta activity by Ki-1 positive lymphoma cells and analysis of its role in the regulation of Ki-1 positive lymphoma growth. Am. J. Pathol. 1988, 131, 569–577. [Google Scholar] [PubMed]
- Newcom, S.R.; Kadin, M.E.; Ansari, A.A.; Diehl, V. L-428 nodular sclerosing Hodgkin’s cell secretes a unique transforming growth factor-beta active at physiologic pH. J. Clin. Investig. 1988, 82, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Kadin, M.E.; Agnarsson, B.A.; Ellingsworth, L.R.; Newcom, S.R. Immunohistochemical evidence of a role for transforming growth factor beta in the pathogenesis of nodular sclerosing Hodgkin’s disease. Am. J. Pathol. 1990, 136, 1209–1214. [Google Scholar] [PubMed]
- Hsu, S.M.; Lin, J.; Xie, S.S.; Hsu, P.L.; Rich, S. Abundant expression of transforming growth factor-beta 1 and -beta 2 by Hodgkin’s Reed-Sternberg cells and by reactive T lymphocytes in Hodgkin’s disease. Hum. Pathol. 1993, 24, 249–255. [Google Scholar] [CrossRef]
- Maeda, H.; Shiraishi, A. TGF-beta contributes to the shift toward Th2-type responses through direct and IL-10-mediated pathways in tumor-bearing mice. J. Immunol. 1996, 156, 73–78. [Google Scholar] [PubMed]
- Di Renzo, L.; Altiok, A.; Klein, G.; Klein, E. Endogenous TGF-beta contributes to the induction of the EBV lytic cycle in two Burkitt lymphoma cell lines. Int. J. Cancer 1994, 57, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Flemington, E.K. Identification of cellular target genes of the Epstein-Barr virus transactivator Zta: Activation of transforming growth factor beta igh3 (TGF-beta igh3) and TGF-beta 1. J. Virol. 1995, 69, 4206–4212. [Google Scholar] [PubMed]
- Xu, J.; Menezes, J.; Prasad, U.; Ahmad, A. Elevated serum levels of transforming growth factor beta1 in Epstein-Barr virus-associated nasopharyngeal carcinoma patients. Int. J. Cancer 1999, 84, 396–399. [Google Scholar] [CrossRef]
- Hu, C.; Wei, W.; Chen, X.; Woodman, C.B.; Yao, Y.; Nicholls, J.M.; Joab, I.; Sihota, S.K.; Shao, J.Y.; Derkaoui, K.D.; et al. A global view of the oncogenic landscape in nasopharyngeal carcinoma: An integrated analysis at the genetic and expression levels. PLoS ONE 2012, 7, e41055. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tian, W.D.; Xu, X.; Nie, B.; Lu, J.; Liu, X.; Zhang, B.; Dong, Q.; Sunwoo, J.B.; Li, G.; et al. Epstein-Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer 2014, 120, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Naef, M.; Ishiwata, T.; Friess, H.; Buchler, M.W.; Gold, L.I.; Korc, M. Differential localization of transforming growth factor-beta isoforms in human gastric mucosa and overexpression in gastric carcinoma. Int. J. Cancer 1997, 71, 131–137. [Google Scholar] [CrossRef]
- Shukla, S.K.; Khatoon, J.; Prasad, K.N.; Rai, R.P.; Singh, A.K.; Kumar, S.; Ghoshal, U.C.; Krishnani, N. Transforming growth factor beta 1 (TGF-β1) modulates Epstein-Barr virus reactivation in absence of Helicobacter pylori infection in patients with gastric cancer. Cytokine 2016, 77, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shen, Z.; Wang, K.; Ha, Y.; Lei, H.; Jia, Y.; Ding, R.; Wu, D.; Gan, S.; Li, R.; et al. High FMNL3 expression promotes nasopharyngeal carcinoma cell metastasis: Role in TGF-beta1-induced epithelia-to-mesenchymal transition. Sci. Rep. 2017, 7, 42507. