Astrogliosis Releases Pro-Oncogenic Chitinase 3-Like 1 Causing MAPK Signaling in Glioblastoma

 ,
,
Abstract
:1. Introduction
2. Results
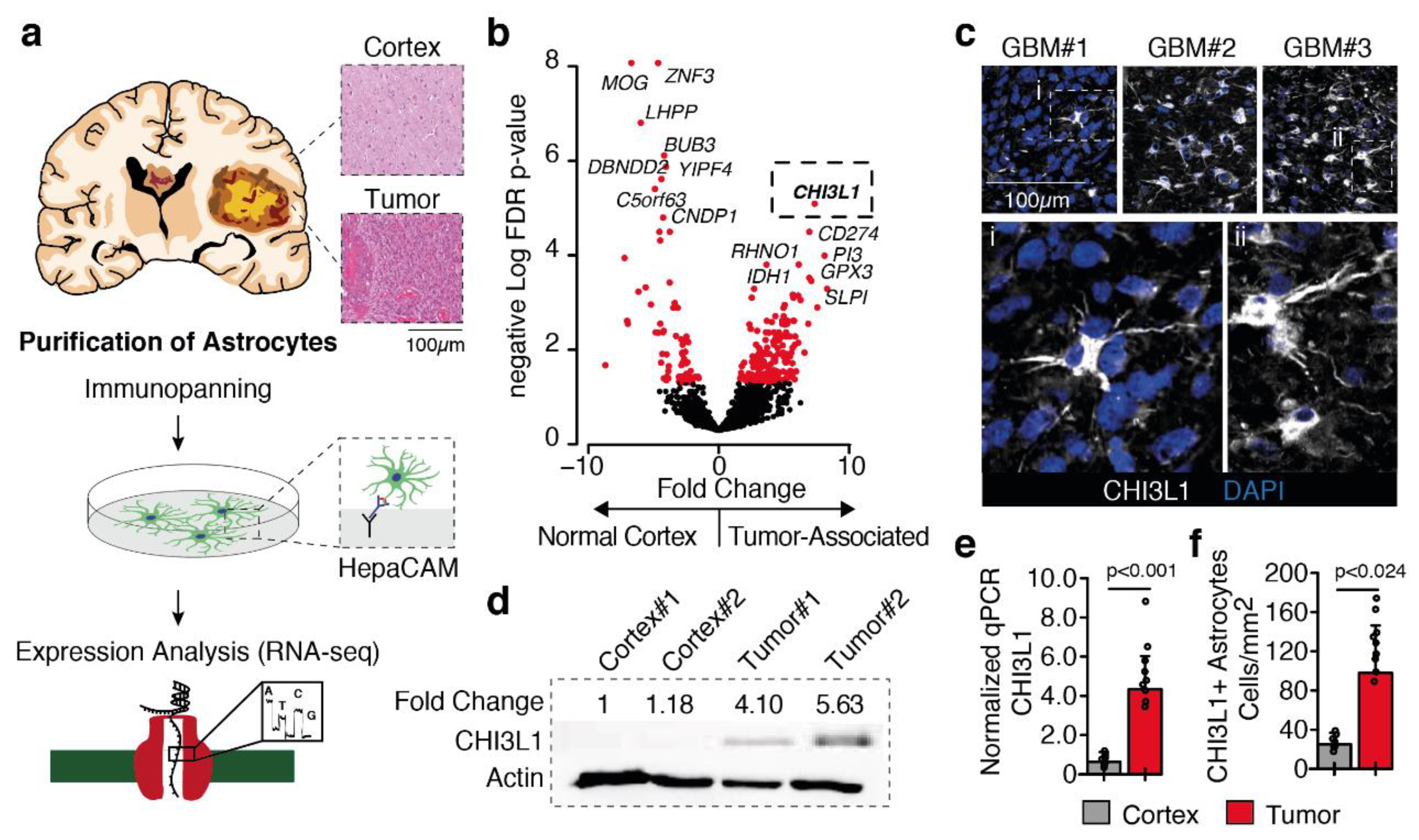
2.1. Tumor-Astrocyte Marked by Chitinase 3-Like 1 Expression
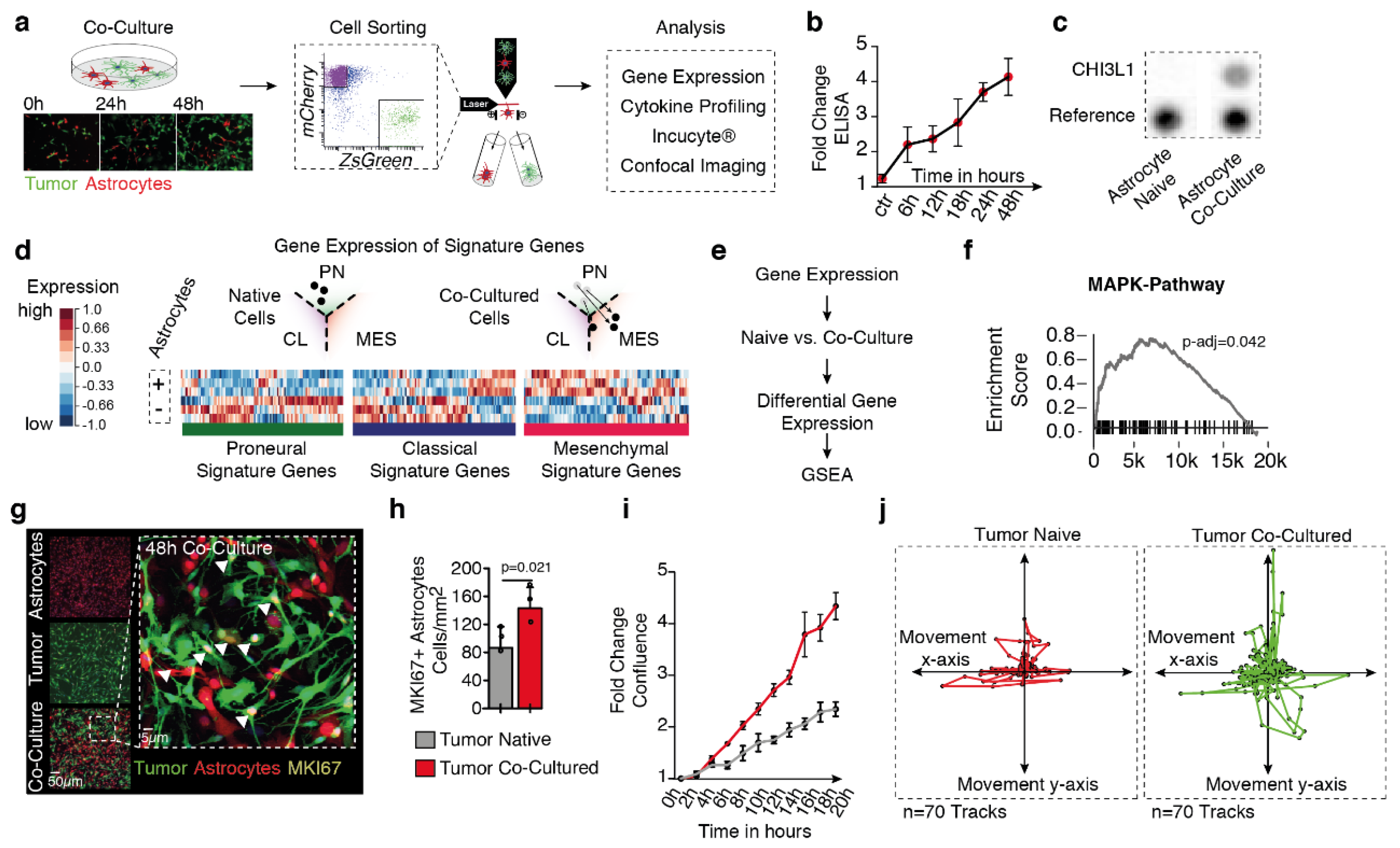
2.2. Tumor-Astrocyte Co-Culture Model Confirmed CHI3L1 Release
2.3. Astrocytic Co-Culture Drive MAPK Pathway Activity and Proliferation in Glioblastoma
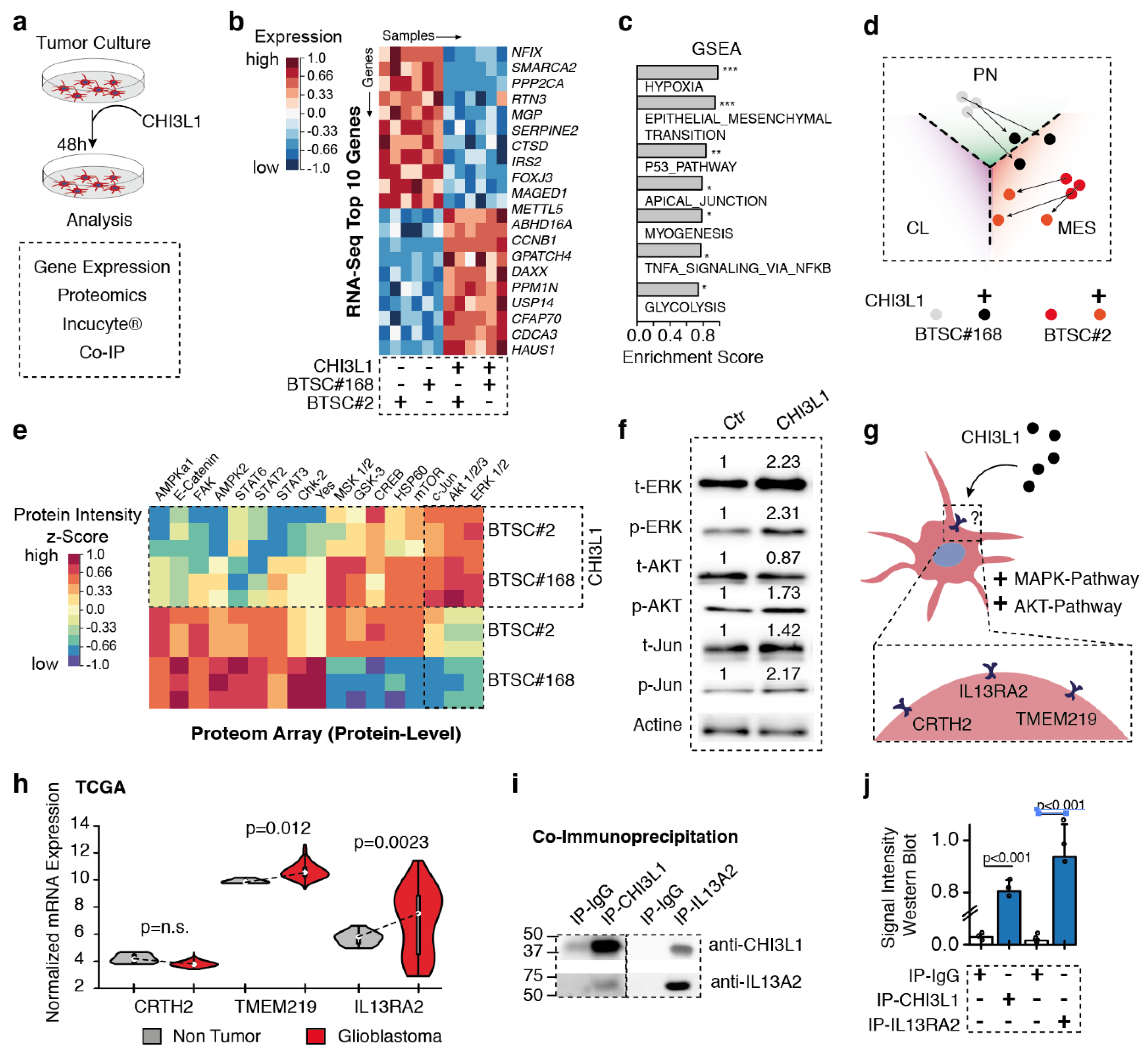
2.4. CHI3L1 Treatment Causes AKT and MAPK Activity and Mesenchymal Gene Expression
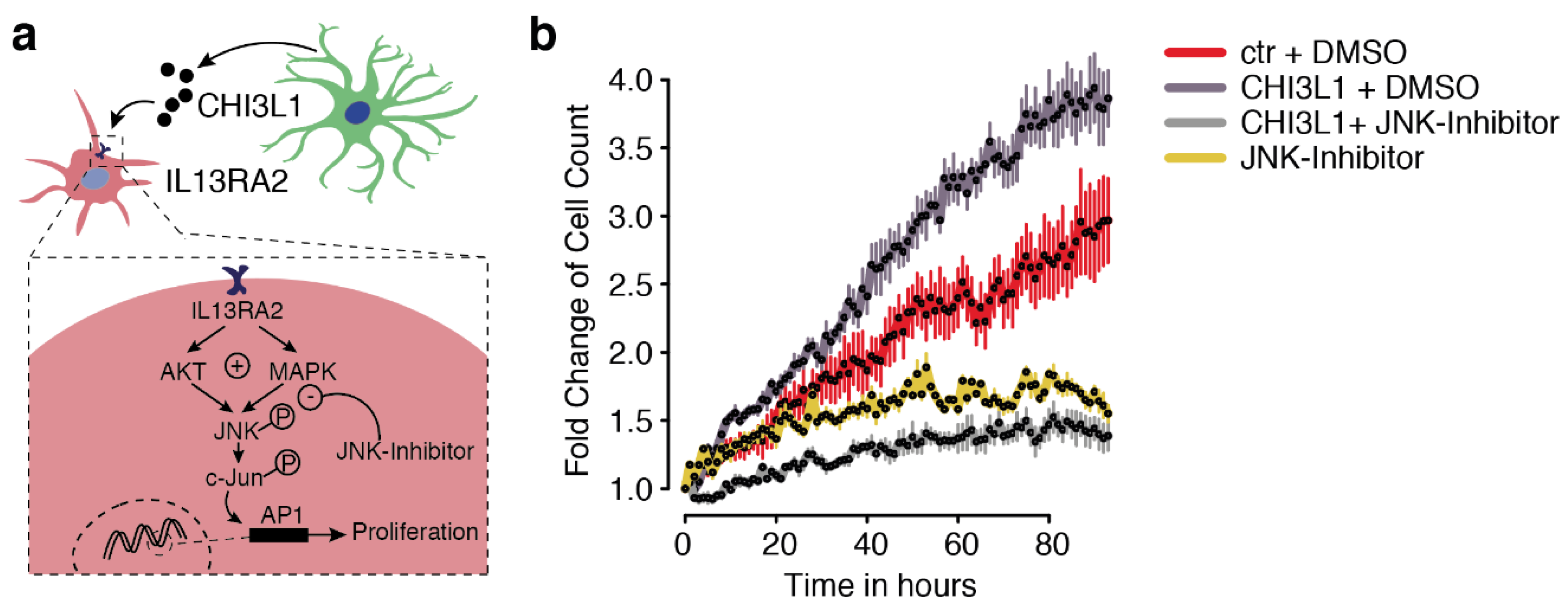
2.5. Interleukin 13Ra2 Is the Binding Partner of CHI3L1
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
4.2. Cell Culture and Co-Culture Model
4.3. Viral Transduction by Constitutive Reporter Lentiviral Vectors
4.4. Cell Purification by Immunopanning
4.5. Flow Cytometry and Cell Sorting
4.6. Secretome Profiling
4.7. Proteome Profiling
4.8. Immunohistochemistry
4.9. Quantitative Real-Time PCR
4.10. Immunoblotting
4.11. Co-Immunoprecipitation
4.12. Proliferation Measurement by IncuCyte©
4.13. RNA Sequencing
4.14. Microarray Gene Expression
4.15. Expression Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verkhratsky, A.; Parpura, V. Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol. Dis. 2016, 85, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón, Y.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tian, E.; Chen, X.; Chao, J.; Klein, J.; Qu, Q.; Sun, G.; Sun, G.; Huang, Y.; Warden, C.D.; et al. GFAP Mutations in Astrocytes Impair Oligodendrocyte Progenitor Proliferation and Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell 2018, 23, 239–251.