An Integrated Bioinformatics Analysis Repurposes an Antihelminthic Drug Niclosamide for Treating HMGA2-Overexpressing Human Colorectal Cancer


Abstract
:1. Introduction
2. Results
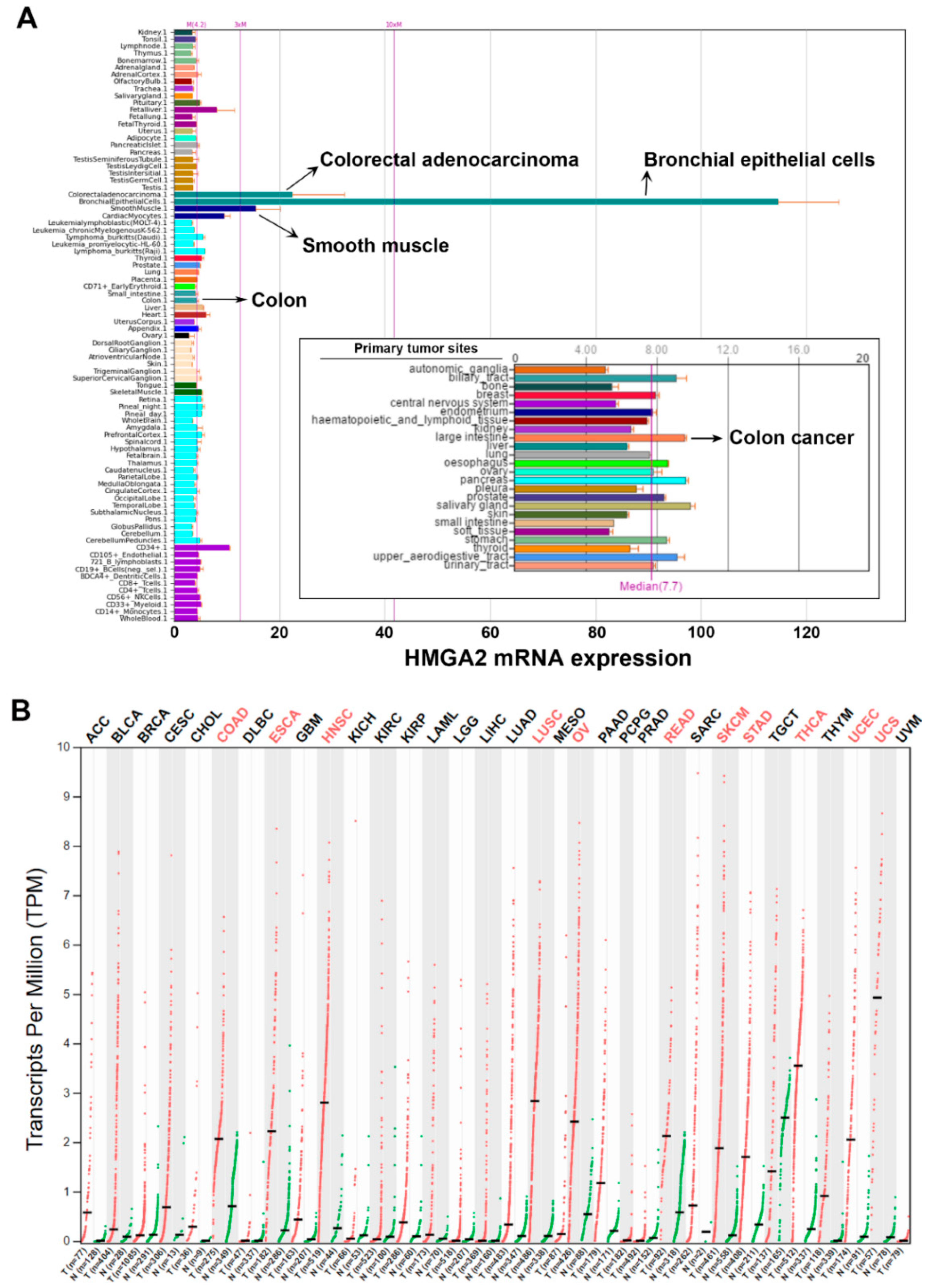
2.1. Pan-Cancer Analyses of HMGA2 Gene Expression
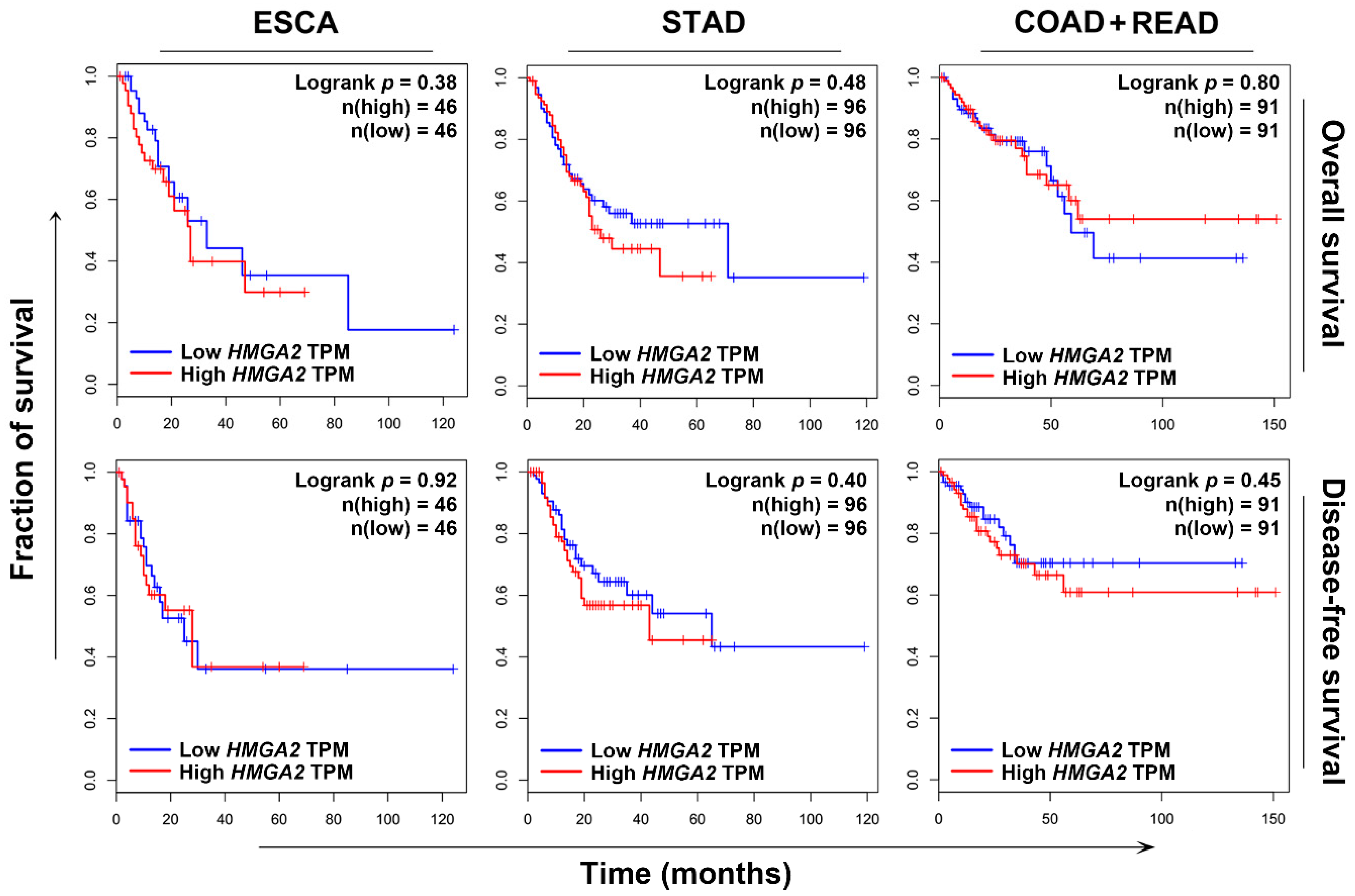
2.2. HMGA2 Is Not a Suitable Prognosis Marker and Therapeutic Target
2.3. S100A4 Is a Potential Surrogate Therapeutic Target for HMGA2-Overexpressing Colorectal Cancer
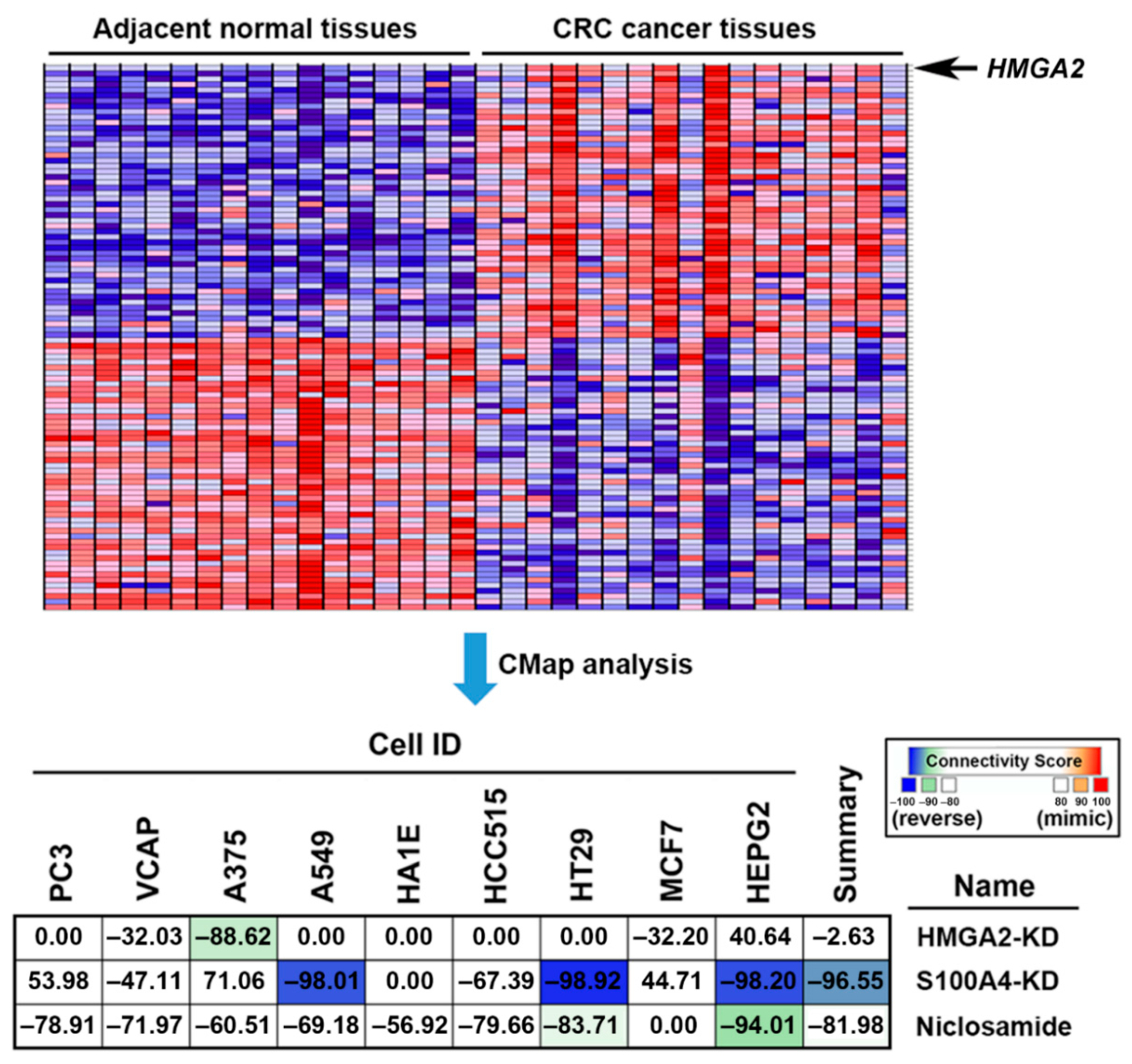
2.4. Connectivity Map (CMap) Analysis Identified that S100A4 Inhibition Reverses the HMGA2-Driven Gene Signature
2.5. Connectivity Map (CMap) Analysis Identified that S100A4 Inhibition by Niclosamide is Clinically Relevant in Colorectal Cancer
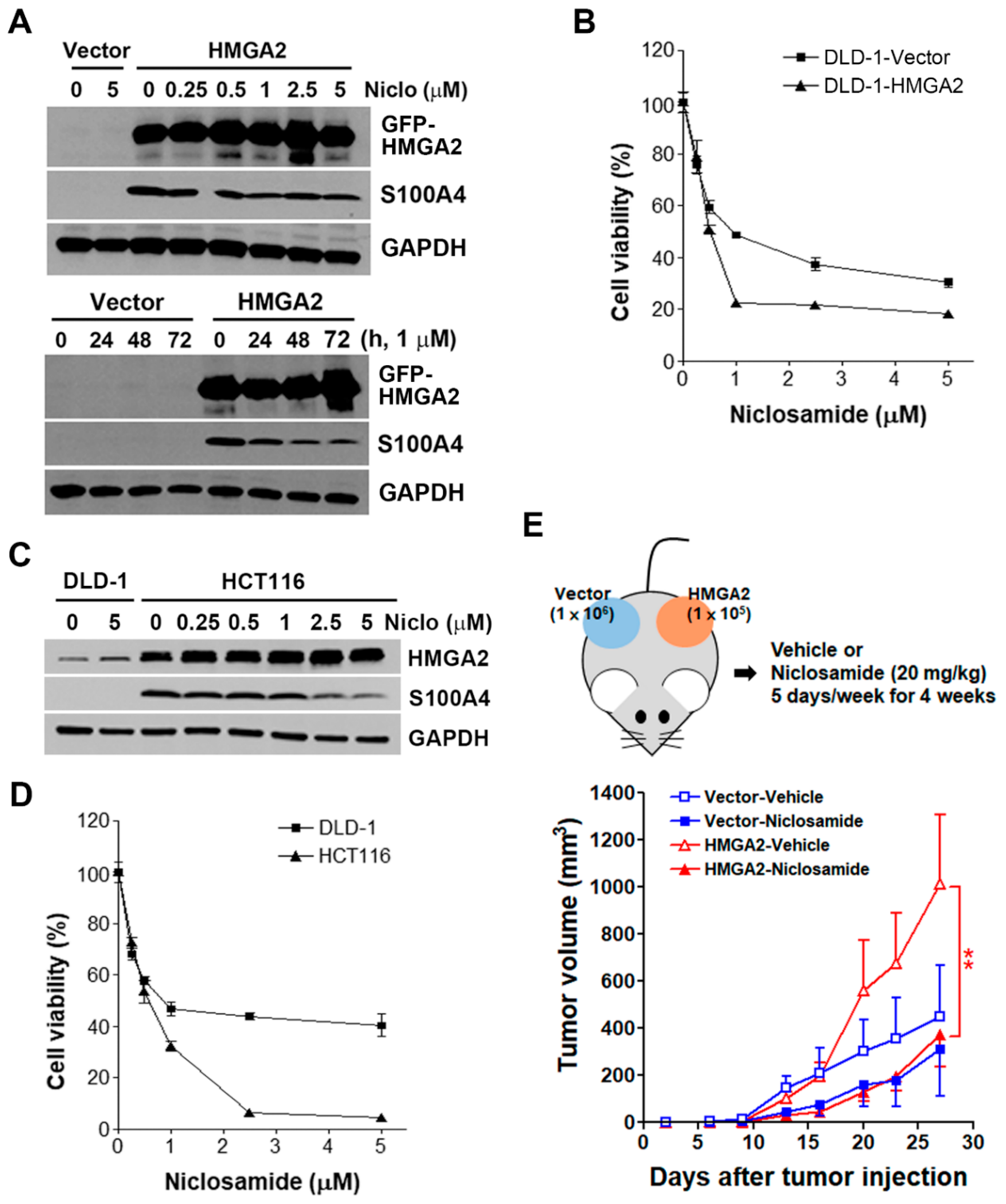
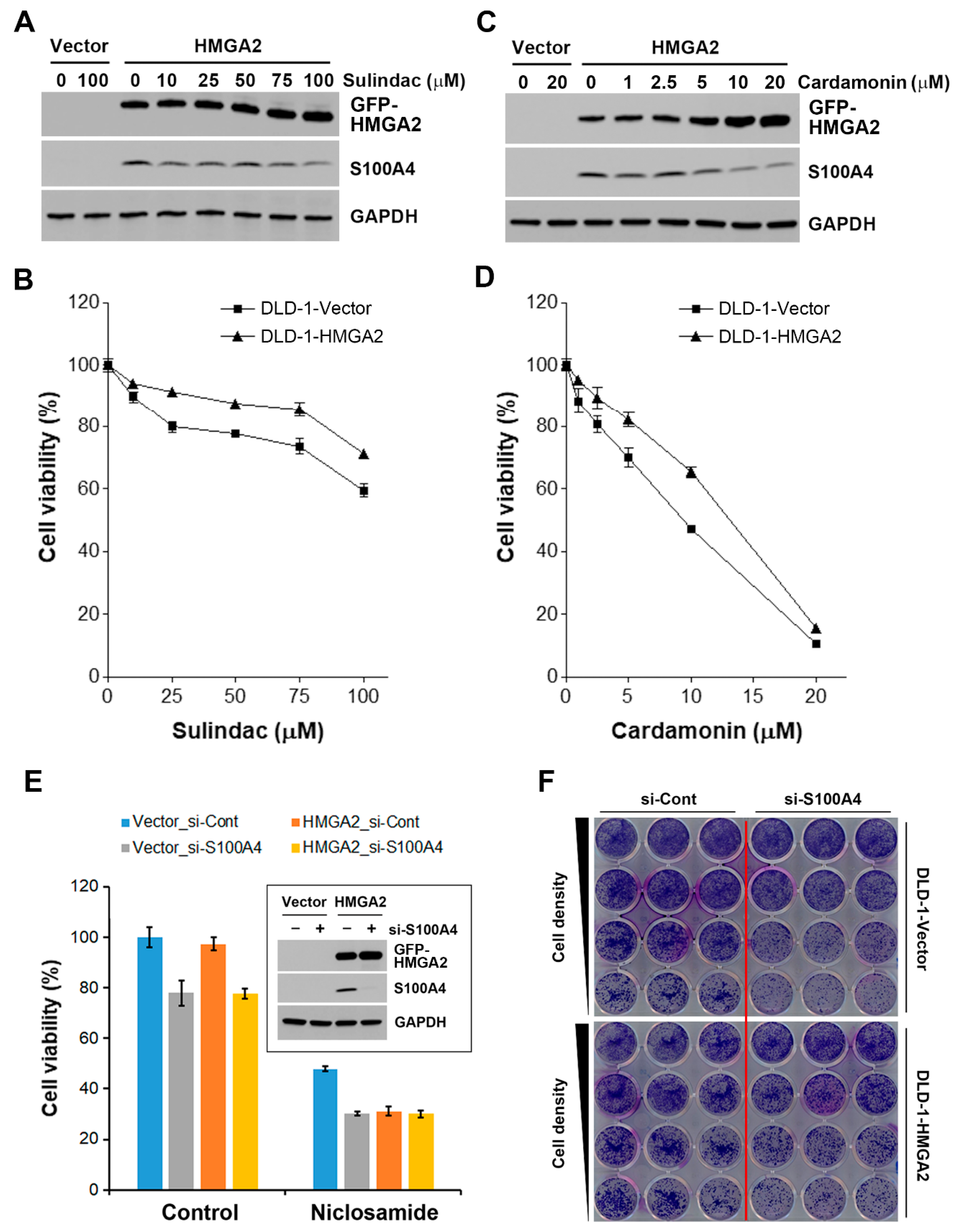
2.6. HMGA2-Overexpressing Colorectal Cancer Cells Are More Susceptible to Niclosamide
2.7. Inhibition of S100A4 Is Not Sufficient to Selectively Kill HMGA2-Overexpressing Colorectal Cancer Cells
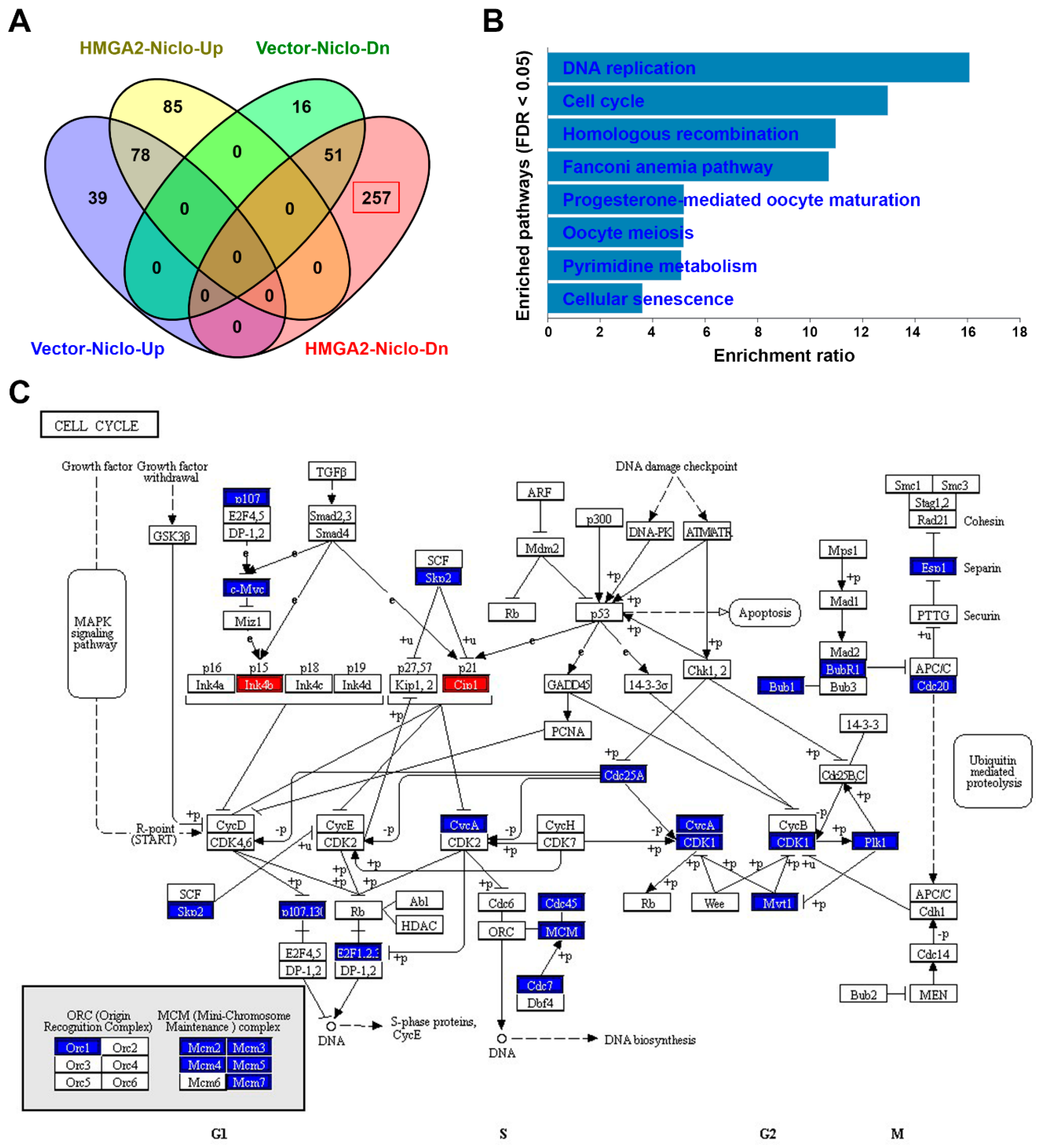
2.8. RNA Sequencing Identifies that Niclosamide Inhibits Cell Cycle-Related Genes in HMGA2-Overexpressing Colorectal Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analysis of Public Data
4.2. Microarray
4.3. Gene Set Enrichment Analysis (GSEA) and Connectivity Map (CMap)
4.4. RNA Sequencing and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
4.5. Materials
4.6. Cell Culture and Transfection
4.7. Determination of Cell Proliferation and Cell Viability
4.8. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
4.9. Western Blot Analysis and Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Mice Tumor Xenograft Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ozturk, N.; Singh, I.; Mehta, A.; Braun, T.; Barreto, G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev. Biol. 2014, 2, e5. [Google Scholar] [CrossRef] [PubMed]