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Ni, J.; Feng, W.T.; Yao, R.; Yue, S.; Zhu, Y.N.; Tang, H.Y.; Lv, L.Y.; Feng, J.F.; Zhu, W.G. High YBX1 expression indicates poor prognosis and promotes cell migration and invasion in nasopharyngeal carcinoma. Exp. Cell Res. 2017, 361, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Cui, Y.; Xiao, H.; Mai, M.; Wang, C.; Xie, S.; Yang, J.; Wu, S.; Li, J.; Song, L.; et al. Upregulation of flotillin-1 promotes invasion and metastasis by activating TGF-beta signaling in nasopharyngeal carcinoma. Oncotarget 2016, 7, 4252–4264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lin, L.; Pan, C.; Shi, M.; Liao, Y.; Bin, J.; Liao, W. Flotillin-2 promotes nasopharyngeal carcinoma metastasis and is necessary for the epithelial-mesenchymal transition induced by transforming growth factor-β. Oncotarget 2015, 6, 9781–9793. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.Y.; Yin, L.; Jiang, N.; Guo, W.J.; Tian, H.; Jiang, X.S.; Wu, J.; Chen, M.; Wu, J.Z.; He, X. Downregulating HMGA2 attenuates epithelial-mesenchymal transition-induced invasion and migration in nasopharyngeal cancer cells. Biochem. Biophys. Res. Commun. 2015, 463, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Chen, W.; Zhang, W.; Tian, X.K.; Wang, T.; Wu, J.; Gu, J.; Zhang, N.; Lu, Z.W.; Qian, L.X.; et al. TGF-β regulates the ERK/MAPK pathway independent of the SMAD pathway by repressing miRNA-124 to increase MALAT1 expression in nasopharyngeal carcinoma. Biomed. Pharmacother. 2018, 99, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Brunen, D.; Willems, S.M.; Kellner, U.; Midgley, R.; Simon, I.; Bernards, R. TGF-β: An emerging player in drug resistance. Cell cycle 2013, 12, 2960–2968. [Google Scholar] [CrossRef] [PubMed]
- Bissey, P.A.; Law, J.H.; Bruce, J.P.; Shi, W.; Renoult, A.; Chua, M.L.K.; Yip, K.W.; Liu, F.F. Dysregulation of the MiR-449b target TGFBI alters the TGFbeta pathway to induce cisplatin resistance in nasopharyngeal carcinoma. Oncogenesis 2018, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, P.; Hofler, P.; Bauer, G. Epstein-barr virus induction by a serum factor: IV. Ubiquitous occurrence of the factor within vertebrates and its interaction with defined lymphoid cell lines. Int. J. Cancer 1982, 30, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Chasserot-Golaz, S.; Schuster, C.; Dietrich, J.B.; Beck, G.; Lawrence, D.A. Antagonistic action of RU38486 on the activity of transforming growth factor-β in fibroblasts and lymphoma cells. J. Steroid Biochem. 1988, 30, 381–385. [Google Scholar] [CrossRef]
- Fahmi, H.; Cochet, C.; Hmama, Z.; Opolon, P.; Joab, I. Transforming growth factor beta 1 stimulates expression of the Epstein-Barr virus BZLF1 immediate-early gene product ZEBRA by an indirect mechanism which requires the MAPK kinase pathway. J. Virol. 2000, 74, 5810–5818. [Google Scholar] [CrossRef] [PubMed]
- Oussaief, L.; Hippocrate, A.; Ramirez, V.; Rampanou, A.; Zhang, W.; Meyers, D.; Cole, P.; Khelifa, R.; Joab, I. Phosphatidylinositol 3-kinase/Akt pathway targets acetylation of Smad3 through Smad3/CREB-binding protein interaction: Contribution to transforming growth factor beta1-induced Epstein-Barr virus reactivation. J. Biol. Chem. 2009, 284, 23912–23924. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.L.; Chen, J.L.; Hsu, Y.P.; Ou, J.T.; Chang, Y.S. Epstein-Barr virus BZLF1 gene is activated by transforming growth factor-beta through cooperativity of Smads and c-Jun/c-Fos proteins. J. Biol. Chem. 2002, 277, 23345–23357. [Google Scholar] [CrossRef] [PubMed]
