The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Discovery of Tumor Heterogeneity
2. Current Models of Tumor Evolution
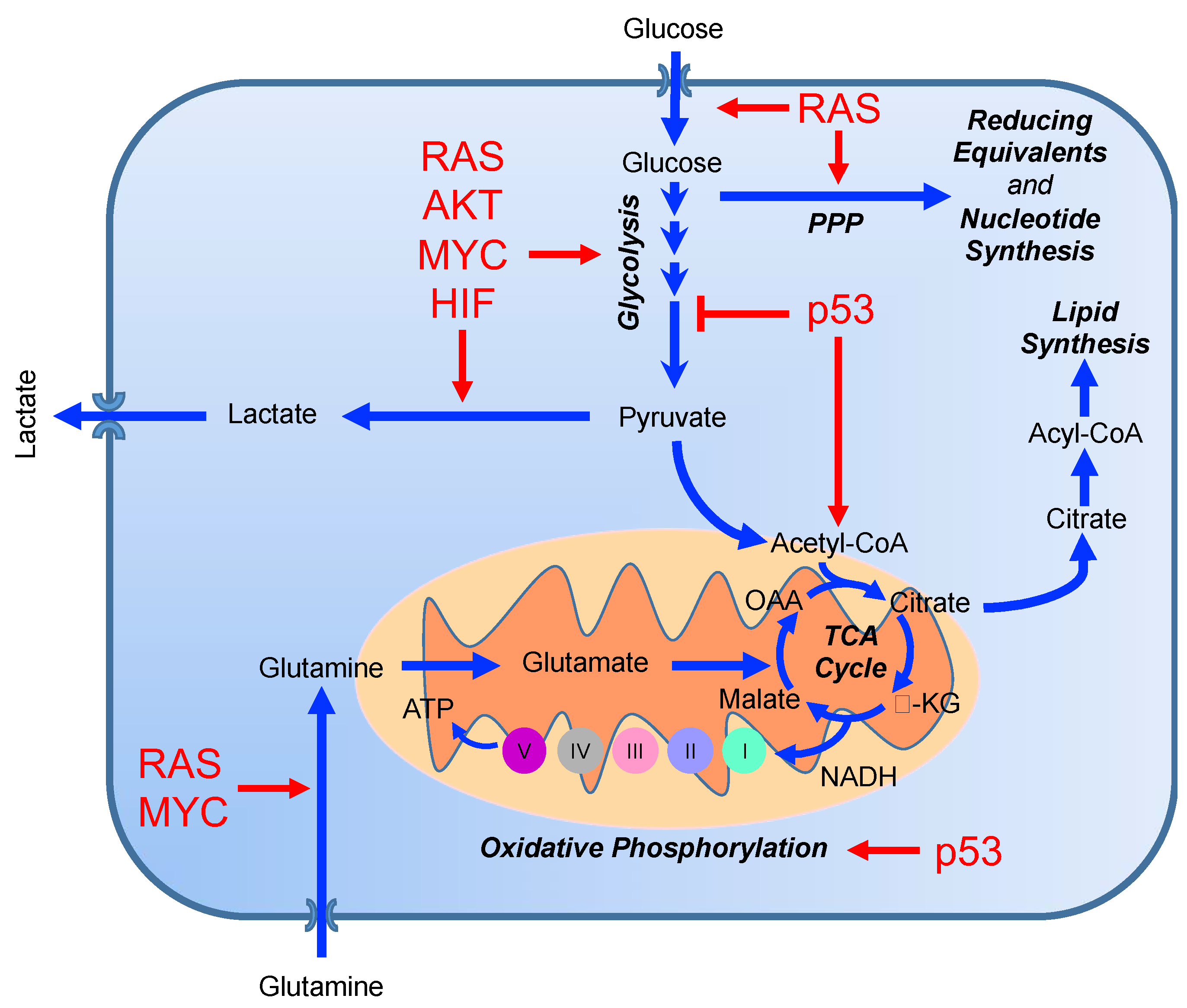
3. From Genomics to Metabolomics: The Role of Genes in Reprogramming Tumor Metabolism
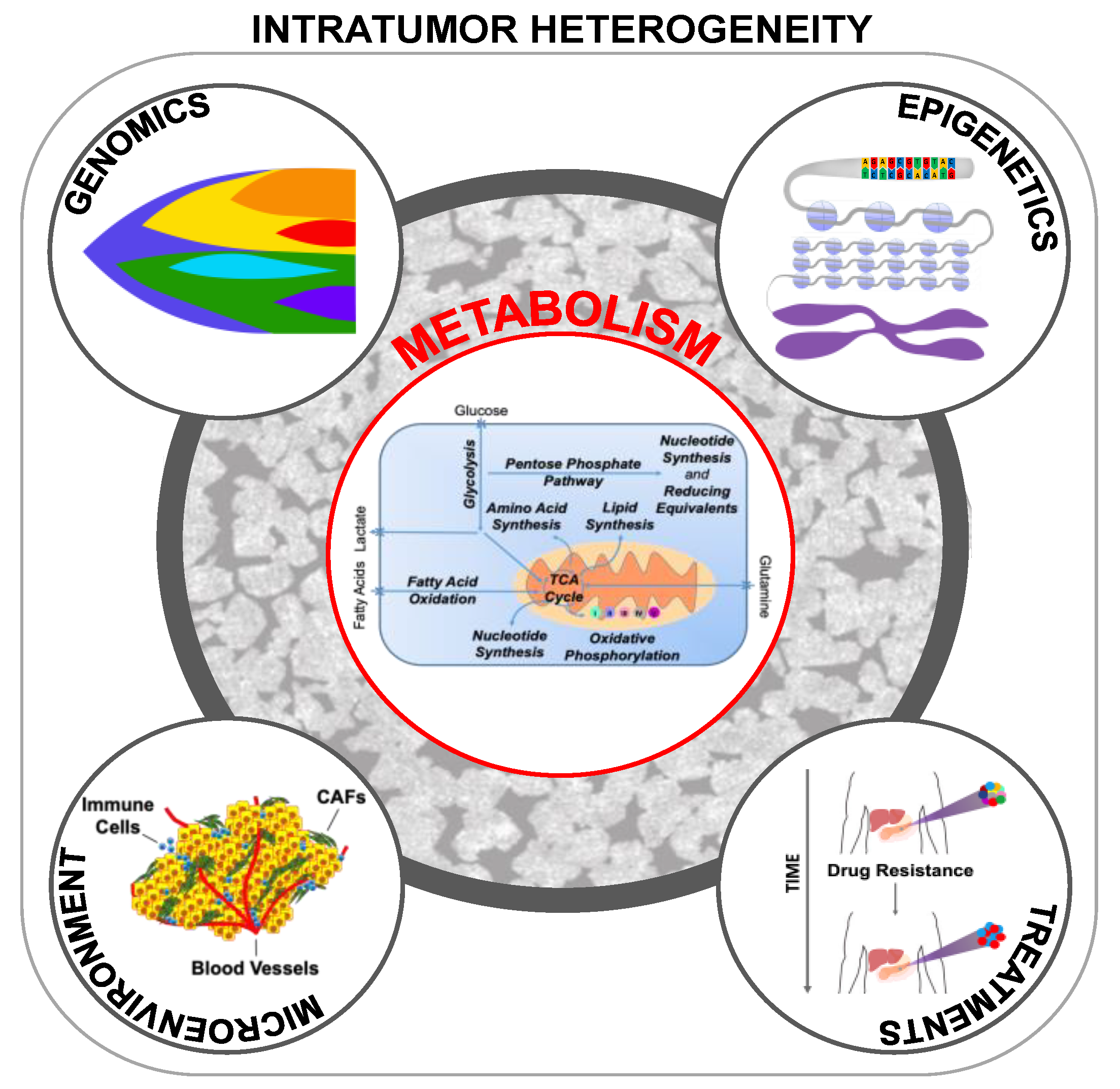
4. Metabolic Intratumor Heterogeneity
5. Metabolic Heterogeneity in the Tumor Microenvironment
5.1. Cancer Associated-Fibroblasts (CAFs)
5.2. Endothelial Cells
6. Metabolic Heterogeneity of Cancer Treatment Responses
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muller, J. Ueber den Feinern Bau und die Formen der Krankhaften Geschwülste; G. Reimer: Berlin, Germany, 1838. [Google Scholar]
- Muller, J. On the nature and structural characteristics of cancer: General observations on the minute structure of morbid growths. CA Cancer J. Clin. 1973, 23, 307–312. [Google Scholar] [CrossRef]
- Hajdu, S.I. A note from history: The first tumor pathologist. Ann. Clin. Lab. Sci. 2004, 34, 355–356. [Google Scholar] [PubMed]
- Virchow, R. Cellular Pathology as Based upon Physiological and Pathological Histology; J. B. Lippincott: Philadelphia, PA, USA, 1863. [Google Scholar] [CrossRef]
- Silver, G.A. Virchow, the heroic model in medicine: Health policy by accolade. Am. J. Public Health 1987, 77, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Furth, J. The Transmission of Leukemia of Mice with a Single Cell. Cancer Res. 1937, 31, 276–282. [Google Scholar] [CrossRef]
- Southam, C. Quantitative studies of autotransplantation of human cancer. Cancer 1961, 14, 971–978. [Google Scholar] [CrossRef]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Hamilton, S.R.; Vogelstein, B. Clonal analysis of human colorectal tumors. Science 1987, 238, 193–197. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Dexter, D.L.; Kowalski, H.M.; Blazar, B.A.; Fligiel, Z.; Vogel, R.; Heppner, G.H. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. 1978, 38, 3174–3181. [Google Scholar]
- Calabresi, P.; Dexter, D.L.; Heppner, G.H. Clinical and pharmacological implications of cancer cell differentiation and heterogeneity. Biochem. Pharmacol. 1979, 28, 1933–1941. [Google Scholar] [CrossRef]
- Hager, J.C.; Heppner, G.H. Heterogeneity of expression and induction of mouse mammary tumor virus antigens in mouse mammary tumors. Cancer Res. 1982, 42, 4325–4329. [Google Scholar] [PubMed]
- Heppner, G.H.; Dexter, D.L.; DeNucci, T.; Miller, F.R.; Calabresi, P. Heterogeneity in drug sensitivity among tumor cell subpopulations of a single mammary tumor. Cancer Res. 1978, 38, 3758–3763. [Google Scholar] [PubMed]
- Miller, B.E.; Miller, F.R.; Leith, J.; Heppner, G.H. Growth interaction in vivo between tumor subpopulations derived from a single mouse mammary tumor. Cancer Res. 1980, 40, 3977–3981. [Google Scholar]
