Development of a MEL Cell-Derived Allograft Mouse Model for Cancer Research


 , and
, and 
Abstract
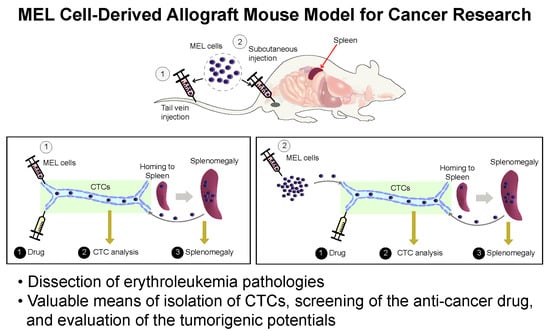
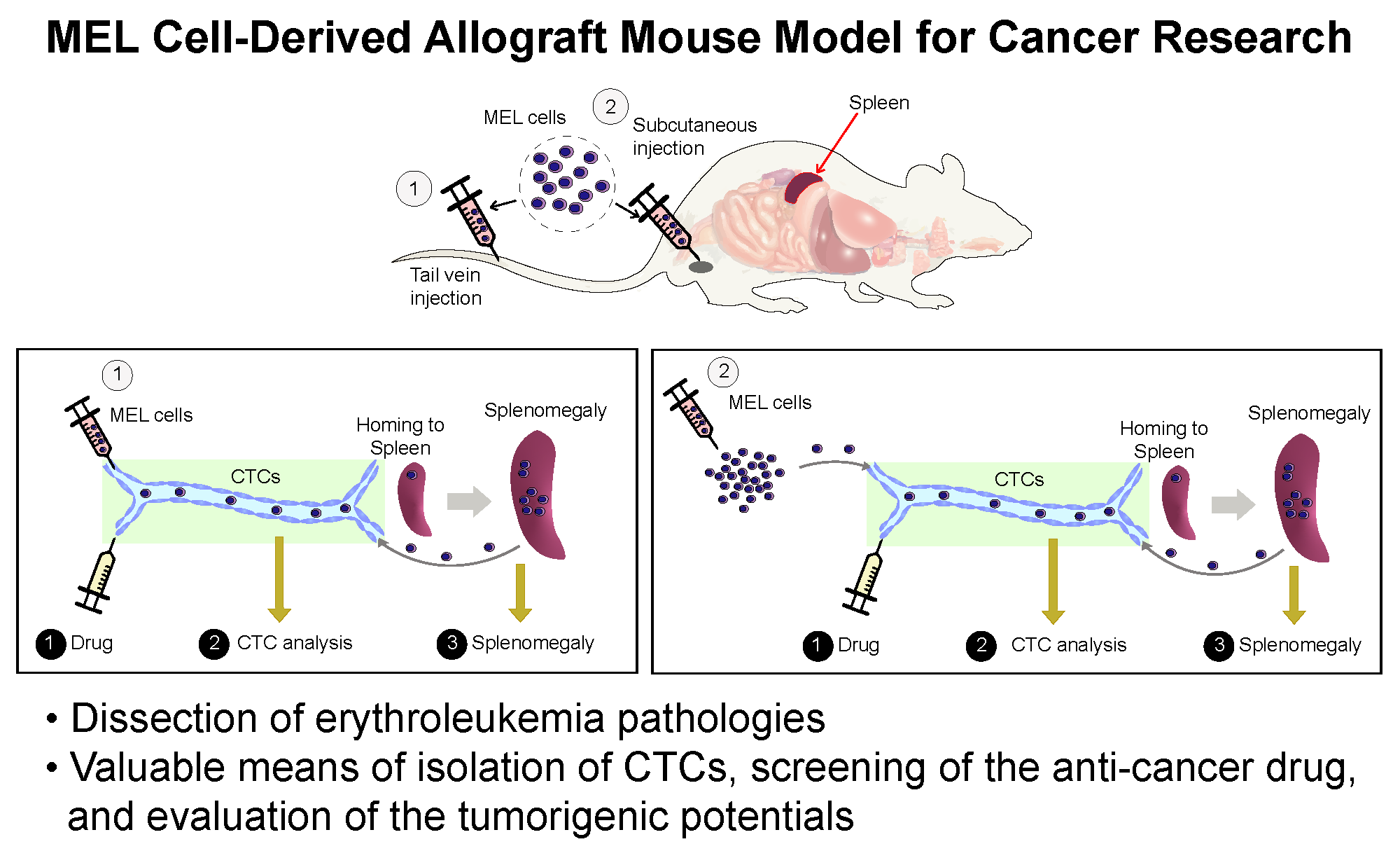
