Tumour Microenvironment and Immune Evasion in EGFR Addicted NSCLC: Hurdles and Possibilities

 ,
,  and
and 
Abstract
:1. Introduction
2. PD-L1 Expression in EGFR Mutated Patients
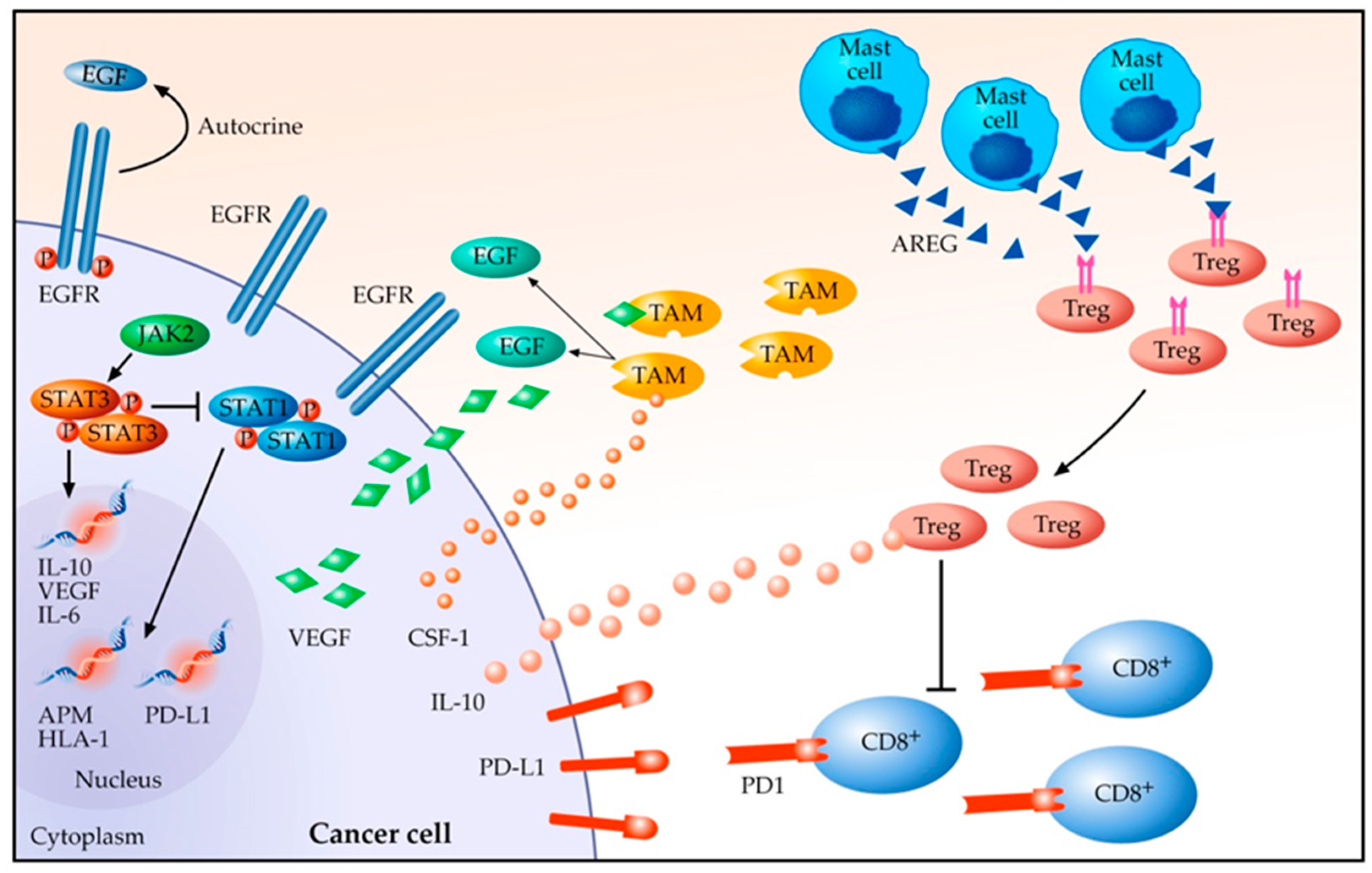
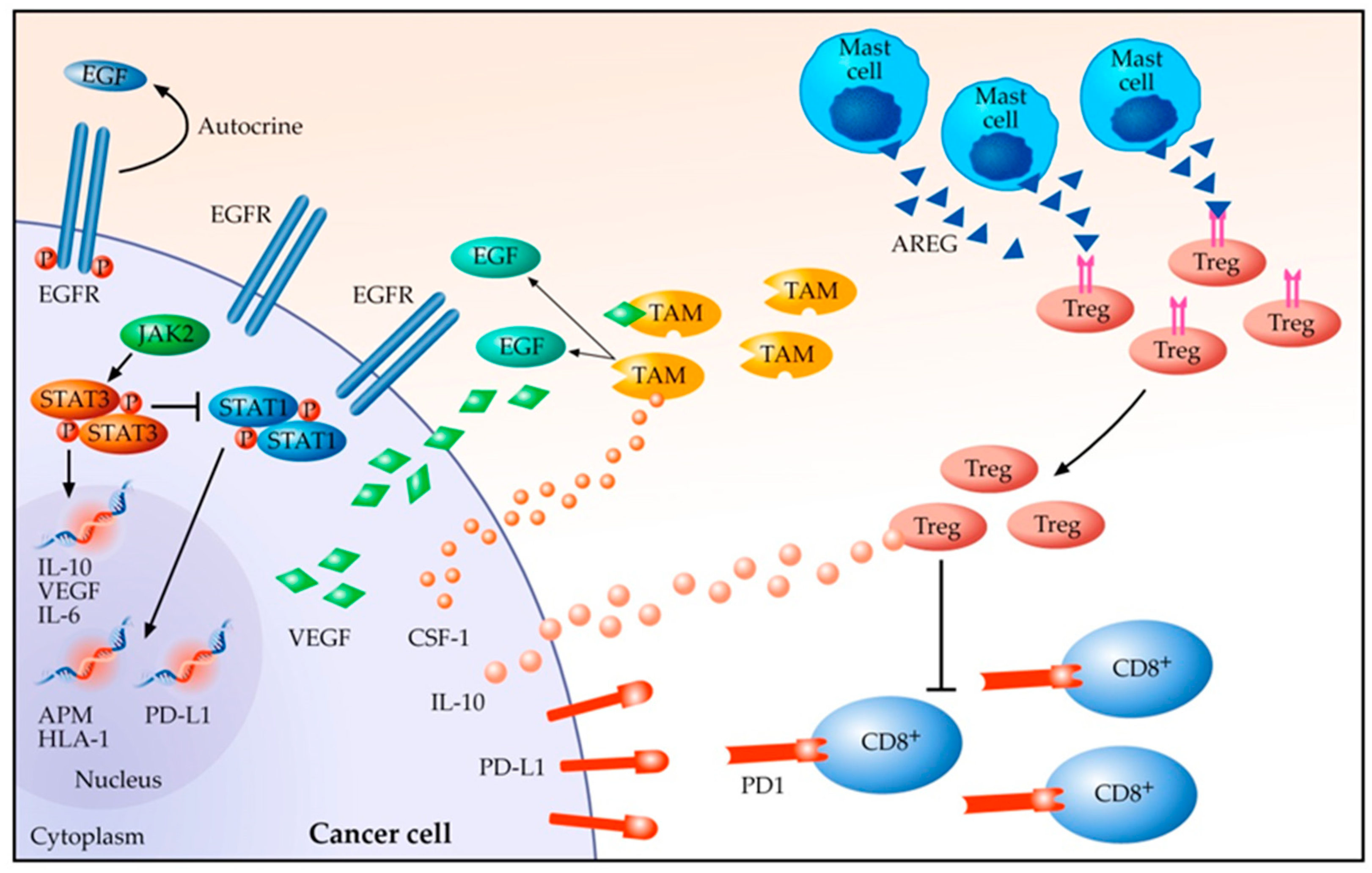
3. Tumour Microenvironment
4. Immunocheckpoint Inhibitors (ICIs) Treatment Failure
5. Tumour Mutational Burden (TMB) and Non-Synonymous Mutations
6. Role of Radiotherapy
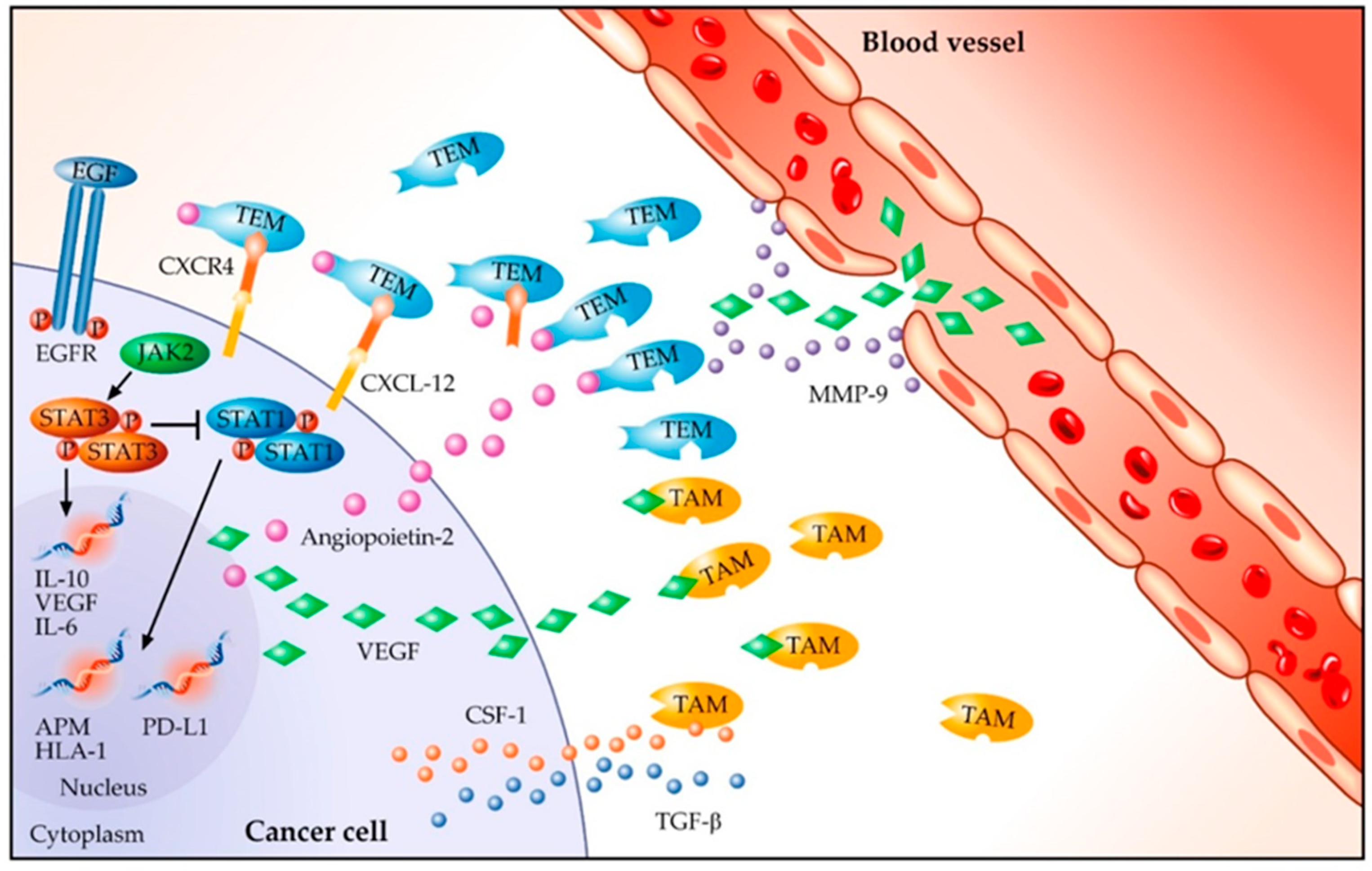
7. Role of Angiogenesis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Hughes, B.G.M. Targeted Therapy for Non-Small Cell Lung Cancer: Current Standards and the Promise of the Future. Transl. Lung Cancer Res. 2015. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus Chemotherapy as First-Line Treatment for Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2011. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus Standard Chemotherapy as First-Line Treatment for European Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2012. [Google Scholar] [CrossRef]
- Goss, G.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Lee, J.S.; Chang, G.C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for Pretreated EGFR Thr790Met-Positive Advanced Non-Small-Cell Lung Cancer (AURA2): A Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet Oncol. 2016. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Wu, Y.L.; Hirsh, V.; O’Byrne, K.; Yamamoto, N.; Mok, T.; Popat, S.; Sequist, L.V.; Massey, D.; Zazulina, V.; et al. First-Line Afatinib versus Chemotherapy in Patients with Non-Small Cell Lung Cancer and Common Epidermal Growth Factor Receptor Gene Mutations and Brain Metastases. J. Thorac. Oncol. 2016. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.H.; Lee, K.H.; Lu, S.; et al. Afatinib versus Gefitinib as First-Line Treatment of Patients with EGFR Mutation-Positive Non-Small-Cell Lung Cancer (LUX-Lung 7): A Phase 2B, Open-Label, Randomised Controlled Trial. Lancet Oncol. 2016. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Quatrale, A.E.; Porcelli, L.; Silvestris, N.; Colucci, G.; Angelo, A.; Azzariti, A. EGFR Tyrosine Kinases Inhibitors in Cancer Treatment: In Vitro and in Vivo Evidence. Front. Biosci. (Landmark Ed.) 2011, 16, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Nixon, N.A.; Blais, N.; Ernst, S.; Kollmannsberger, C.; Bebb, G.; Butler, M.; Smylie, M.; Verma, S. Current Landscape of Immunotherapy in the Treatment of Solid Tumours, with Future Opportunities and Challenges. Curr. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- D’Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 Expression in Molecularly Selected Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2015. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus Docetaxel for Previously Treated, PD-L1-Positive, Advanced Non-Small-Cell Lung Cancer (KEYNOTE-010): A Randomised Controlled Trial. Lancet 2016. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus Docetaxel for Patients with Previously Treated Non-Small-Cell Lung Cancer (POPLAR): A Multicentre, Open-Label, Phase 2 Randomised Controlled Trial. Lancet 2016. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus Docetaxel in Patients with Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.M.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.E.; Juergens, R.A.; Shepherd, F.A.; et al. Nivolumab Plus Erlotinib in Patients With EGFR-Mutant Advanced NSCLC. J. Thorac. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.; Cervantes, A.; Dowlati, A.; Besse, B.; Ma, B.; Costa, D.; Schmid, P.; Heist, R.; Villaflor, V.; Sarkar, I.; et al. MA15.02 Long-Term Safety and Clinical Activity Results from a Phase Ib Study of Erlotinib Plus Atezolizumab in Advanced NSCLC. J. Thorac. Oncol. 2018. [Google Scholar] [CrossRef]
- Creelan, B.C.; Yeh, T.; Kim, S.-W.; Nogami, N.; Kim, D.-W.; Chow, L.Q.; Kanda, S.; Taylor, R.; Tang, W.; Tang, M.; et al. 84OPhase I Study of Gefitinib (G) + Durvalumab (D) for Locally Advanced/Metastatic Non-Small Cell Lung Cancer (NSCLC) Harbouring Epidermal Growth Factor Receptor (EGFR) Sensitising Mutations. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Ahn, M.-J.; Yang, J.; Yu, H.; Saka, H.; Ramalingam, S.; Goto, K.; Kim, S.-W.; Yang, L.; Walding, A.; Oxnard, G.R. 136O: Osimertinib Combined with Durvalumab in EGFR-Mutant Non-Small Cell Lung Cancer: Results from the TATTON Phase Ib Trial. J. Thorac. Oncol. 2016, 11, S115. [Google Scholar] [CrossRef]