- Miller, B.E.; Miller, F.R.; Wilburn, D.; Heppner, G.H. Dominance of a tumor subpopulation line in mixed heterogeneous mouse mammary tumors. Cancer Res. 1988, 48, 5747–5753. [Google Scholar] [PubMed]
- Miller, F.R.; Heppner, G.H. Immunologic heterogeneity of tumor cell subpopulations from a single mouse mammary tumor. J. Natl. Cancer Inst. 1979, 63, 1457–1463. [Google Scholar] [PubMed]
- Yamashina, K.; Heppner, G.H. Correlation of frequency of induced mutation and metastatic potential in tumor cell lines from a single mouse mammary tumor. Cancer Res. 1985, 45, 4015–4019. [Google Scholar] [PubMed]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar]
- Welch, D.R. Tumor Heterogeneity—A ‘Contemporary Concept’ Founded on Historical Insights and Predictions. Cancer Res. 2016, 76, 4–6. [Google Scholar] [CrossRef]
- Foulds, L. The natural history of cancer. J. Chronic Dis. 1958, 8, 2–37. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.C.; Torres, M.; Real, F.X. Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 2016, 16, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Hu, Z.; Curtis, C. Big Bang Tumor Growth and Clonal Evolution. Cold Spring Harb. Perspect. Med. 2018, 8, a028381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.M.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Seth, S.; Li, C.Y.; Ho, I.L.; Corti, D.; Loponte, S.; Sapio, L.; Del Poggetto, E.; Yen, E.Y.; Robinson, F.S.; Peoples, M.; et al. Pre-existing Functional Heterogeneity of Tumorigenic Compartment as the Origin of Chemoresistance in Pancreatic Tumors. Cell Rep. 2019, 26, 1518–1532. [Google Scholar] [CrossRef]
- Gallaher, J.A.; Enriquez-Navas, P.M.; Luddy, K.A.; Gatenby, R.A.; Anderson, A.R.A. Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res. 2018, 78, 2127–2139. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb. Perspect. Med. 2016, 6, a026583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science (New York N.Y.) 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Haleem, A.M.; Lewis, N.E.; Jamshidi, N.; Mineta, K.; Gao, X.; Gojobori, T. The Emerging Facets of Non-Cancerous Warburg Effect. Front. Endocrinol. (Lausanne) 2017, 8, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Burgess, D. MYC provides a global boost. Nat. Rev. Genet. 2012, 13, 758. [Google Scholar] [CrossRef]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Najafov, A.; Alessi, D.R. Uncoupling the Warburg effect from cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 19135–19136. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Bolanos, J.P.; Moncada, S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc. Natl. Acad. Sci. USA 2010, 107, 738–741. [Google Scholar] [CrossRef]
