The Tip of an Iceberg: Replication-Associated Functions of the Tumor Suppressor p53



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Early Evidences of Transcription-Independent Roles of p53 in DNA Replication, Recombination and Repair
2. Current View: The Impact of p53 on DNA Replication
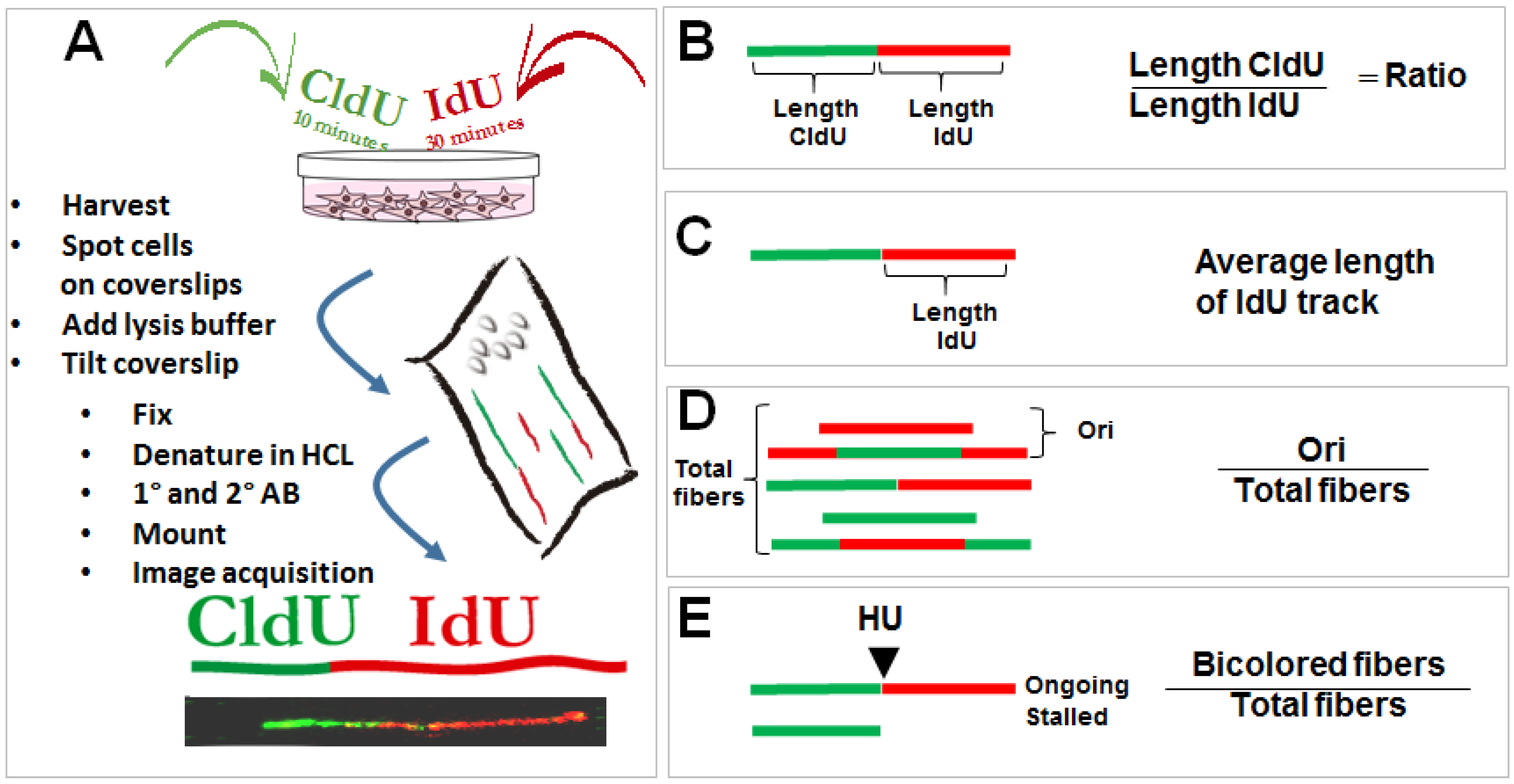
2.1. The Control of Nascent DNA Elongation by p53
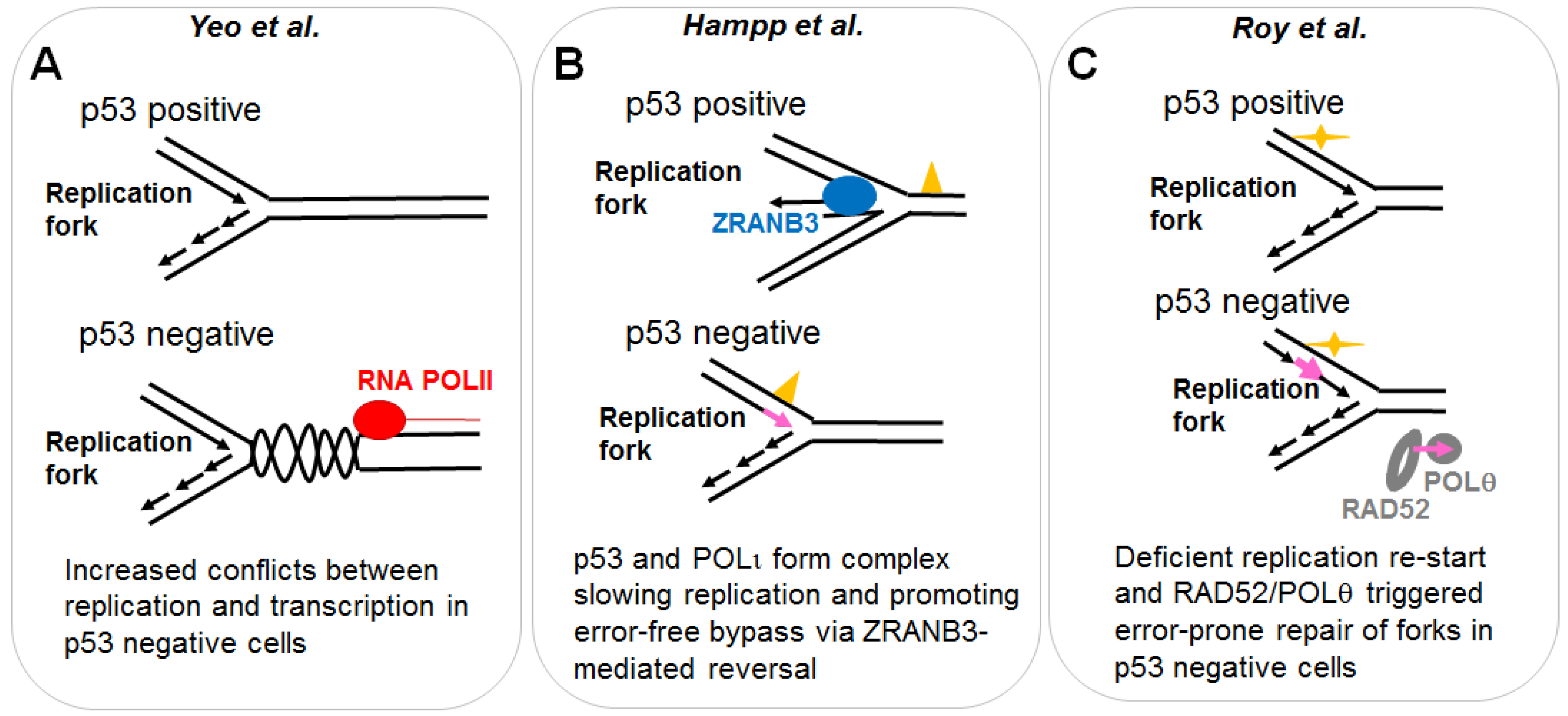
2.1.1. p53 Favors the Coupling between Transcription and Replication Fork Progression
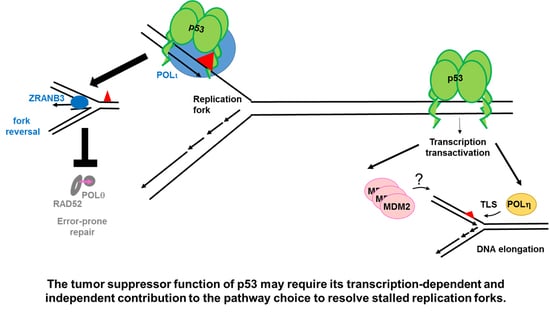
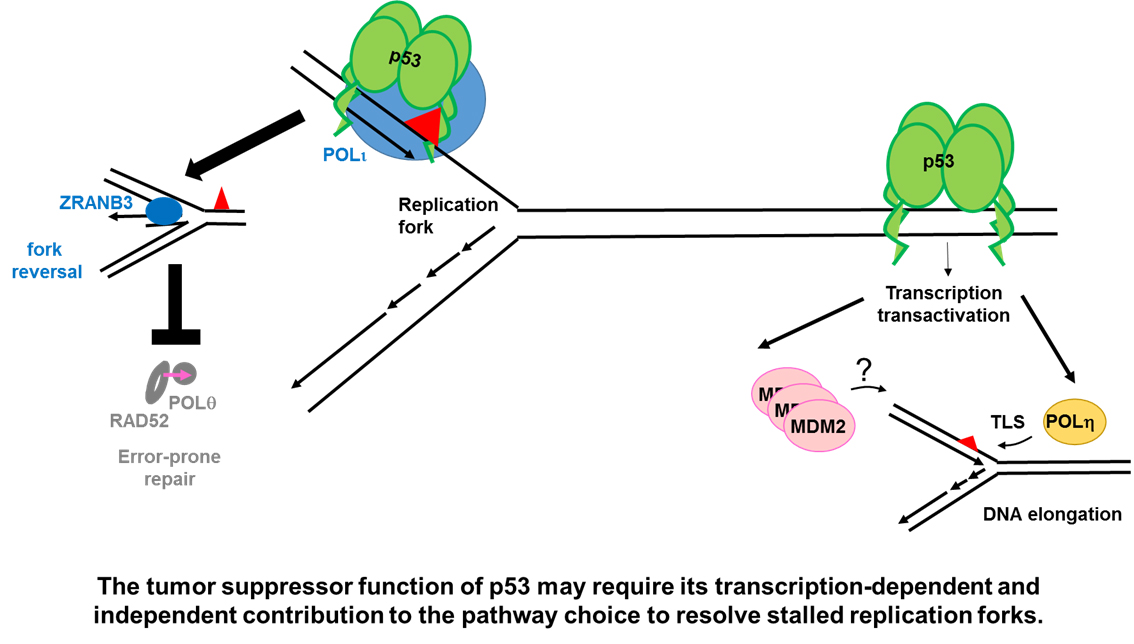
2.1.2. p53 Participates in the Choice of a Tolerance Pathway
2.1.3. p53 Promotes Replication Re-Start Preventing Mutagenic RAD52-Mediated DNA Repair
2.2. Biological Relevance of the Participation of p53 in the Replisome
2.3. The Control of Nascent DNA Elongation by p53 Targets
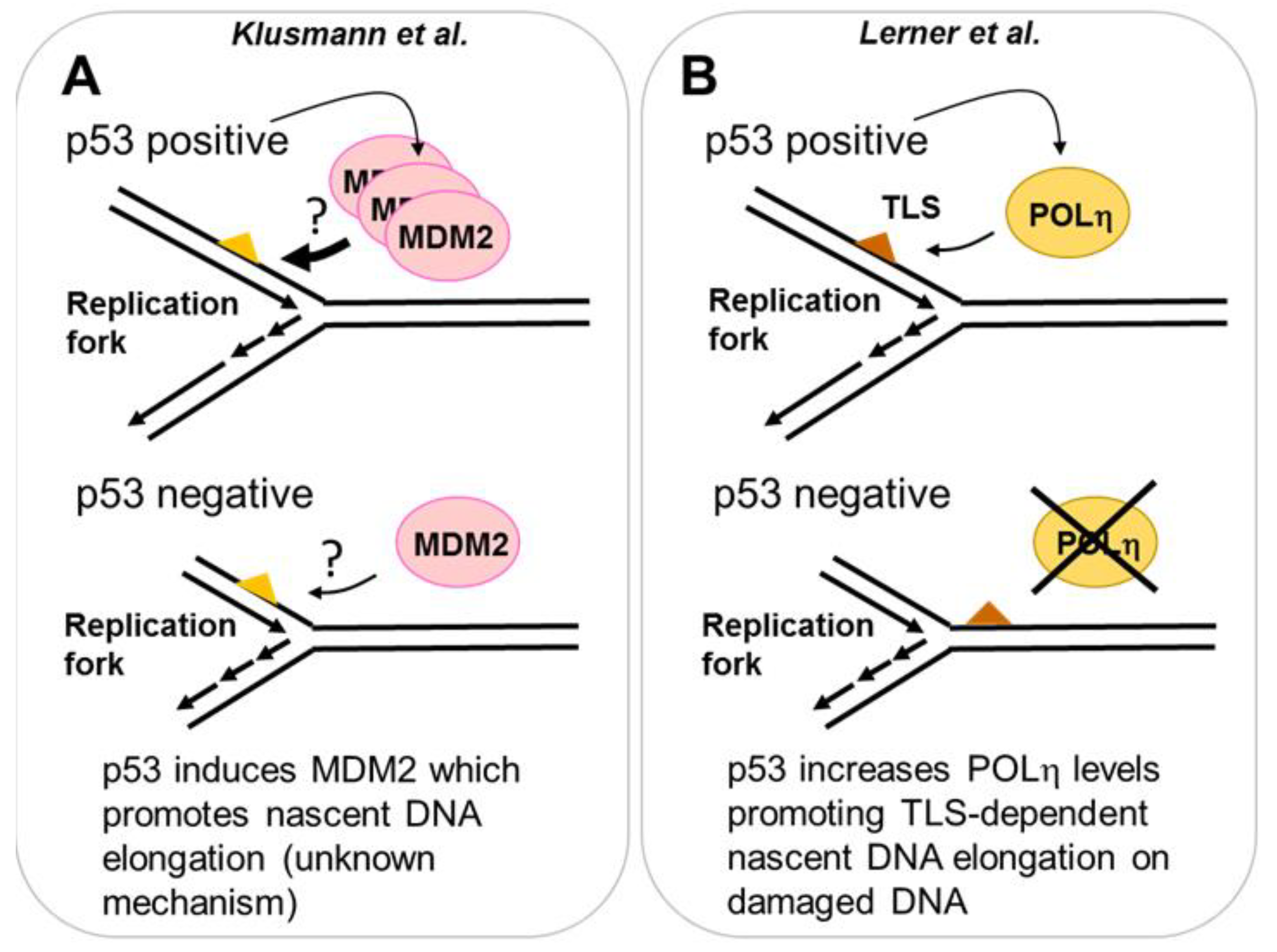
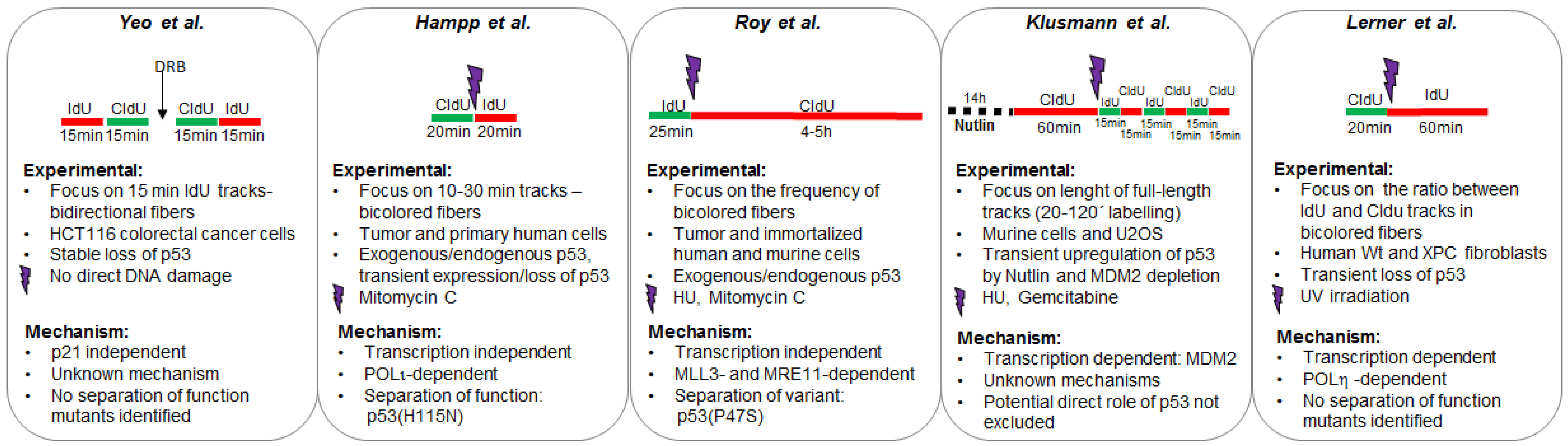
2.3.1. MDM2 Is a p53 Target That Promotes Nascent DNA Elongation
2.3.2. POLη Is a Target of p53 That Promotes Nascent DNA Elongation
2.4. Biological Relevance of the Contribution of p53 Targets to DNA Elongation
3. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The p53 pathway: Origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Attardi, L.D. Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol. 2018, 51, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Saintigny, Y.; Lopez, B.S. P53’s double life: Transactivation-independent repression of homologous recombination. Trends Genet. TIG 2004, 20, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Harris, C.C. P53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Gatz, S.A.; Wiesmuller, L. P53 in recombination and repair. Cell Death Differ. 2006, 13, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Mummenbrauer, T.; Janus, F.; Muller, B.; Wiesmuller, L.; Deppert, W.; Grosse, F. P53 protein exhibits 3′-to-5′ exonuclease activity. Cell 1996, 85, 1089–1099. [Google Scholar] [CrossRef]