- Pfannkuche, K.; Summer, H.; Li, O.; Hescheler, J.; Droge, P. The high mobility group protein HMGA2: A co-regulator of chromatin structure and pluripotency in stem cells? Stem Cell Rev. 2009, 5, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Battista, S.; Kenyon, L.; Baldassarre, G.; Fidanza, V.; Klein-Szanto, A.J.; Parlow, A.F.; Visone, R.; Pierantoni, G.M.; Outwater, E.; et al. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene 2002, 21, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, M.; Fusco, A. HMGA and cancer. BBA-Gene Regul. Mech. 2010, 1799, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Shen, G.; Liu, S.; Meng, Q. Downregulation of HMGA2 inhibits cellular proliferation and invasion, improves cellular apoptosis in prostate cancer. Tumour Biol. 2016, 37, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Di Cello, F.; Hillion, J.; Hristov, A.; Wood, L.J.; Mukherjee, M.; Schuldenfrei, A.; Kowalski, J.; Bhattacharya, R.; Ashfaq, R.; Resar, L.M. HMGA2 participates in transformation in human lung cancer. Mol. Cancer Res. 2008, 6, 743–750. [Google Scholar] [CrossRef]
- Malek, A.; Bakhidze, E.; Noske, A.; Sers, C.; Aigner, A.; Schafer, R.; Tchernitsa, O. HMGA2 gene is a promising target for ovarian cancer silencing therapy. Int. J. Cancer 2008, 123, 348–356. [Google Scholar] [CrossRef]
- Morishita, A.; Zaidi, M.R.; Mitoro, A.; Sankarasharma, D.; Szabolcs, M.; Okada, Y.; D’Armiento, J.; Chada, K. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013, 73, 4289–4299. [Google Scholar] [CrossRef]
- Zhang, K.; Gao, H.; Wu, X.; Wang, J.; Zhou, W.; Sun, G.; Wang, J.; Wang, Y.; Mu, B.; Kim, C.; et al. Frequent overexpression of HMGA2 in human atypical teratoid/rhabdoid tumor and its correlation with let-7a3/let-7b miRNA. Clin. Cancer Res. 2014, 20, 1179–1189. [Google Scholar] [CrossRef]
- Lee, C.T.; Wu, T.T.; Lohse, C.M.; Zhang, L. High-mobility group AT-hook 2: An independent marker of poor prognosis in intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 2334–2340. [Google Scholar] [CrossRef]
- Motoyama, K.; Inoue, H.; Nakamura, Y.; Uetake, H.; Sugihara, K.; Mori, M. Clinical significance of high mobility group A2 in human gastric cancer and its relationship to let-7 microRNA family. Clin. Cancer Res. 2008, 14, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, X.; Zhang, W.; Wei, Y.; Li, Y.; Zhang, Q.; Dong, R.; Kwon, J.S.; Liu, Z.; Zheng, W.; et al. Overexpression and oncogenic function of HMGA2 in endometrial serous carcinogenesis. Am. J. Cancer Res. 2016, 6, 249–259. [Google Scholar] [PubMed]