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recklies, A.D.; Ling, H.; White, C.; Bernier, S.M. Inflammatory cytokines induce production of CHI3L1 by articular chondrocytes. J. Biol. Chem. 2005, 280, 41213–41221. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-M.; He, C.H.; Nour, A.M.; Zhou, Y.; Ma, B.; Park, J.W.; Kim, K.H.; Dela Cruz, C.; Sharma, L.; Nasr, M.L.; et al. IL-13Rα2 uses TMEM219 in chitinase 3-like-1-induced signalling and effector responses. Nat. Commun. 2016, 7, 12752. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Lee, C.G.; Dela Cruz, C.S.; Lee, C.-M.; Zhou, Y.; Ahangari, F.; Ma, B.; Herzog, E.L.; Rosenberg, S.A.; Li, Y.; et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor α2. Cell Rep. 2013, 4, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Lee, Y.K.; Ryu, J.; Jeong, J.Y.; Choi, J.; Eun, K.M.; Shin, H.Y.; Kim, D.G.; Hwang, E.M.; Yoo, J.C.; et al. CHI3L1 (YKL-40) is expressed in human gliomas and regulates the invasion, growth and survival of glioma cells. Int. J. Cancer 2011, 128, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Libreros, S.; Garcia-Areas, R.; Iragavarapu-Charyulu, V. CHI3L1 plays a role in cancer through enhanced production of pro-inflammatory/pro-tumorigenic and angiogenic factors. Immunol. Res. 2013, 57, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deluche, E.; Bessette, B.; Durand, S.; Caire, F.; Rigau, V.; Robert, S.; Chaunavel, A.; Forestier, L.; Labrousse, F.; Jauberteau, M.