- Lisberg, A.; Cummings, A.; Goldman, J.W.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ledezma, B.; Mendenhall, M.; Hunt, J.; et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC. J. Thorac. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy-Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 Overexpression with Activating EGFR Mutations in Surgically Resected Nonsmall-Cell Lung Cancer. Ann. Oncol. 2014. [Google Scholar] [CrossRef]
- Tang, Y.; Fang, W.; Zhang, Y.; Hong, S.; Kang, S.; Yan, Y.; Chen, N.; Zhan, J.; He, X.; Qin, T.; et al. The Association between PD-L1 and EGFR Status and the Prognostic Value of PD-L1 in Advanced Non-Small Cell Lung Cancer Patients Treated with EGFR-TKIs. Oncotarget 2015. [Google Scholar] [CrossRef] [PubMed]
- Soo, R.A.; Lim, S.M.; Syn, N.L.; Teng, R.; Soong, R.; Mok, T.S.K.; Cho, B.C. Immune Checkpoint Inhibitors in Epidermal Growth Factor Receptor Mutant Non-Small Cell Lung Cancer: Current Controversies and Future Directions. Lung Cancer. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.Y.; Zhang, J.T.; Liu, S.Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.Y.; Xu, C.R.; Yan, L.X.; et al. EGFR Mutation Correlates with Uninflamed Phenotype and Weak Immunogenicity, Causing Impaired Response to PD-1 Blockade in Non-Small Cell Lung Cancer. Oncoimmunology 2017. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.; Garattini, S.K.; Bonotto, M.; Ongaro, E.; Casagrande, M.; Cattaneo, M.; Fanotto, V.; De Carlo, E.; Loupakis, F.; Urbano, F.; et al. Immunotherapy for Colorectal Cancer: Where Are We Heading? Expert Opin. Biol. Ther. 2017, 17, 709–721. [Google Scholar] [CrossRef]
- Whiteside, T.L. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene 2008. [Google Scholar] [CrossRef]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res. 2004. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune Escape to PD-L1/PD-1 Blockade: Seven Steps to Success (or Failure). Ann. Oncol. 2016. [Google Scholar] [CrossRef]
- Zaiss, D.M.; Yang, L.; Shah, P.R.; Kobie, J.J.; Urban, J.F.; Mosmann, T.R. Amphiregulin, a T H 2 Cytokine Enhancing Resistance to Nematodes. Science 2006. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.J.; Gröne, A.; Sibilia, M.; van Bergen en Henegouwen, P.M.P.