- Feng, L.; Achanta, G.; Pelicano, H.; Zhang, W.; Plunkett, W.; Huang, P. Role of p53 in cellular response to anticancer nucleoside analog-induced DNA damage. Int. J. Mol. Med. 2000, 5, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Skalski, V.; Lin, Z.Y.; Choi, B.Y.; Brown, K.R. Substrate specificity of the p53-associated 3′-5′ exonuclease. Oncogene 2000, 19, 3321–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhanashvili, M. Exonucleolytic proofreading by p53 protein. Eur. J. Biochem. 2001, 268, 2047–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakked, Z.; Yavnilovitch, M.; Kalb Gilboa, A.J.; Kessler, N.; Wolkowicz, R.; Rotter, V.; Haran, T.E. DNA binding and 3′-5′ exonuclease activity in the murine alternatively-spliced p53 protein. Oncogene 2002, 21, 5117–5126. [Google Scholar] [CrossRef] [PubMed]
- Melle, C.; Nasheuer, H.P. Physical and functional interactions of the tumor suppressor protein p53 and DNA polymerase alpha-primase. Nucl. Acids Res. 2002, 30, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Poyurovsky, M.V.; Baptiste, N.; Beckerman, R.; Cain, C.; Mattia, M.; McKinney, K.; Zhou, J.; Zupnick, A.; Gottifredi, V.; et al. Dissection of the sequence-specific DNA binding and exonuclease activities reveals a superactive yet apoptotically impaired mutant p53 protein. Cell Cycle 2009, 8, 1603–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janus, F.; Albrechtsen, N.; Knippschild, U.; Wiesmuller, L.; Grosse, F.; Deppert, W. Different regulation of the p53 core domain activities 3′-to-5′ exonuclease and sequence-specific DNA binding. Mol. Cell. Biol. 1999, 19, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Zink, D.; Mayr, C.; Janz, C.; Wiesmuller, L. Association of p53 and MSH2 with recombinative repair complexes during s phase. Oncogene 2002, 21, 4788–4800. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Linke, S.P.; Pedeux, R.; Yang, Q.; Farnsworth, J.; Garfield, S.H.; Valerie, K.; Shay, J.W.; Ellis, N.A.; Wasylyk, B.; et al. BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J. 2003, 22, 1210–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Restle, A.; Farber, M.; Baumann, C.; Bohringer, M.; Scheidtmann, K.H.; Muller-Tidow, C.; Wiesmuller, L. Dissecting the role of p53 phosphorylation in homologous recombination provides new clues for gain-of-function mutants. Nucl. Acids Res. 2008, 36, 5362–5375. [Google Scholar] [CrossRef] [PubMed]
- Bohringer, M.; Obermeier, K.; Griner, N.; Waldraff, D.; Dickinson, E.; Eirich, K.; Schindler, D.; Hagen, M.; Jerry, D.J.; Wiesmuller, L. SiRNA screening identifies differences in the fanconi anemia pathway in BALB/c-Trp53+/- with susceptibility versus C57BL/6-Trp53+/- mice with resistance to mammary tumors. Oncogene 2013, 32, 5458–5470. [Google Scholar] [CrossRef] [PubMed]
- Livneh, Z. Keeping mammalian mutation load in check: Regulation of the activity of error-prone DNA polymerases by p53 and p21. Cell Cycle 2006, 5, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Avkin, S.; Sevilya, Z.; Toube, L.; Geacintov, N.; Chaney, S.G.; Oren, M.; Livneh, Z. P53 and p21 regulate error-prone DNA repair to yield a lower mutation load. Mol. Cell 2006, 22, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Tomaszowski, K.H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. P53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and poltheta pathways. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.A.; Pombo, A. Replicon clusters are stable units of chromosome structure: Evidence that nuclear organization contributes to the efficient activation and propagation of s phase in human cells. J. Cell Biol. 1998, 140, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Vindigni, A.; Lopes, M. Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics. Biophys. Chem. 2017, 225, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Bertolin, A.P.; Mansilla, S.F.; Gottifredi, V. The identification of translesion DNA synthesis regulators: Inhibitors in the spotlight. DNA Repair 2015, 32, 158–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, J.A.; Conti, C.; Syed, A.; Aladjem, M.I.; Pommier, Y. The intra-s-phase checkpoint affects both DNA replication initiation and elongation: Single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007, 27, 5806–5818. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, C.Q.X.; Alexander, I.; Lin, Z.; Lim, S.; Aning, O.A.; Kumar, R.; Sangthongpitag, K.; Pendharkar, V.; Ho, V.H.B.; Cheok, C.F. P53 maintains genomic stability by preventing interference between transcription and replication. Cell Rep. 2016, 15, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain g2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Grosse, F. Human dhx9 helicase preferentially unwinds RNA-containing displacement loops (r-loops) and g-quadruplexes. DNA Repair 2011, 10, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ireno, I.C.; Wiehe, R.S.; Stahl, A.I.; Hampp, S.; Aydin, S.; Troester, M.A.; Selivanova, G.; Wiesmuller, L. Modulation of the poly (ADP-ribose) polymerase inhibitor response and DNA recombination in breast cancer cells by drugs affecting endogenous wild-type p53. Carcinogenesis 2014, 35, 2273–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of li-fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Fraser, N.W.; Sehgal, P.B.; Darnell, J.E., Jr. Multiple discrete sites for premature RNA chain termination late in adenovirus-2 infection: Enhancement by 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole. Proc. Natl. Acad. Sci. USA 1979, 76, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Hampp, S.; Kiessling, T.; Buechle, K.; Mansilla, S.F.; Thomale, J.; Rall, M.; Ahn, J.; Pospiech, H.; Gottifredi, V.; Wiesmuller, L. DNA damage tolerance pathway involving DNA polymerase iota and the tumor suppressor p53 regulates DNA replication fork progression. Proc. Natl. Acad. Sci. USA 2016, 113, E4311–E4319. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.E.; Woodgate, R. Insights into the cellular role of enigmatic DNA polymerase iota. DNA Repair 2009, 8, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Eckert, K.A. The proofreading 3′→5′ exonuclease activity of DNA polymerases: A kinetic barrier to translesion DNA synthesis. Mutat. Res. 2002, 510, 45–54. [Google Scholar] [CrossRef]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.W.; Holland, C.L.; Zwicky, K.; et al. Replication fork slowing and reversal upon DNA damage require pcna polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell 2017, 67, 882–890.e5. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Alvino, G.M.; Collingwood, D.; Murphy, J.M.; Delrow, J.; Brewer, B.J.; Raghuraman, M.K. Replication in hydroxyurea: It’s a matter of time. Mol. Cell. Biol. 2007, 27, 6396–6406. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Luzwick, J.W.; Schlacher, K. SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J. Cell Biol. 2018, 217, 1521–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquel, C.; Jacob, S.; Grandjouan, S.; Aime, A.; Viguier, J.; Sabourin, J.C.; Sarasin, A.; Duval, A.; Praz, F. Frequent alteration of DNA damage signalling and repair pathways in human colorectal cancers with microsatellite instability. Oncogene 2007, 26, 5919–5926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Q.; Scorah, J.; Phear, G.; Rodgers, G.; Rodgers, S.; Meuth, M. A mutant allele of mre11 found in mismatch repair-deficient tumor cells suppresses the cellular response to DNA replication fork stress in a dominant negative manner. Mol. Biol. Cell 2008, 19, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Jennis, M.; Kung, C.P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An african-specific polymorphism in the tp53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.E.; Liu, S.; Yao, S.; Huo, D.; Liu, Q.; Dolfi, S.C.; Hirshfield, K.M.; Hong, C.C.; Hu, Q.; Olshan, A.F.; et al. A functionally significant SNP in tp53 and breast cancer risk in African-American women. NPJ Breast Cancer 2017, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Felley-Bosco, E.; Weston, A.; Cawley, H.M.; Bennett, W.P.; Harris, C.C. Functional studies of a germ-line polymorphism at codon 47 within the p53 gene. Am. J. Hum. Genet. 1993, 53, 752–759. [Google Scholar] [PubMed]