- Colombo, S.L.; Palacios-Callender, M.; Frakich, N.; De Leon, J.; Schmitt, C.A.; Boorn, L.; Davis, N.; Moncada, S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 18868–18873. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Mathupala, S.P.; Heese, C.; Pedersen, P.L. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997, 272, 22776–22780. [Google Scholar] [CrossRef] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Boidot, R.; Vegran, F.; Meulle, A.; Le Breton, A.; Dessy, C.; Sonveaux, P.; Lizard-Nacol, S.; Feron, O. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012, 72, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chretien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef]
- Lebedeva, M.A.; Eaton, J.S.; Shadel, G.S. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim. Biophys. Acta 2009, 1787, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floter, J.; Kaymak, I.; Schulze, A. Regulation of Metabolic Activity by p53. Metabolites 2017, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Clarkson, B.; Fried, J.; Strife, A.; Sakai, Y.; Ota, K.; Okita, T. Studies of cellular proliferation in human leukemia. 3. Behavior of leukemic cells in three adults with acute leukemia given continuous infusions of 3H-thymidine for 8 or 10 days. Cancer 1970, 25, 1237–1260. [Google Scholar] [CrossRef]
- Clarkson, B.D. Review of recent studies of cellular proliferation in acute leukemia. Natl. Cancer Inst. Monogr. 1969, 30, 81–120. [Google Scholar]
- Viale, A.; Pelicci, P. Regulation of Self-Renewing Division in Normal and Leukaemia Stem Cells; Springer: New York, NY, USA, 2010. [Google Scholar]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of iPSCs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef] [Green Version]
- Hochedlinger, K.; Blelloch, R.; Brennan, C.; Yamada, Y.; Kim, M.; Chin, L.; Jaenisch, R. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004, 18, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Mintz, B.; Illmensee, K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 1975, 72, 3585–3589. [Google Scholar] [CrossRef]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Lissanu Deribe, Y.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Korbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Draetta, G.F. Metabolic Features of Cancer Treatment Resistance. Recent Results Cancer Res. 2016, 207, 135–156. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, I.S.; Heiles, S.; Pieterse, C.L. Metabolic Imaging at the Single-Cell Scale: Recent Advances in Mass Spectrometry Imaging. Annu. Rev. Anal. Chem. (Palo Alto Calif.) 2019, 12, 201–224. [Google Scholar] [CrossRef]
- O’Connor, J.P.; Rose, C.J.; Waterton, J.C.; Carano, R.A.; Parker, G.J.; Jackson, A. Imaging intratumor heterogeneity: Role in therapy response, resistance, and clinical outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 249–257. [Google Scholar] [CrossRef]