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J.A.M. Amphiregulin Enhances Regulatory T Cell-Suppressive Function via the Epidermal Growth Factor Receptor. Immunity 2013. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging Functions of Amphiregulin in Orchestrating Immunity, Inflammation, and Tissue Repair. Immunity 2015. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies That Are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-Induced Perturbations of Cytokines and Immune Cell Networks. Biochim. Biophys. Acta Rev. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Rubin Grandis, J.; Drenning, S.D.; Chakraborty, A.; Zhou, M.Y.; Zeng, Q.; Pitt, A.S.; Tweardy, D.J. Requirement of Stat3 but Not Stat1 Activation for Epidermal Growth Factor Receptor-Mediated Cell Growth in Vitro. J. Clin. Investig. 1998, 102, 1385–1392. [Google Scholar] [CrossRef]
- Schrump, D.S.; Nguyen, D.M. Targets for Molecular Intervention in Multistep Pulmonary Carcinogenesis. World J. Surg. 2001. [Google Scholar] [CrossRef]
- Kijima, T.; Niwa, H.; Steinman, R.A.; Drenning, S.D.; Gooding, W.E.; Wentzel, A.L.; Xi, S.; Grandis, J.R. STAT3 Activation Abrogates Growth Factor Dependence and Contributes to Head and Neck Squamous Cell Carcinoma Tumor Growth in Vivo. Cell Growth Differ. 2002, 13, 355–362. [Google Scholar]
- Concha-Benavente, F.; Srivastava, R.M.; Trivedi, S.; Lei, Y.; Chandran, U.; Seethala, R.R.; Freeman, G.J.; Ferris, R.L. Identification of the Cell-Intrinsic and -Extrinsic Pathways Downstream of EGFR and IFNγ That Induce PD-L1 Expression in Head and Neck Cancer. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Leibowitz, M.S.; Srivastava, R.M.; Filho, P.A.A.; Egloff, A.M.; Wang, L.; Seethala, R.R.; Ferrone, S.; Ferris, R.L. SHP2 Is Overexpressed and Inhibits PSTAT1-Mediated APM Component Expression, T-Cell Attracting Chemokine Secretion, and CTL Recognition in Head and Neck Cancer Cells. Clin. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of Vascular Endothelial Growth Factor by Human Tumors Inhibits the Functional Maturation of Dendritic Cells. Nat. Med. 1996. [Google Scholar] [CrossRef]
- Yang, A.S.; Lattime, E.C. Tumor-Induced Interleukin 10 Suppresses the Ability of Splenic Dendritic Cells to Stimulate CD4 and CD8 T-Cell Responses. Cancer Res. 2003, 63, 2150–2157. [Google Scholar]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C.H. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non–Small Cell Lung Cancer—A Meta-Analysis. J. Thorac. Oncol. 2017. [Google Scholar] [CrossRef]