- Iempridee, T.; Das, S.; Xu, I.; Mertz, J.E. Transforming growth factor beta-induced reactivation of Epstein-Barr virus involves multiple Smad-binding elements cooperatively activating expression of the latent-lytic switch BZLF1 gene. J. Virol. 2011, 85, 7836–7848. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-N.; Liu, S.-T.; Chang, Y.-S. Identification of a novel promoter located within the Bam HI Q region of the Epstein-Barr virus genome for the EBNA 1 gene. DNA Cell Biol. 1995, 14, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lee, J.M.; Wang, Y.; Huang, D.P.; Ambinder, R.F.; Hayward, S.D. The Epstein-Barr virus latency BamHI-Q promoter is positively regulated by STATs and Zta interference with JAK/STAT activation leads to loss of BamHI-Q promoter activity. Proc. Natl. Acad. Sci. USA 1999, 96, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.M.; Zhang, G.; Seto, E.; Takada, K.; Deng, W.; Yip, Y.L.; Man, C.; Hau, P.M.; Chen, H.; Cao, Y.; et al. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: Regulation of infection and phenotypic characterization. Int. J. Cancer 2010, 127, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Ohashi, M.; Yoshiyama, H.; Ohba, Y. The Role of Transforming Growth Factor beta in Cell-to-Cell Contact-Mediated Epstein-Barr Virus Transmission. Front. Microbiol. 2018, 9, 984. [Google Scholar] [CrossRef] [PubMed]
- Poh, Y.W.; Gan, S.Y.; Tan, E.L. Effects of IL-6, IL-10 and TGF-beta on the expression of survivin and apoptosis in nasopharyngeal carcinoma TW01 cells. Exp. Oncol. 2012, 34, 85–89. [Google Scholar] [PubMed]
- Iglesias, M.; Frontelo, P.; Gamallo, C.; Quintanilla, M. Blockade of Smad4 in transformed keratinocytes containing a Ras oncogene leads to hyperactivation of the Ras-dependent Erk signalling pathway associated with progression to undifferentiated carcinomas. Oncogene 2000, 19, 4134–4145. [Google Scholar] [CrossRef] [PubMed]
- Huntley, S.P.; Davies, M.; Matthews, J.B.; Thomas, G.; Marshall, J.; Robinson, C.M.; Eveson, J.W.; Paterson, I.C.; Prime, S.S. Attenuated type II TGF-β receptor signalling in human malignant oral keratinocytes induces a less differentiated and more aggressive phenotype that is associated with metastatic dissemination. Int. J. Cancer 2004, 110, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.; Kim, S.W.; McKeller, M.R.; Dahia, P.L.; Aguiar, R.C. Targeting of SMAD5 links microRNA-155 to the TGF-beta pathway and lymphomagenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3111–31116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Aguiar, R.C. MicroRNA-155 controls RB phosphorylation in normal and malignant B lymphocytes via the noncanonical TGF-β1/SMAD5 signaling module. Blood 2014, 123, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Stelling, A.; Hashwah, H.; Bertram, K.; Manz, M.G.; Tzankov, A.; Muller, A. The tumor suppressive TGF-beta/SMAD1/S1PR2 signaling axis is recurrently inactivated in diffuse large B-cell lymphoma. Blood 2018, 131, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Lo, K.W.; Wei, W.; Tsao, S.W.; Chung, G.T.Y.; Ibrahim, M.H.; Dawson, C.W.; Murray, P.G.; Paterson, I.C.; Yap, L.F. Oncogenic S1P signalling in EBV-associated nasopharyngeal carcinoma activates AKT and promotes cell migration through S1P receptor 3. J. Pathol. 