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
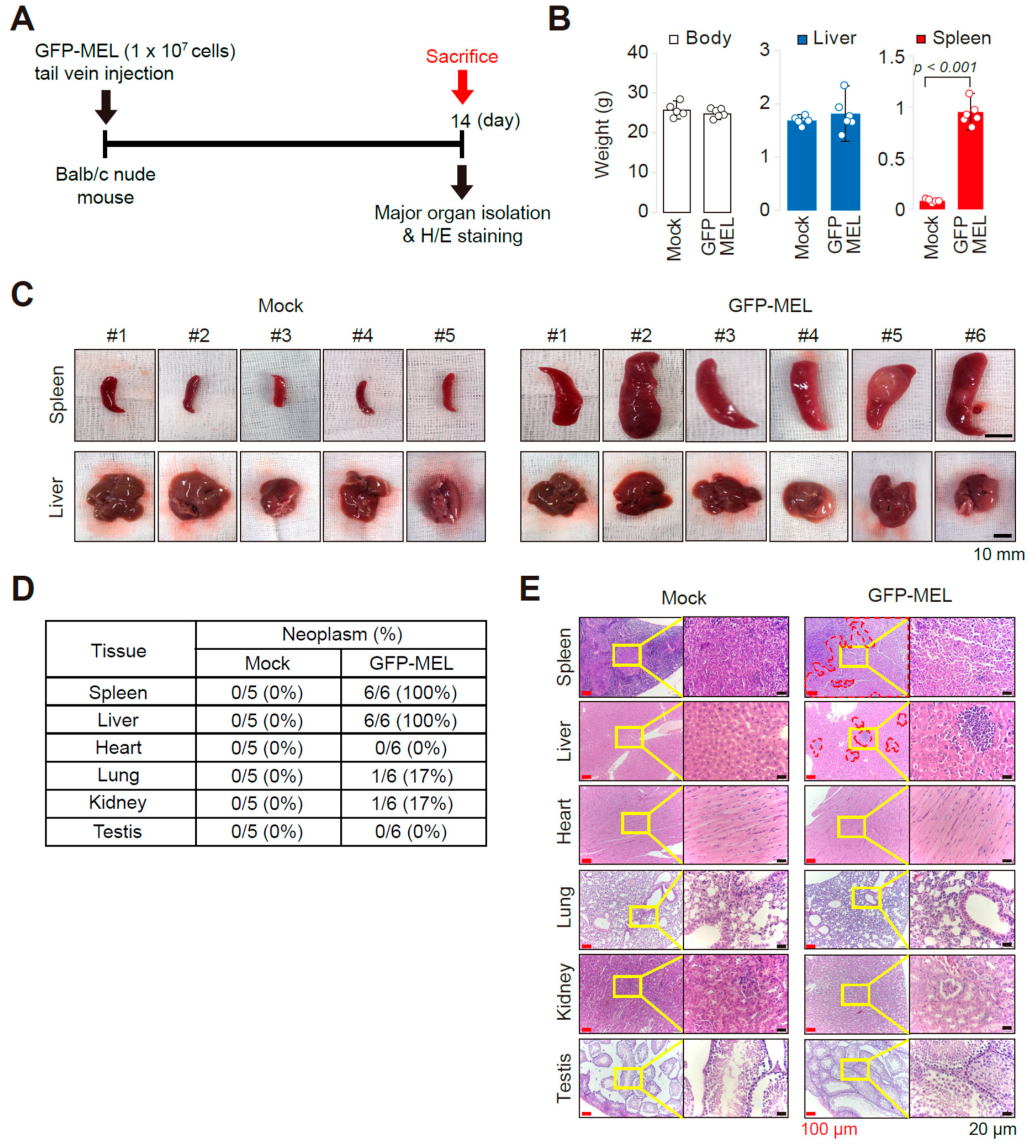