- Garassino, M.C.; Cho, B.C.; Kim, J.H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Corral Jaime, J.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Durvalumab as Third-Line or Later Treatment for Advanced Non-Small-Cell Lung Cancer (ATLANTIC): An Open-Label, Single-Arm, Phase 2 Study. Lancet Oncol. 2018. [Google Scholar] [CrossRef]
- Cavanna, L.; Citterio, C.; Orlandi, E. Immune Checkpoint Inhibitors in EGFR-Mutation Positive TKI-Treated Patients with Advanced Non-Small-Cell Lung Cancer Network Meta-Analysis. Oncotarget 2019, 10, 209–215. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The Gut Microbiota Shapes Intestinal Immune Responses during Health and Disease. Nat. Rev. Immunol. 2009. [Google Scholar] [CrossRef]
- Yu, G.; Gail, M.H.; Consonni, D.; Carugno, M.; Humphrys, M.; Pesatori, A.C.; Caporaso, N.E.; Goedert, J.J.; Ravel, J.; Landi, M.T. Characterizing Human Lung Tissue Microbiota and Its Relationship to Epidemiological and Clinical Features. Genome Biol. 2016. [Google Scholar] [CrossRef]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.X. Recurrent Antibiotic Exposure May Promote Cancer Formation-Another Step in Understanding the Role of the Human Microbiota? Eur. J. Cancer 2015. [Google Scholar] [CrossRef]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative Association of Antibiotics on Clinical Activity of Immune Checkpoint Inhibitors in Patients with Advanced Renal Cell and Non-Small-Cell Lung Cancer. Ann. Oncol. 2018. [Google Scholar] [CrossRef]
- Noronha, V.; Joshi, A.; Patil, V.M.; Chougule, A.; Mahajan, A.; Janu, A.; Purandare, N.; Kumar, R.; More, S.; Goud, S.; et al. Phase III Randomized Trial Comparing Gefitinib to Gefitinib with Pemetrexed-Carboplatin Chemotherapy in Patients with Advanced Untreated EGFR Mutant Non-Small Cell Lung Cancer (Gef vs Gef+C). J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science 2016. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat. Rev. Cancer 2016. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients with Non-Small-Cell Lung Cancer Profiled with Targeted next-Generation Sequencing. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Carbone, D.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.; Ciuleanu, T.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015. [Google Scholar] [CrossRef]