2017, 242, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Rodon, L.; Folch, G.; Carmona, M.A.; Prieto-Sanchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Wakisaka, N.; Muramatsu, M.; Zen, Y.; Endo, K.; Murono, S.; Sugimoto, H.; Yamaoka, S.; Pagano, J.S.; Yoshizaki, T. Epstein-Barr virus latent membrane protein 1 induces cancer stem/progenitor-like cells in nasopharyngeal epithelial cell lines. J. Virol. 2011, 85, 11255–11264. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.L.; Hu, L.J.; Cao, J.Y.; Huang, Y.J.; Xu, L.H.; Liang, Y.; Xiong, D.; Guan, S.; Guo, B.H.; Mai, H.Q.; et al. Epstein-Barr virus-encoded LMP2A induces an epithelial-mesenchymal transition and increases the number of side population stem-like cancer cells in nasopharyngeal carcinoma. PLoS Pathog. 2010, 6, e1000940. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; MacIsaac, K.D.; Zhou, T.; Huang, P.Y.; Xin, C.; Dobson, J.R.; Yu, K.; Chiang, D.Y.; Fan, Y.; Pelletier, M.; et al. Genomic Analysis of Nasopharyngeal Carcinoma Reveals TME-Based Subtypes. Mol. Cancer Res. 2017, 15, 1722–1732. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Malignancy | %EBV+ Cases | Latency | EBV Latent Genes |
|---|---|---|---|
| Endemic Burkitt Lymphoma | 100% | I | EBNA1, EBER1, EBER2, BARTs, miR-BARTs |
| Sporadic Burkitt Lymphoma | 10–85% | ||
| HIV-associated Burkitt Lymphoma | 30–40% | ||
| T/NK cell lymphoma | 100% | II | EBNA1, LMP1, LMP2A, EBER1, EBER2, BARTs, miR-BARTs |
| Hodgkin’s Lymphoma | 80–90% | ||
| Hodgkin’s Lymphoma (Nodular sclerosing) | 15–20% | ||
| HIV-associated Hodgkin’s Lymphoma | <90% | ||
| Diffuse large B-cell lymphoma (Pythorax lymphoma) | 100% | II/III | EBNA1, LMP1, LMP2A, EBER1, EBER2, BARTs, miR-BARTs and/or EBNA2, 3A, 3B, 3C, LP |
| Diffuse large B-cell lymphoma (in Elderly patients) | >50% | ||
| Diffuse large B-cell lymphoma (late post-transplant) | >50% | ||
| HIV-associated diffuse large B-cell lymphoma | 30% | ||
| Post-transplant B-lymphoproliferative disorder | 100% | III | EBNA 1, 2, 3A, 3B, 3C, LP, LMP1, LMP2A, LMP2B, EBER1, EBER2, BARTs, miRNAs-BARTs, BHRF1 |
| HIV-associated B-lymphoproliferative disease | 100% | ||
| Nasopharyngeal carcinoma | 98% | II | EBNA1, LMP1, LMP2A, EBER1, EBER2, BARTs, miR-BARTs, BARF1 |
| EBV-associated gastric cancer | 10% |
| Cancer | Total Number of Cases | EBV Status | Genes | Alterations | Number of Cases with Alterations | References |
|---|---|---|---|---|---|---|
| NPC | 56 primary tumours | Positive | SMAD3 | Missense mutation | 1 (primary tumour) | [118] |
| NPC |
| Positive | TGF-β1 | Missense mutation | 1 (primary tumour) | [119] |
| TGF-β2 | Missense mutation | 1 (primary tumour) | ||||
| TGFBR2 | Missense mutation | 1 (primary tumour) | ||||
| NPC |
| Positive | TGF-β1 | Missense mutation | 1 (primary tumour) | [120] |
| TGF-β1 | Nonsense mutation | 1 (local recurrent tumour) | ||||
| TGF-β1 | Silent mutation | 1 (primary tumour) | ||||
| TGF-β2 | Frame shift deletion | 1 (local recurrent tumour) | ||||
| TGF-β2 | Inversion | 1 (primary tumour) | ||||
| TGFBR1 | Missense mutation | 1 (primary tumour) 1 (local recurrent tumour) | ||||
| TGFBR2 | Inter chromosomal translocation | 1 (primary tumour) | ||||
| SMAD3 | Silent mutation | 1 (local recurrent tumour) | ||||
| SMAD4 | Missense mutation | 1 (primary tumour) | ||||