-O.; et al. CHI3L1, NTRK2, 1p/19q and IDH status predicts prognosis in glioma. Cancers (Basel) 2019, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Pelloski, C.E.; Mahajan, A.; Maor, M.; Chang, E.L.; Woo, S.; Gilbert, M.; Colman, H.; Yang, H.; Ledoux, A.; Blair, H.; et al. YKL-40 expression is associated with poorer response to radiation and shorter overall survival in glioblastoma. Clin. Cancer Res. 2005, 11, 3326–3334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Heiland, D.H.; Gaebelein, A.; Börries, M.; Wörner, J.; Pompe, N.; Franco, P.; Heynckes, S.; Bartholomae, M.; hAilín, D.Ó.; Carro, M.S.; et al. Microenvironment-Derived Regulation of HIF Signaling Drives Transcriptional Heterogeneity in Glioblastoma Multiforme. Mol. Cancer Res. 2018, 16, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Kling, T.; Ferrarese, R.; Ó hAilín, D.; Johansson, P.; Heiland, D.H.; Dai, F.; Vasilikos, I.; Weyerbrock, A.; Jörnsten, R.; Carro, M.S.; et al. Integrative Modeling Reveals Annexin A2-mediated Epigenetic Control of Mesenchymal Glioblastoma. EBioMedicine 2016, 12, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Wasilewski, D.; Priego, N.; Fustero-Torre, C.; Valiente, M. Reactive astrocytes in brain metastasis. Front. Oncol. 2017, 7, 298. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, S.; Deora, A.B.; Annes, J.P.; Osoria, J.; Rifkin, D.B.; Hajjar, K.A. Annexin II-mediated plasmin generation activates TGF-beta3 during epithelial-mesenchymal transformation in the developing avian heart. Dev. Biol. 2004, 265, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide Gene/Primer | Sequence | Source |