- Spigel, D.R.; Schrock, A.B.; Fabrizio, D.; Frampton, G.M.; Sun, J.; He, J.; Gowen, K.; Johnson, M.L.; Bauer, T.M.; Kalemkerian, G.P.; et al. Total Mutation Burden (TMB) in Lung Cancer (LC) and Relationship with Response to PD-1/PD-L1 Targeted Therapies. J. Clin. Oncol. 2016, 34 (Suppl. 15), 9017. [Google Scholar] [CrossRef]
- Miyauchi, E.; Matsuda, T.; Kiyotani, K.; Low, S.K.; Hsu, Y.W.; Tsukita, Y.; Ichinose, M.; Sakurada, A.; Okada, Y.; Saito, R.; et al. Significant Differences in T Cell Receptor Repertoires in Lung Adenocarcinomas with and without Epidermal Growth Factor Receptor Mutations. Cancer Sci. 2019. [Google Scholar] [CrossRef]
- Jonna, S.; Vanderwalde, A.M.; Nieva, J.J.; Poorman, K.A.; Saul, M.; von Buttlar, X.; Hu, J.Y.; Liu, S.V. Impact of Prior Chemotherapy or Radiation Therapy on Tumor Mutation Burden in NSCLC. J. Clin. Oncol. 2019, 37 (Suppl. 15), 2627. [Google Scholar] [CrossRef]
- Hastings, K.; Yu, H.; Wei, W.; Sanchez-Vega, F.; DeVeaux, M.; Choi, J.; Rizvi, H.; Lisberg, A.; Truini, A.; Lydon, C.A.; et al. EGFR Mutation Subtypes and Response to Immune Checkpoint Blockade Treatment in Non-Small Cell Lung Cancer. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor Immune Microenvironment and Nivolumab Efficacy in EGFR Mutation-Positive Non-Small-Cell Lung Cancer Based on T790M Status after Disease Progression during EGFR-TKI Treatment. Ann. Oncol. 2017. [Google Scholar] [CrossRef]
- Yamada, T.; Hirai, S.; Katayama, Y.; Yoshimura, A.; Shiotsu, S.; Watanabe, S.; Kikuchi, T.; Hirose, K.; Kubota, Y.; Chihara, Y.; et al. Retrospective Efficacy Analysis of Immune Checkpoint Inhibitors in Patients with EGFR-mutated Non-small Cell Lung Cancer. Cancer Med. 2019, 8, 1521–1529. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.E.; Mhanna, L.; Milia, J.; Lusque, A.; Cortot, A.B.; Mezquita, L.; Thai, A.; Couraud, S.; Veillon, R.; et al. Efficacy of Immune-Checkpoint Inhibitors (ICI) in Non-Small Cell Lung Cancer (NSCLC) Patients Harboring Activating Molecular Alterations (ImmunoTarget). J. Clin. Oncol. 2018, 36 (Suppl. 15), 9010. [Google Scholar] [CrossRef]