| SMAD4 | Nonsense mutation | 1 (primary tumour) | ||||
| SMAD7 | Missense mutation | 1 (local recurrent tumour) | ||||
| EBVaGC | 134 primary tumours |
| TGFBR1 | Nonsynonymous mutation |
| [121] |
| AGS cell line | Before and after EBV infection | Missense mutation | EBV-infected AGS cells | |||
| EBVaGC | 22 primary tumours | Positive | SMAD4 | Missense mutation | 2 | [122] |
| HL | 7 cell lines |
| SMAD9 | Missense mutation | 1 (KMH2) | [123] |
| HL | 5 cell lines | Negative (HDML2, KMH2, UH01, L540, L428) | TGF-β1 | Amplification | 2 (L540, L428) | [124] |
| TGF-β2 | Amplification | 3 (KMH2, L540, L428) | ||||
| Deletion | 1 (UH01) | |||||
| TGFBR2 | Amplification | 3 (KMH2, L540, L428) | ||||
| TGFBR3 | Amplification | 2 (KMH2, L428) | ||||
| SMAD1 | Amplification | 3 (KMH2, L540, L428) | ||||
| Deletion | 2 (HDML2, UH01) | |||||
| SMAD5 | Amplification | 3 (KMH2, L540, L428) | ||||
| DLBCL |
| Unreported | TGF-β1 | Missense mutation | 1 (primary tumours) | [125] |
| TGF-β1 | Intronic mutation | 1 (primary tumours) | ||||
| TGFBR2 | Intronic mutation | 2 (primary tumours) | ||||
| TGFBR3 | Intronic mutation | 2 (primary tumours) | ||||
| SMAD9 | Intronic mutation | 1 (primary tumours) | ||||
| DLBCL | 51 primary tumours & immunochemotherapy-treated tumours | Unreported | TGF-β1 | CNA | 3 (treated tumours) | [126] |
| DLBCL | 6 refractory & 7 responsive tumours to R-Chop | Negative | TGFBR2 | Missense mutation | 1 (refractory tumour) | [127] |
| DLBCL |
| Unreported | TGFBR2 | Missense mutation | 6 (relapsed/refractory tumours) | [128] |
| DLBCL |
| Unreported | TGF-β1 | Missense mutation | 5 (4 ABC, 1 GCB) | [129] |
| Truncated mutation | 2 (ABC) | |||||
| TGF-β2 | Truncated mutation | 2 (1 ABC, 1 GCB) | ||||
| TGF-β3 | Missense mutation | 1 (ABC) | ||||
| TGFBR1 | Missense mutation | 1 (GCB) | ||||
| TGFBR2 | Missense mutation | 2 (1 ABC, 1 GCB) | ||||
| Truncated mutation | 2 (1 GCB, 1 unclassified) | |||||
| TGFBR3 | Missense mutation | 2 (1 ABC, 1 unclassified) | ||||
| Truncated mutation | 1 (GCB) | |||||
| SMAD1 | Missense mutation | 1 (ABC) | ||||
| Truncated mutation | 1 (GCB) | |||||
| SMAD2 | Missense mutation | 3 (1 ABC, 1 GCB, 1 unclassified) | ||||
| SMAD4 | Missense mutation | 3 (2 ABC, 1 unclassified) | ||||
| Truncated mutation | 1 (ABC) | |||||
| SMAD5 | Missense mutation | 5 (4 ABC, 1 GCB) | ||||
| SMAD6 | Missense mutation | 1 (GCB) | ||||
| SMAD7 | Missense mutation | 2 (1 ABC, 1 GCB) | ||||
| SMAD9 | Truncated mutation | 1 (ABC) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velapasamy, S.; Dawson, C.W.; Young, L.S.; Paterson, I.C.; Yap, L.F. The Dynamic Roles of TGF-β Signalling in EBV-Associated Cancers. Cancers 2018, 10, 247. https://doi.org/10.3390/cancers10080247
Velapasamy S, Dawson CW, Young LS, Paterson IC, Yap LF. The Dynamic Roles of TGF-β Signalling in EBV-Associated Cancers. Cancers. 2018; 10(8):247. https://doi.org/10.3390/cancers10080247
Chicago/Turabian StyleVelapasamy, Sharmila, Christopher W. Dawson, Lawrence S. Young, Ian C. Paterson, and Lee Fah Yap. 2018. "The Dynamic Roles of TGF-β Signalling in EBV-Associated Cancers" Cancers 10, no. 8: 247. https://doi.org/10.3390/cancers10080247
APA StyleVelapasamy, S., Dawson, C. W., Young, L. S., Paterson, I. C., & Yap, L. F. (2018). The Dynamic Roles of TGF-β Signalling in EBV-Associated Cancers. Cancers, 10(8), 247. https://doi.org/10.3390/cancers10080247