|---|---|---|
| TIMP1 forward | CTTCTGCAATTCCGACCTCGT | Thermo Fisher Scientific, Carlsbad, CA, USA |
| TIMP1 reverse | ACGCTGGTATAAGGTGGTCTG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| VIM forward | AGTCCACTGAGTACCGGAGAC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| VIM reverse | CATTTCACGCATCTGGCGTTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| STEAP4 forward | GAGAGTTCCGATTTGTCCAGTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| STEAP4 reverse | ATCTCTTCCCACCGTACACCA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CXCL10 forward | GTGGCATTCAAGGAGTACCTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CXCL10 reverse | TGATGGCCTTCGATTCTGGATT | Thermo Fisher Scientific, Carlsbad, CA, USA |
| TGM1 forward | GCACCACACAGACGAGTATGA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| TGM1 reverse | GGTGATGCGATCAGAGGATTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| SLC10A6 forward | GGAAGCTGTGGTCGCACAT | Thermo Fisher Scientific, Carlsbad, CA, USA |
| SLC10A6 reverse | GTAAAAGGCATGAGCCCAAACT | Thermo Fisher Scientific, Carlsbad, CA, USA |
| S100A10 forward | GGCTACTTAACAAAGGAGGACC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| S100A10 reverse | GAGGCCCGCAATTAGGGAAA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| EMP1 forward | GTGCTGGCTGTGCATTCTTG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| EMP1 reverse | CCGTGGTGATACTGCGTTCC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CD14 forward | ACGCCAGAACCTTGTGAGC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CD14 reverse | GCATGGATCTCCACCTCTACTG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CD109 forward | AAGCCAGTGAAAGGAGACGTA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CD109 reverse | CCAGGGGAAGATAGATCCAGG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| SRGN forward | AGGTTATCCTACGCGGAGAG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| SRGN reverse | GTCTTTGGAAAAAGGTCAGTCCT | Thermo Fisher Scientific, Carlsbad, CA, USA |
| GBP2 forward | CATCCGAAAGTTCTTCCCCAA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| GBP2 reverse | CTCTAGGTGAGCAAGGTACTTCT | Thermo Fisher Scientific, Carlsbad, CA, USA |
| AMIGO2 forward | CCTGGGAACCTTTTCAGACTG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| AMIGO2 reverse | GCAAACGATACTGGAATCCACT | Thermo Fisher Scientific, Carlsbad, CA, USA |
| PSMB8 forward | CACGCTCGCCTTCAAGTTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| PSMB8 reverse | AGGCACTAATGTAGGACCCAG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| FBLN5 forward | CTACTCGAACCCCTACTCGAC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| FBLN5 reverse | TCGTGGGATAGTTTGGAGCTG | Thermo Fisher Scientific, Carlsbad, CA, USA |
| TAB1 forward | AACTGCTTCCTGTATGGGGTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| TAB1 reverse | AAGGCGTCGTCAATGGACTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CD44 forward | CCACCCTAATCAAGGAAATGA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CD44 reverse | TGAAATCCAGGTGTTGGGATA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| GFAP forward | CTG CGG CTC GAT CAA CTC A | Thermo Fisher Scientific, Carlsbad, CA, USA |