- Gillies, R.J.; Kinahan, P.E.; Hricak, H. Radiomics: Images Are More than Pictures, They Are Data. Radiology 2016, 278, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Aerts, H.J.; Velazquez, E.R.; Leijenaar, R.T.; Parmar, C.; Grossmann, P.; Carvalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; Rietveld, D.; et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 2014, 5, 4006. [Google Scholar] [CrossRef]
- Kurhanewicz, J.; Vigneron, D.B.; Ardenkjaer-Larsen, J.H.; Bankson, J.A.; Brindle, K.; Cunningham, C.H.; Gallagher, F.A.; Keshari, K.R.; Kjaer, A.; Laustsen, C.; et al. Hyperpolarized (13)C MRI: Path to Clinical Translation in Oncology. Neoplasia 2019, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Salzillo, T.C.; Pudakalakatti, S.; Gammon, S.T.; Kaipparettu, B.A.; McAllister, F.; Wagner, S.; Frigo, D.E.; Logothetis, C.J.; Zacharias, N.M.; et al. Assessing Therapeutic Efficacy in Real-time by Hyperpolarized Magnetic Resonance Metabolic Imaging. Cells 2019, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Fathi Kazerooni, A.; Bakas, S.; Saligheh Rad, H.; Davatzikos, C. Imaging signatures of glioblastoma molecular characteristics: A radiogenomics review. J. Magn. Reson. Imaging 2019. [Google Scholar] [CrossRef]
- Jeong, W.K.; Jamshidi, N.; Felker, E.R.; Raman, S.S.; Lu, D.S. Radiomics and radiogenomics of primary liver cancers. Clin. Mol. Hepatol. 2019, 25, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Reig, B.; Heacock, L.; Geras, K.J.; Moy, L. Machine learning in breast MRI. J. Magn. Reson. Imaging 2019. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Bousamra, M., II; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef]
- Hensley, C.T.; DeBerardinis, R.J. In vivo analysis of lung cancer metabolism: Nothing like the real thing. J. Clin. Investig. 2015, 125, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Achreja, A.; Zhao, H.; Yang, L.; Yun, T.H.; Marini, J.; Nagrath, D. Exo-MFA—A 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab. Eng. 2017, 43, 156–172. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Ketone body utilization drives tumor growth and metastasis. Cell Cycle 2012, 11, 3964–3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Meyer, K.A.; Neeley, C.K.; Baker, N.A.; Washabaugh, A.R.; Flesher, C.G.; Nelson, B.S.; Frankel, T.L.; Lumeng, C.N.; Lyssiotis, C.A.; Wynn, M.L.; et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem. Biophys. Rep. 2016, 7, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Kryczek, I.; Dostal, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timosenko, E.; Hadjinicolaou, A.V.; Cerundolo, V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy 2017, 9, 83–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Bruning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid beta-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894. [Google Scholar] [CrossRef]