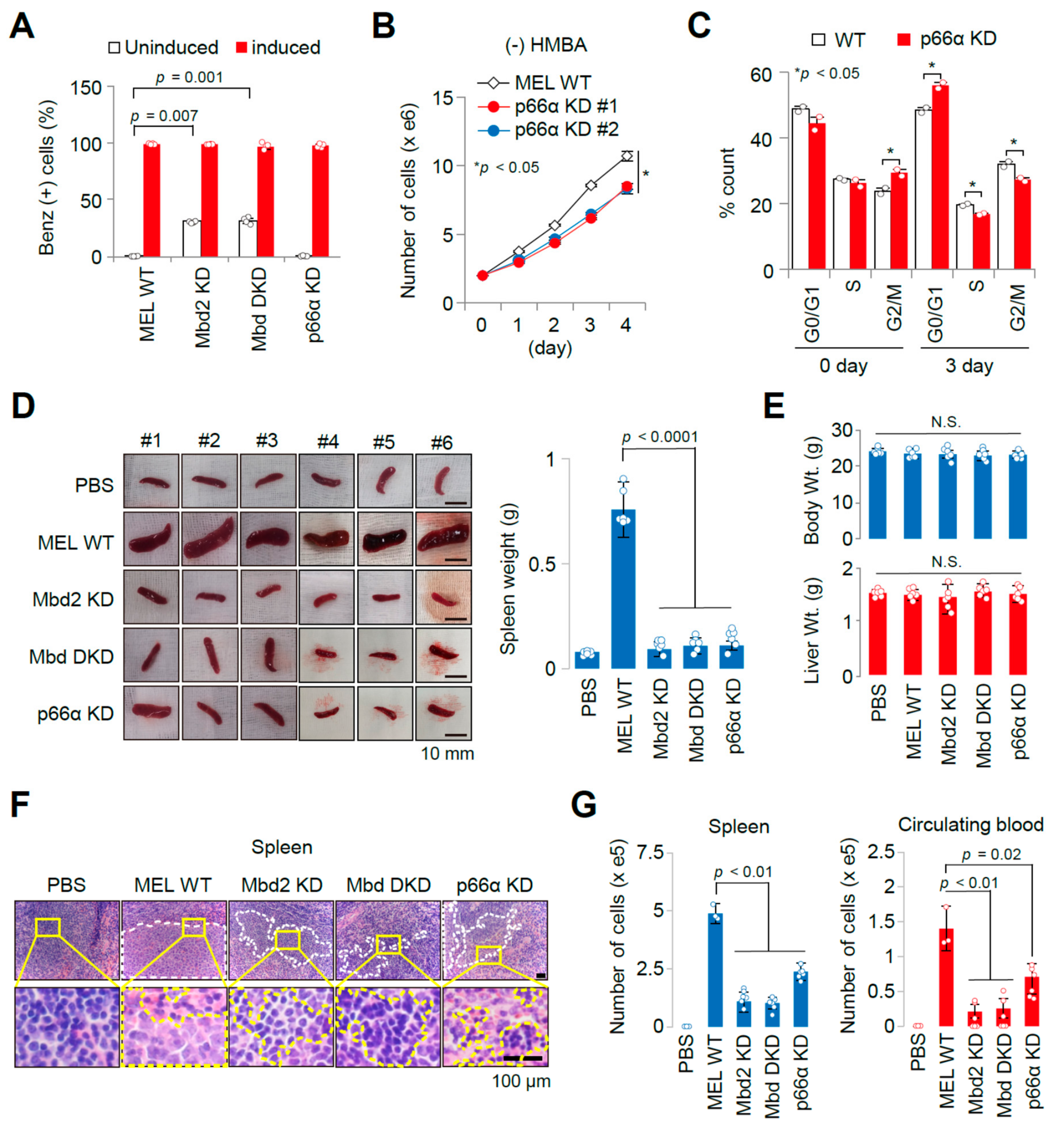
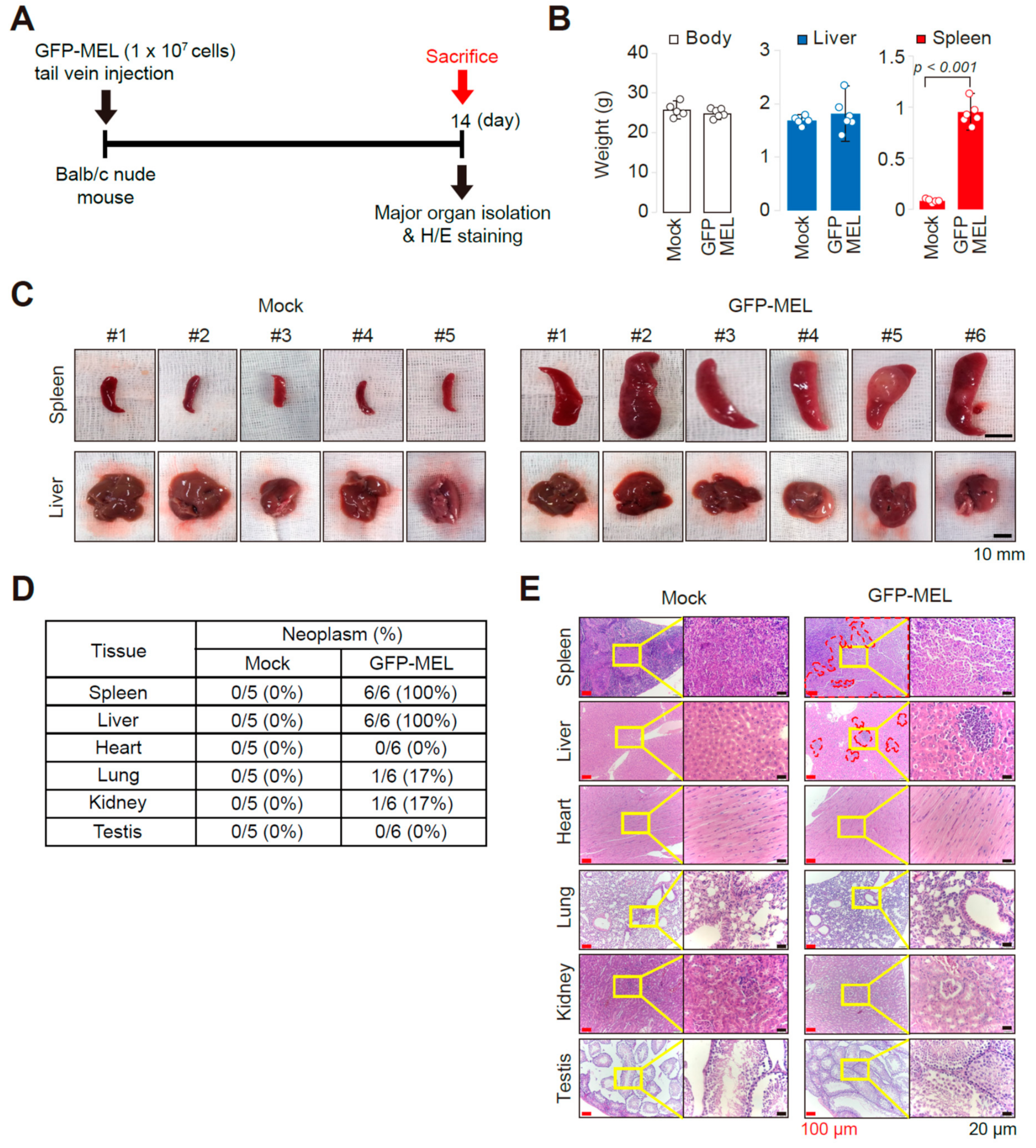
2.1. Establishment of an