- Romanova, L.Y.; Willers, H.; Blagosklonny, M.V.; Powell, S.N. The interaction of p53 with replication protein a mediates suppression of homologous recombination. Oncogene 2004, 23, 9025–9033. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, G.; Podhajcer, O.; Prives, C.; Gottifredi, V. P21Cip1/WAF1 downregulation is required for efficient PCNA ubiquitination after UV irradiation. Oncogene 2006, 25, 2829–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, H.D. Regulating post-translational modifications of the eukaryotic replication clamp pcna. DNA Repair 2009, 8, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Gottifredi, V. PCNA-coupled p21 degradation after DNA damage: The exception that confirms the rule? DNA Repair 2010, 9, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansilla, S.F.; Soria, G.; Vallerga, M.B.; Habif, M.; Martinez-Lopez, W.; Prives, C.; Gottifredi, V. UV-triggered p21 degradation facilitates damaged-DNA replication and preserves genomic stability. Nucl. Acids Res. 2013, 41, 6942–6951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansilla, S.F.; Bertolin, A.P.; Bergoglio, V.; Pillaire, M.J.; Gonzalez Besteiro, M.A.; Luzzani, C.; Miriuka, S.G.; Cazaux, C.; Hoffmann, J.S.; Gottifredi, V. Cyclin kinase-independent role of p21(CDKN1A) in the promotion of nascent DNA elongation in unstressed cells. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wienken, M.; Moll, U.M.; Dobbelstein, M. MDM2 as a chromatin modifier. J. Mol. Cell Biol. 2017, 9, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Klusmann, I.; Rodewald, S.; Muller, L.; Friedrich, M.; Wienken, M.; Li, Y.; Schulz-Heddergott, R.; Dobbelstein, M. P53 activity results in DNA replication fork processivity. Cell Rep. 2016, 17, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Batista, L.F.; Kaina, B.; Meneghini, R.; Menck, C.F. How DNA lesions are turned into powerful killing structures: Insights from UV-induced apoptosis. Mutat. Res. 2009, 681, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.T.; Johnson, R.E.; Prakash, L.; Prakash, S. Accuracy of lesion bypass by yeast and human DNA polymerase eta. Proc. Natl. Acad. Sci. USA 2001, 98, 8355–8360. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E. Common pathways for ultraviolet skin carcinogenesis in the repair and replication defective groups of xeroderma pigmentosum. J. Dermatol. Sci. 2000, 23, 1–11. [Google Scholar] [CrossRef]
- Liu, G.; Chen, X. DNA polymerase eta, the product of the xeroderma pigmentosum variant gene and a target of p53, modulates the DNA damage checkpoint and p53 activation. Mol. Cell. Biol. 2006, 26, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Lerner, L.K.; Francisco, G.; Soltys, D.T.; Rocha, C.R.; Quinet, A.; Vessoni, A.T.; Castro, L.P.; David, T.I.; Bustos, S.O.; Strauss, B.E.; et al. Predominant role of DNA polymerase eta and p53-dependent translesion synthesis in the survival of ultraviolet-irradiated human cells. Nucl. Acids Res. 2017, 45, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Chung, M.; Kudo, T.; Meyer, T. Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 2017, 549, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Vallerga, M.B.; Mansilla, S.F.; Federico, M.B.; Bertolin, A.P.; Gottifredi, V. RAD51 recombinase prevents Mre11 nuclease-dependent degradation and excessive primpol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E6624–E6633. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Larroque, M.; Ribeyre, C.; Bec, N.; Larroque, C.; Decaillet, C.; Gari, K.; Constantinou, A. Fancd2 binds mcm proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol. Cell 2013, 51, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Mizutani, S.; Lamorte, G.; Goi, K.; Iwata, S.; Pierotti, M.A. P53 activity and chemotherapy. Nat. Med. 1996, 2, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V. Converting p53 from a killer into a healer. Nat. Med. 2002, 8, 1196–1198. [Google Scholar] [CrossRef] [PubMed]