- Lovisa, S.; Kalluri, R. Fatty Acid Oxidation Regulates the Activation of Endothelial-to-Mesenchymal Transition. Trends Mol. Med. 2018, 24, 432–434. [Google Scholar] [CrossRef]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Menga, A.; Martin-Perez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquiere, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Bruning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Paudel, B.B.; Quaranta, V. Metabolic plasticity meets gene regulation. Proc. Natl. Acad. Sci. USA 2019, 116, 3370–3372. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.M.; Bardeesy, N. Pancreatic Cancer Metabolism: Breaking It Down to Build It Back Up. Cancer Discov. 2015, 5, 1247–1261. [Google Scholar] [CrossRef]
- Warburg, O.; Minami, S. Versuche an Überlebendem Carcinom-gewebe. Klin. Wochenschr. 1923, 2, 776–777. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]
- Birsoy, K.; Sabatini, D.M.; Possemato, R. Untuning the tumor metabolic machine: Targeting cancer metabolism: A bedside lesson. Nat. Med. 2012, 18, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Tennant, D.A.; Durán, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In Vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- El Mjiyad, N.; Caro-Maldonado, A.; Ramírez-Peinado, S.; Muñoz-Pinedo, C. Sugar-free approaches to cancer cell killing. Oncogene 2010, 30, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Zhang, Y.; Chen, T.; Wang, Y.; Xue, J.; Zhang, Y.; Xiao, W.; Mo, X.; Lu, Y. Efficacy of RNAi targeting of pyruvate kinase M2 combined with cisplatin in a lung cancer model. J. Cancer Res. Clin. Oncol. 2011, 137, 65–72. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. (Lond. Engl.) 2010, 6, 551–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thupari, J.N.; Pinn, M.L.; Kuhajda, F.P. Fatty Acid Synthase Inhibition in Human Breast Cancer Cells Leads to Malonyl-CoA-Induced Inhibition of Fatty Acid Oxidation and Cytotoxicity. Biochem. Biophys. Res. Commun. 2001, 285, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Angeles, T.S.; Hudkins, R.L. Recent advances in targeting the fatty acid biosynthetic pathway using fatty acid synthase inhibitors. Expert Opin. Drug Discov. 2016, 11, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-S.; Lei, J.-P.; Wei, G.-Q.; Chen, H.; Ma, C.-Y.; Jiang, H.-Z. Natural fatty acid synthase inhibitors as potent therapeutic agents for cancers: A review. Pharm. Biol. 2016, 54, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2015, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J. Med. Chem. 2019, 62, 1096–1115. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. NCT03894540. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03894540?term=NCT03894540&rank=1 (accessed on 27 August 2019).