Allograft Model and Evaluation of the Methyl-CpG-Binding Domain 2 (MBD2) and p66α Roles in Normal Erythroid Differentiation and Their Tumorigenic Potential in This Model
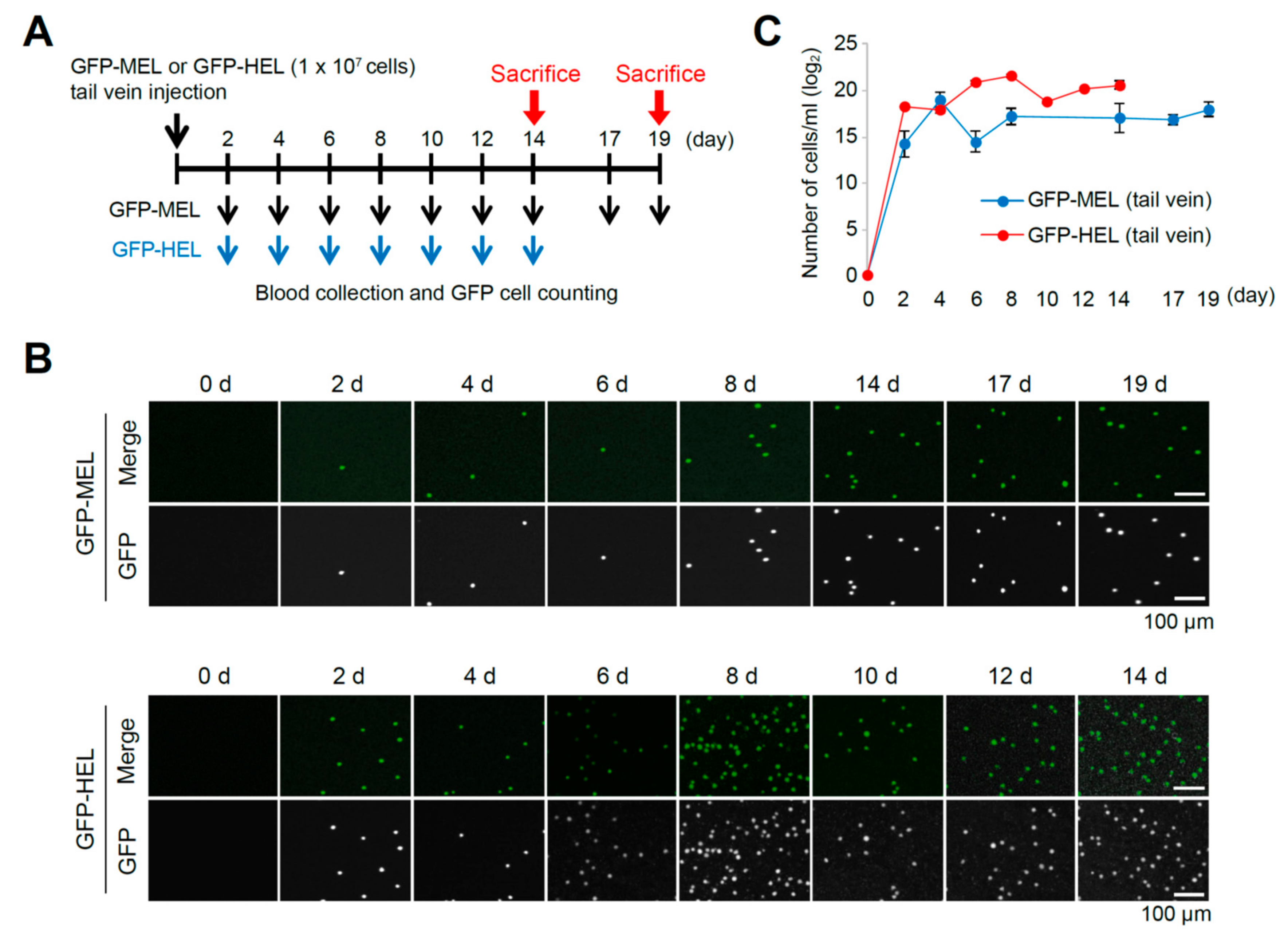