| GFAP reverse | TCCAGCGACTCAATCTTCCTC | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CHI3L1 forward | CCA CCC TAA TCA AGG AAA TGA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| CHI3L1 reverse | TGA AAT CCA GGT GTT GGG ATA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| 18S forward | TTT GCG AGT ACT CAA CAC CA | Thermo Fisher Scientific, Carlsbad, CA, USA |
| 18S reverse | CCA CAC CCC TTA ATG GCA | Thermo Fisher Scientific, Carlsbad, CA, USA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wurm, J.; Behringer, S.P.; Ravi, V.M.; Joseph, K.; Neidert, N.; Maier, J.P.; Doria-Medina, R.; Follo, M.; Delev, D.; Pfeifer, D.; et al. Astrogliosis Releases Pro-Oncogenic Chitinase 3-Like 1 Causing MAPK Signaling in Glioblastoma. Cancers 2019, 11, 1437. https://doi.org/10.3390/cancers11101437
Wurm J, Behringer SP, Ravi VM, Joseph K, Neidert N, Maier JP, Doria-Medina R, Follo M, Delev D, Pfeifer D, et al. Astrogliosis Releases Pro-Oncogenic Chitinase 3-Like 1 Causing MAPK Signaling in Glioblastoma. Cancers. 2019; 11(10):1437. https://doi.org/10.3390/cancers11101437
Chicago/Turabian StyleWurm, Julian, Simon P. Behringer, Vidhya M. Ravi, Kevin Joseph, Nicolas Neidert, Julian P. Maier, Roberto Doria-Medina, Marie Follo, Daniel Delev, Dietmar Pfeifer, and et al. 2019. "Astrogliosis Releases Pro-Oncogenic Chitinase 3-Like 1 Causing MAPK Signaling in Glioblastoma" Cancers 11, no. 10: 1437. https://doi.org/10.3390/cancers11101437