- Zhang, S.; Mo, Q.; Wang, X. Oncological role of HMGA2 (Review). Int. J. Oncol. 2019, 55, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Weinbach, E.C.; Garbus, J. Mechanism of action of reagents that uncouple oxidative phosphorylation. Nature 1969, 221, 1016–1018. [Google Scholar] [CrossRef]
- Li, Y.; Li, P.K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Kobelt, D.; Lemm, M.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. Novel effect of antihelminthic niclosamide on S100A4-mediated metastatic progression in colon cancer. J. Natl. Cancer Inst. 2011, 103, 1018–1036. [Google Scholar] [CrossRef]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The NIKOLO trial. BMC Cancer 2018, 18, e297. [Google Scholar] [CrossRef]
- Wu, C.; Jin, X.; Tsueng, G.; Afrasiabi, C.; Su, A.I. BioGPS: Building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2016, 44, 313–316. [Google Scholar] [CrossRef]
- Wu, C.; Macleod, I.; Su, A.I. BioGPS and MyGene.info: Organizing online, gene-centric information. Nucleic Acids Res. 2013, 41, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W.; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, e130. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Alonso, N.; Guillen, R.; Chambers, J.W.; Leng, F. A rapid and sensitive high-throughput screening method to identify compounds targeting protein-nucleic acids interactions. Nucleic Acids Res. 2015, 43, e52. [Google Scholar] [CrossRef]
- Miao, Y.; Cui, T.; Leng, F.; Wilson, W.D. Inhibition of high-mobility-group A2 protein binding to DNA by netropsin: A biosensor-surface plasmon resonance assay. Anal. Biochem. 2008, 374, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, M.; Kobelt, D.; Walther, W.; Mudduluru, G.; Stein, U. S100A4 in cancer metastasis: Wnt signaling-driven interventions for metastasis restriction. Cancers (Basel) 2016, 8, e59. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kruger, A. Molecular targets and pathways involved in liver metastasis of colorectal cancer. Clin. Exp. Metastasis 2015, 32, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Qu, J.; Zhang, M.; Li, Y.; Zhang, S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget 2017, 8, 73219–73239. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J. The connectivity map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452, e1417. [Google Scholar] [CrossRef]
- Liu, L.; Qi, L.; Knifley, T.; Piecoro, D.W.; Rychahou, P.; Liu, J.; Mitov, M.I.; Martin, J.; Wang, C.; Wu, J.; et al. S100A4 alters metabolism and promotes invasion of lung cancer cells by up-regulating mitochondrial complex I protein NDUFS2. J. Biol. Chem. 2019, 294, 7516–7527. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, 991–995. [Google Scholar] [CrossRef]
- Grigorian, M.; Tulchinsky, E.; Burrone, O.; Tarabykina, S.; Georgiev, G.; Lukanidin, E. Modulation of mts1 expression in mouse and human normal and tumor cells. Electrophoresis 1994, 15, 463–468. [Google Scholar] [CrossRef]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Aumann, J.; Lemos, C.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. S100A4-induced cell motility and metastasis is restricted by the Wnt/beta-catenin pathway inhibitor calcimycin in colon cancer cells. Mol. Biol. Cell 2011, 22, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Arlt, F.; Smith, J.; Sack, U.; Herrmann, P.; Walther, W.; Lemm, M.; Fichtner, I.; Shoemaker, R.H.; Schlag, P.M. Intervening in beta-catenin signaling by sulindac inhibits S100A4-dependent colon cancer metastasis. Neoplasia 2011, 13, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Garrett, S.C.; Hodgson, L.; Rybin, A.; Toutchkine, A.; Hahn, K.M.; Lawrence, D.S.; Bresnick, A.R. A biosensor of S100A4 metastasis factor activation: Inhibitor screening and cellular activation dynamics. Biochemistry (Mosc.) 2008, 47, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Malashkevich, V.N.; Dulyaninova, N.G.; Ramagopal, U.A.; Liriano, M.A.; Varney, K.M.; Knight, D.; Brenowitz, M.; Weber, D.J.; Almo, S.C.; Bresnick, A.R. Phenothiazines inhibit S100A4 function by inducing protein oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 8605–8610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulyaninova, N.G.; Hite, K.M.; Zencheck, W.D.; Scudiero, D.A.; Almo, S.C.; Shoemaker, R.H.; Bresnick, A.R. Cysteine 81 is critical for the interaction of S100A4 and myosin-IIA. Biochemistry (Mosc.) 2011, 50, 7218–7227. [Google Scholar] [CrossRef] [PubMed]