- Vatner, R.E.; Cooper, B.T.; Vanpouille-Box, C.; Demaria, S.; Formenti, S.C. Combinations of Immunotherapy and Radiation in Cancer Therapy. Front. Oncol. 2014. [Google Scholar] [CrossRef]
- Ko, E.C.; Raben, D.; Formenti, S.C. The Integration of Radiotherapy with Immunotherapy for the Treatment of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Dos Reis, M. How to Calculate the Non-Synonymous to Synonymous Rate Ratio of Protein-Coding Genes under the Fisher-Wright Mutation-Selection Framework. Biol. Lett. 2015. [Google Scholar] [CrossRef]
- Moschini, I.; Dell’Anna, C.; Losardo, P.L.; Bordi, P.; D’abbiero, N.; Tiseo, M. Radiotherapy of Non-Small-Cell Lung Cancer in the Era of EGFR Gene Mutations and EGF Receptor Tyrosine Kinase Inhibitors. Future Oncol. 2015, 11, 2329–2342. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell. Physiol. Biochem. 2017. [Google Scholar] [CrossRef]
- Dirkx, A.E.M.; Oude Egbrink, M.G.A.; Kuijpers, M.J.E.; Van der Niet, S.T.; Heijnen, V.V.T.; Bouma-ter Steege, J.C.A.; Wagstaff, J.; Griffioen, A.W. Tumor Angiogenesis Modulates Leukocyte-Vessel Wall Interactions in Vivo by Reducing Endothelial Adhesion Molecule Expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of Tumor Associated Macrophages in Tumor Angiogenesis and Lymphangiogenesis. Front. Physiol. 2014. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages Regulate the Angiogenic Switch in a Mouse Model of Breast Cancer. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Trial | Phase | Treatment Arms | Line of Treatment | Number of Patients Mutated Versus Wild Type | Results Mutated Versus Wild Type (95% C.I. Range) | Reference |
|---|---|---|---|---|---|---|
| CheckMate 057 | III | Docetaxel vs. Nivolumab | ≥2 | 82 versus 340 | HR OS 1.18 (0.69–2.00) HR PFS 1.45 (0.90–2.37) versus HR OS 0.66 (0.51–0.86) HR PFS 0.83 (0.65–1.06) | [15] |
| KEYNOTE-010 | II-III | Docetaxel vs. Pembrolizumab | ≥2 | 86 versus 875 | HR OS 0.88 (0.45–1.70) HR PFS 1.79 (0.94–3.42) versus HR OS 0.66 (0.55–0.80) HR PFS 0.83 (0.73–0.98) | [16] |
| POPLAR | II | Docetaxel vs. Atezolizumab | ≥2 | 19 versus 147 | HR OS 0.99 (0.29–3.40) HR PFS N/A versus HR OS 0.73 (0.53–0.99) HR PFS N/A | [17] |