- Kranz, D.; Dobbelstein, M. A killer promoting survival: P53 as a selective means to avoid side effects of chemotherapy. Cell Cycle 2012, 11, 2053–2054. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Vaughan, C.A.; Frum, R.A.; Grossman, S.R.; Deb, S.; Palit Deb, S. Mutant p53 establishes targetable tumor dependency by promoting unscheduled replication. J. Clin. Investig. 2017, 127, 1839–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polotskaia, A.; Xiao, G.; Reynoso, K.; Martin, C.; Qiu, W.G.; Hendrickson, R.C.; Bargonetti, J. Proteome-wide analysis of mutant p53 targets in breast cancer identifies new levels of gain-of-function that influence parp, pcna, and mcm4. Proc. Natl. Acad. Sci. USA 2015, 112, E1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Ghatak, D.; Das, S.; Banerjee, T.; Paul, A.; Butti, R.; Gorain, M.; Ghuwalewala, S.; Roychowdhury, A.; Alam, S.K.; et al. P53 gain-of-function mutations increase CDC7-dependent replication initiation. EMBO Rep. 2017, 18, 2030–2050. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with tp53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Ivkov, R.; Bunz, F. Pathways to chromothripsis. Cell Cycle 2015, 14, 2886–2890. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Larsen, A.R.; Chen, E.; Bunz, F. A ptch1 homolog transcriptionally activated by p53 suppresses hedgehog signaling. J. Biol. Chem. 2014, 289, 33020–33031. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gottifredi, V.; Wiesmüller, L. The Tip of an Iceberg: Replication-Associated Functions of the Tumor Suppressor p53. Cancers 2018, 10, 250. https://doi.org/10.3390/cancers10080250
Gottifredi V, Wiesmüller L. The Tip of an Iceberg: Replication-Associated Functions of the Tumor Suppressor p53. Cancers. 2018; 10(8):250. https://doi.org/10.3390/cancers10080250
Chicago/Turabian StyleGottifredi, Vanesa, and Lisa Wiesmüller. 2018. "The Tip of an Iceberg: Replication-Associated Functions of the Tumor Suppressor p53" Cancers 10, no. 8: 250. https://doi.org/10.3390/cancers10080250
APA StyleGottifredi, V., & Wiesmüller, L. (2018). The Tip of an Iceberg: Replication-Associated Functions of the Tumor Suppressor p53. Cancers, 10(8), 250. https://doi.org/10.3390/cancers10080250