- Sabnis, H.S.; Somasagara, R.R.; Bunting, K.D. Targeting MYC Dependence by Metabolic Inhibitors in Cancer. Genes 2017, 8, 114. [Google Scholar] [CrossRef]
- Kalaany, N.Y.; Sabatini, D.M. Tumours with PI3K activation are resistant to dietary restriction. Nature 2009, 458, 725–731. [Google Scholar] [CrossRef]
- Ma, L.; Tao, Y.; Duran, A.; Llado, V.; Galvez, A.; Barger, J.F.; Castilla, E.A.; Chen, J.; Yajima, T.; Porollo, A.; et al. Control of nutrient stress-induced metabolic reprogramming by PKCζ in tumorigenesis. Cell 2013, 152, 599–611. [Google Scholar] [CrossRef]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; et al. Dietary downregulation of mutant p53 levels via glucose restriction: Mechanisms and implications for tumor therapy. Cell Cycle (Georget. Tex.) 2012, 11, 4436–4446. [Google Scholar] [CrossRef]
- Walker-Samuel, S.; Ramasawmy, R.; Torrealdea, F.; Rega, M.; Rajkumar, V.; Johnson, S.P.; Richardson, S.; Gonçalves, M.; Parkes, H.G.; Arstad, E.; et al. In vivo imaging of glucose uptake and metabolism in tumors. Nat. Med. 2013, 19, 1067–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Park, I.; Nelson, S.J. Imaging tumor metabolism using in vivo magnetic resonance spectroscopy. Cancer J. (Sudbury Mass.) 2015, 21, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, K.A.; Styczynski, M.P. Applications of metabolomics in cancer research. J. Carcinog. 2013, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, H.; Bradley, H.L.; Bunting, S.T.; Cooper, T.M.; Bunting, K.D. Capillary nano-immunoassay for Akt 1/2/3 and 4EBP1 phosphorylation in acute myeloid leukemia. J. Transl. Med. 2014, 12, 166. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Chang, C.-P.B.; Tsao, C.-C.; Xu, J. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J. Biol. Chem. 2010, 285, 12647–12654. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.; Ciro, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3beta-MCL-1 Axis. Cancer Cell 2019, 35, 798–815. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef]
- Muir, A.; Danai, L.V.; Vander Heiden, M.G. Microenvironmental regulation of cancer cell metabolism: Implications for experimental design and translational studies. Dis. Models Mech. 2018, 11, dmm035758. [Google Scholar] [CrossRef]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell metab. 2019, 30, 201–211 e206. [Google Scholar] [CrossRef]
- Robertson-Tessi, M.; Gillies, R.J.; Gatenby, R.A.; Anderson, A.R. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer res. 2015, 75, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Smallbone, K.; Gatenby, R.A.; Gillies, R.J.; Maini, P.K.; Gavaghan, D.J. Metabolic changes during carcinogenesis: Potential impact on invasiveness. J. Theor. Biol. 2007, 244, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loponte, S.; Lovisa, S.; Deem, A.K.; Carugo, A.; Viale, A. The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind? Cancers 2019, 11, 1574. https://doi.org/10.3390/cancers11101574
Loponte S, Lovisa S, Deem AK, Carugo A, Viale A. The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind? Cancers. 2019; 11(10):1574. https://doi.org/10.3390/cancers11101574
Chicago/Turabian StyleLoponte, Sara, Sara Lovisa, Angela K. Deem, Alessandro Carugo, and Andrea Viale. 2019. "The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind?" Cancers 11, no. 10: 1574. https://doi.org/10.3390/cancers11101574