- Khamas, A.; Ishikawa, T.; Shimokawa, K.; Mogushi, K.; Iida, S.; Ishiguro, M.; Mizushima, H.; Tanaka, H.; Uetake, H.; Sugihara, K. Screening for epigenetically masked genes in colorectal cancer Using 5-Aza-2’-deoxycytidine, microarray and gene expression profile. Cancer Genom. Proteom. 2012, 9, 67–75. [Google Scholar]
- Kadri, H.; Lambourne, O.A.; Mehellou, Y. Niclosamide, a Drug with many (Re)purposes. Chemmedchem 2018, 13, 1088–1091. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, 353–361. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, 590–595. [Google Scholar] [CrossRef]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based gene set analysis toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Mito, J.K.; Agoston, A.T.; Dal Cin, P.; Srivastava, A. Prevalence and significance of HMGA2 expression in oesophageal adenocarcinoma. Histopathology 2017, 71, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Zhang, L.; Guo, Q.; Mao, X. High mobility group protein A2 overexpression indicates poor prognosis for cancer patients: A meta-analysis. Oncotarget 2018, 9, 1237–1247. [Google Scholar] [CrossRef]
- Pallante, P.; Sepe, R.; Puca, F.; Fusco, A. High mobility group a proteins as tumor markers. Front Med. (Lausanne) 2015, 2, e15. [Google Scholar] [CrossRef]
- Palumbo, A., Jr.; Da Costa, N.M.; Esposito, F.; De Martino, M.; D’Angelo, D.; de Sousa, V.P.; Martins, I.; Nasciutti, L.E.; Fusco, A.; Ribeiro Pinto, L.F. HMGA2 overexpression plays a critical role in the progression of esophageal squamous carcinoma. Oncotarget 2016, 7, 25872–25884. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Li, A.Y.; Chen, L.; Lai, L.; Lin, H.H.; Hu, S.; Yao, L.; Peng, J.; Loera, S.; et al. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clin. Cancer Res. 2011, 17, 2570–2580. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Xu, S.; Hao, Y. Clinicopathological and prognostic significance of HMGA2 overexpression in gastric cancer: A meta-analysis. Oncotarget 2017, 8, 100478–100489. [Google Scholar] [CrossRef]