| OAK | III | Docetaxel vs. Atezolizumab | ≥2 | 85 versus 628 | HR OS 1.24 (0.71–2.18) HR PFS N/A versus HR OS 0.69 (0.57–0.83) HR PFS N/A | [18] |
| ATLANTIC trial (cohort 1) | II | Durvalumab | ≥3 | 97 | 12% ORR (PD-L1 ≥25%) 4% (PD-L1 <25%) ** | [48] |
| NCT02879994 | II | Pembrolizumab | 1 | 11 * | No Responses ** | [24] |
| TKI Treatment | ICI Treatment | Inclusion Criteria | Number of Patients | ORR | Grade 3–4 TRAEs | Discontinuation Rate Due to TRAEs | Status | Reference |
|---|---|---|---|---|---|---|---|---|
| Erlotinib | Nivolumab | EGFR +, TKI treated, or naive | 21 | 15% | 10% diarrhoea | N/A | Completed | [46] |
| Erlotinib | Atezolizumab | EGFR+, TKI naïve, or previously treated but not with TKI | 28 | 75% | 7% hypertransa-minasemia, 7% rash, 7% fever | N/A | Active, Not Recruiting | [47] |
| Gefitinib | Durvalumab | EGFR+, TKI naive | 20 | N/A * | 55% hepatic | 20% | Active, Not Recruiting | [48] |
| Osimertinib | Durvalumab | EGFR +, TKI treated, or naive | 34 | N/A * | 15% ILD | 59% | Active, Not Recruiting | [20] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santaniello, A.; Napolitano, F.; Servetto, A.; De Placido, P.; Silvestris, N.; Bianco, C.; Formisano, L.; Bianco, R. Tumour Microenvironment and Immune Evasion in EGFR Addicted NSCLC: Hurdles and Possibilities. Cancers 2019, 11, 1419. https://doi.org/10.3390/cancers11101419
Santaniello A, Napolitano F, Servetto A, De Placido P, Silvestris N, Bianco C, Formisano L, Bianco R. Tumour Microenvironment and Immune Evasion in EGFR Addicted NSCLC: Hurdles and Possibilities. Cancers. 2019; 11(10):1419. https://doi.org/10.3390/cancers11101419
Chicago/Turabian StyleSantaniello, Antonio, Fabiana Napolitano, Alberto Servetto, Pietro De Placido, Nicola Silvestris, Cataldo Bianco, Luigi Formisano, and Roberto Bianco. 2019. "Tumour Microenvironment and Immune Evasion in EGFR Addicted NSCLC: Hurdles and Possibilities" Cancers 11, no. 10: 1419. https://doi.org/10.3390/cancers11101419
APA StyleSantaniello, A., Napolitano, F., Servetto, A., De Placido, P., Silvestris, N., Bianco, C., Formisano, L., & Bianco, R. (2019). Tumour Microenvironment and Immune Evasion in EGFR Addicted NSCLC: Hurdles and Possibilities. Cancers, 11(10), 1419. https://doi.org/10.3390/cancers11101419