- Huang, B.; Yang, J.; Cheng, Q.; Xu, P.; Wang, J.; Zhang, Z.; Fan, W.; Wang, P.; Yu, M. Prognostic value of HMGA2 in human cancers: A meta-analysis based on literatures and TCGA datasets. Front Physiol. 2018, 9, e776. [Google Scholar] [CrossRef]
- Esmailzadeh, S.; Mansoori, B.; Mohammadi, A.; Shanehbandi, D.; Baradaran, B. SiRNA-mediated silencing of HMGA2 induces apoptosis and cell cycle arrest in human colorectal carcinoma. J. Gastrointest. Cancer 2017, 48, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, Y.; Deng, H.; Liu, C.; Wu, J.; Lai, M. HMGA2 enhances 5-fluorouracil chemoresistance in colorectal cancer via the Dvl2/Wnt pathway. Oncotarget 2018, 9, 9963–9974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Liang, Y.; Shen, L.; Shen, L. MicroRNA-204 modulates colorectal cancer cell sensitivity in response to 5-fluorouracil-based treatment by targeting high mobility group protein A2. Biol. Open 2016, 5, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.Y.; Tu, Y.; Deng, Y.; Guo, C.; Ning, J.; Zhu, Y.; Lv, X.; Ye, H. MiR-4500 is epigenetically downregulated in colorectal cancer and functions as a novel tumor suppressor by regulating HMGA2. Cancer Biol. Ther. 2016, 17, 1149–1157. [Google Scholar] [CrossRef] [Green Version]
- Kao, C.Y.; Yang, P.M.; Wu, M.H.; Huang, C.C.; Lee, Y.C.; Lee, K.H. Heat shock protein 90 is involved in the regulation of HMGA2-driven growth and epithelial-to-mesenchymal transition of colorectal cancer cells. PeerJ 2016, 4, e1683. [Google Scholar] [CrossRef]
- Fong, W.; To, K.K.W. Drug repurposing to overcome resistance to various therapies for colorectal cancer. Cell Mol. Life Sci. 2019, 76, 3383–3406. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Haugk, K.; McKiernan, J.S.; Gulati, R.; Cheng, H.H.; Maes, J.L.; Dumpit, R.F.; Nelson, P.S.; Montgomery, B.; McCune, J.S.; et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE 2018, 13, e0198389. [Google Scholar] [CrossRef]
- Allison, D.B.; Cui, X.; Page, G.P.; Sabripour, M. Microarray data analysis: From disarray to consolidation and consensus. Nat. Rev. Genet. 2006, 7, 55–65. [Google Scholar] [CrossRef]
- Yang, C.; Wei, H. Designing microarray and RNA-Seq experiments for greater systems biology discovery in modern plant genomics. Mol. Plant 2015, 8, 196–206. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierlinski, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.D.; Wu, Z.; Irizarry, R.A.; Leek, J.T. Sequencing technology does not eliminate biological variability. Nat. Biotechnol. 2011, 29, 572–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Description | Fold Change (Log2) |
|---|---|---|
| CASQ1 | calsequestrin 1 (fast-twitch, skeletal muscle) | 5.20 |
| QPRT | quinolinate phosphoribosyltransferase | 4.14 |
| S100A4 | S100 calcium binding protein A4 | 3.33 |
| MYLK | myosin light chain kinase | 3.21 |
| BDNF | brain-derived neurotrophic factor | 3.08 |
| PRKCQ | protein kinase C, theta | 2.86 |
| PCOLCE2 | procollagen C-endopeptidase enhancer 2 | 2.77 |
| PDE2A | phosphodiesterase 2A | 2.70 |
| LCN2 | lipocalin 2 | 2.45 |
| PIK3AP1 | integrin, alpha 1 | 2.32 |
| ZNF711 | zinc finger protein 711 | −5.26 |
| PBDC1 | polysaccharide biosynthesis domain containing 1 | −4.72 |
| NPNT | nephronectin | −4.57 |
| CFTR | cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7) | −4.07 |
| NPTX2 | neuronal pentraxin II | −4.03 |
| KIT | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | −3.99 |
| SH3BGRL | SH3 domain binding glutamic acid-rich protein like | −3.90 |
| METTL7A | methyltransferase like 7A | −3.70 |
| CBWD1 | COBW domain containing 1 | −3.62 |
| CDC27 | cell division cycle 27 | −3.57 |
| Hallmark 1 | Number of Genes in Pathway | Number of Pathway Genes Differentially Expressed (% of total) | NES 2 | p Value | FDR 3 (q Value) |
|---|---|---|---|---|---|
| Epithelial–mesenchymal transition | 197 | 48 (24%) | 1.483 | 0.003 | 0.166 |
| Myogenesis | 198 | 42 (21%) | 1.419 | 0.008 | 0.151 |
| Angiogenesis | 36 | 8 (22%) | 1.373 | 0.083 | 0.154 |
| Gene | Description | Subcellular Localization | Gene Ontology (Biological Process) 1 |
|---|---|---|---|
| MYLK | myosin light chain kinase | stress fiber | positive regulation of cell migration, muscle contraction |
| BDNF | brain-derived neurotrophic factor | extracellular | negative regulation of apoptotic process |
| PCOLCE2 | procollagen C-endopeptidase enhancer 2 | extracellular | positive regulation of peptidase activity |
| S100A4 | S100 calcium binding protein A4 | extracellular | epithelial to mesenchymal transition, positive regulation of I-kappaB kinase/NF-kappaB signaling |
| Drug Name | Drug Type | Mechanism for S100A4 Inhibition | Dose for S100A4 Inhibition | Dose in CMap | Reference |
|---|---|---|---|---|---|
| Niclosamide | anthelminthic agent | inhibition of the Wnt/β-catenin pathway | 1 μM | 20 nM~10 μM | [18] |
| Calcimycin (A23187) | calcium ionophore | 1 μM | N.D. 1 | [40,41] | |
| Sulindac | nonsteroidal anti-inflammatory drug | 100 μM | 100 nM~10 μM | [42] | |
| Trifluoperazine | phenothiazines | disruption of the S100A4/myosin-IIA interaction by sequestering S100A4 via small molecule-induced oligomerization | 50~100 μM | 100 nM~10 μM | [43,44] |
| Prochlorperazine | 10 μM | ||||
| Perphenazine | 10 μM | ||||
| Chlorprothixene | 10 μM | ||||
| Flupentixol | 10 μM | ||||
| Fluphenazine | 100 nM~10 μM | ||||
| NSC-95397 | CDC25 inhibitor | 100 μM | N.D. | [45] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, S.W.; Chou, C.-J.; Huang, T.-C.; Yang, P.-M. An Integrated Bioinformatics Analysis Repurposes an Antihelminthic Drug Niclosamide for Treating HMGA2-Overexpressing Human Colorectal Cancer. Cancers 2019, 11, 1482. https://doi.org/10.3390/cancers11101482
Leung SW, Chou C-J, Huang T-C, Yang P-M. An Integrated Bioinformatics Analysis Repurposes an Antihelminthic Drug Niclosamide for Treating HMGA2-Overexpressing Human Colorectal Cancer. Cancers. 2019; 11(10):1482. https://doi.org/10.3390/cancers11101482
Chicago/Turabian StyleLeung, Stephen Wan, Chia-Jung Chou, Tsui-Chin Huang, and Pei-Ming Yang. 2019. "An Integrated Bioinformatics Analysis Repurposes an Antihelminthic Drug Niclosamide for Treating HMGA2-Overexpressing Human Colorectal Cancer" Cancers 11, no. 10: 1482. https://doi.org/10.3390/cancers11101482