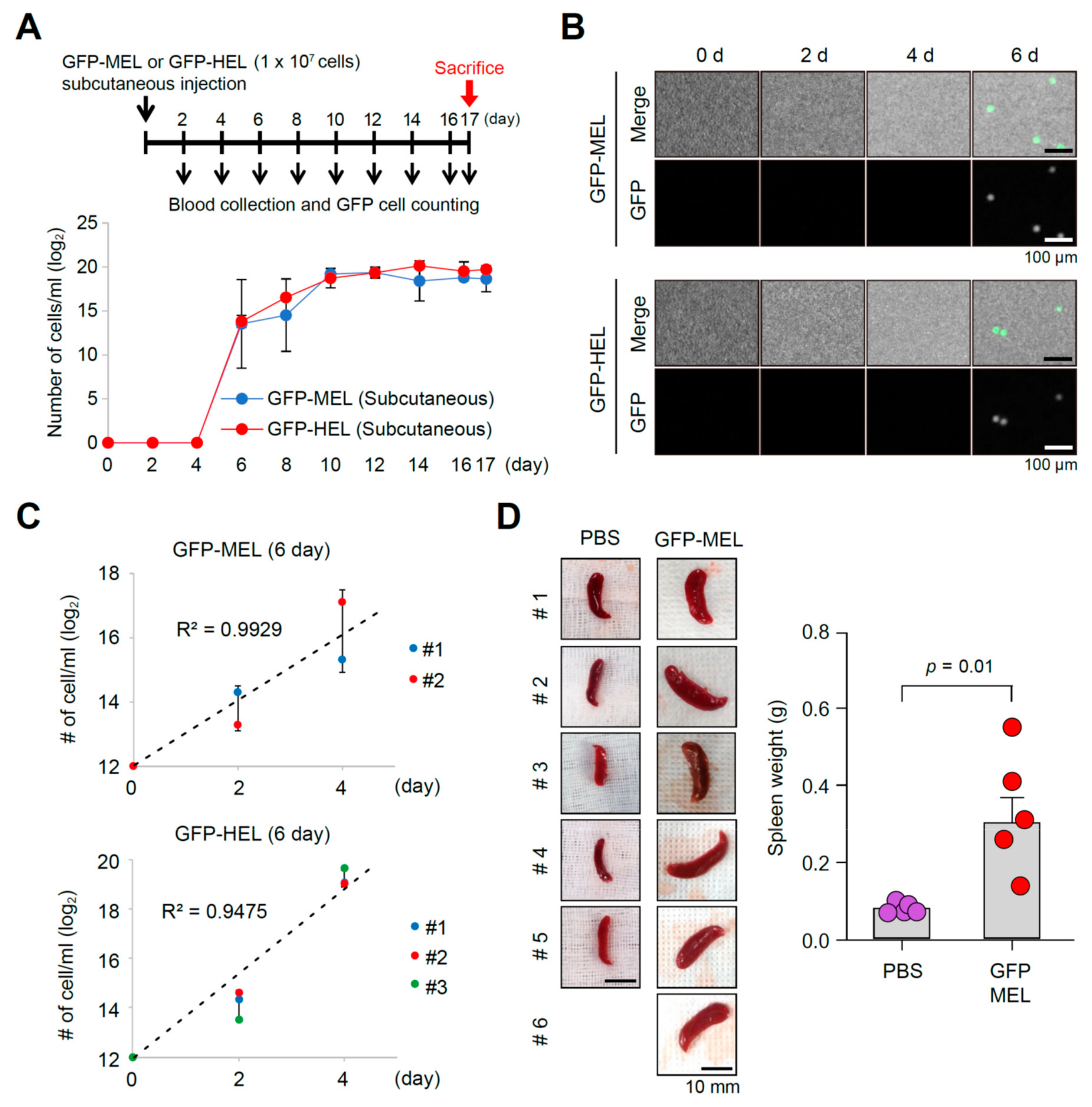
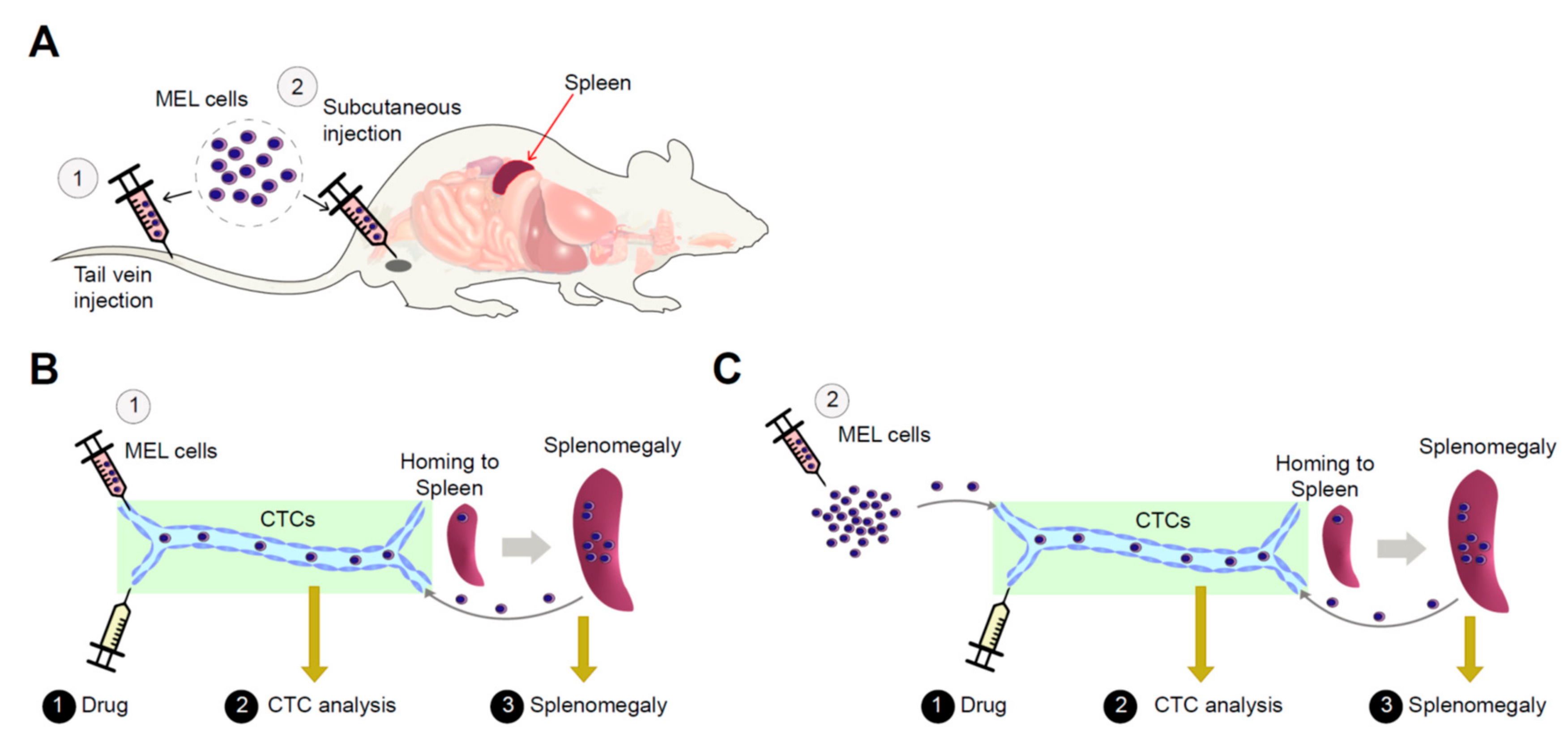
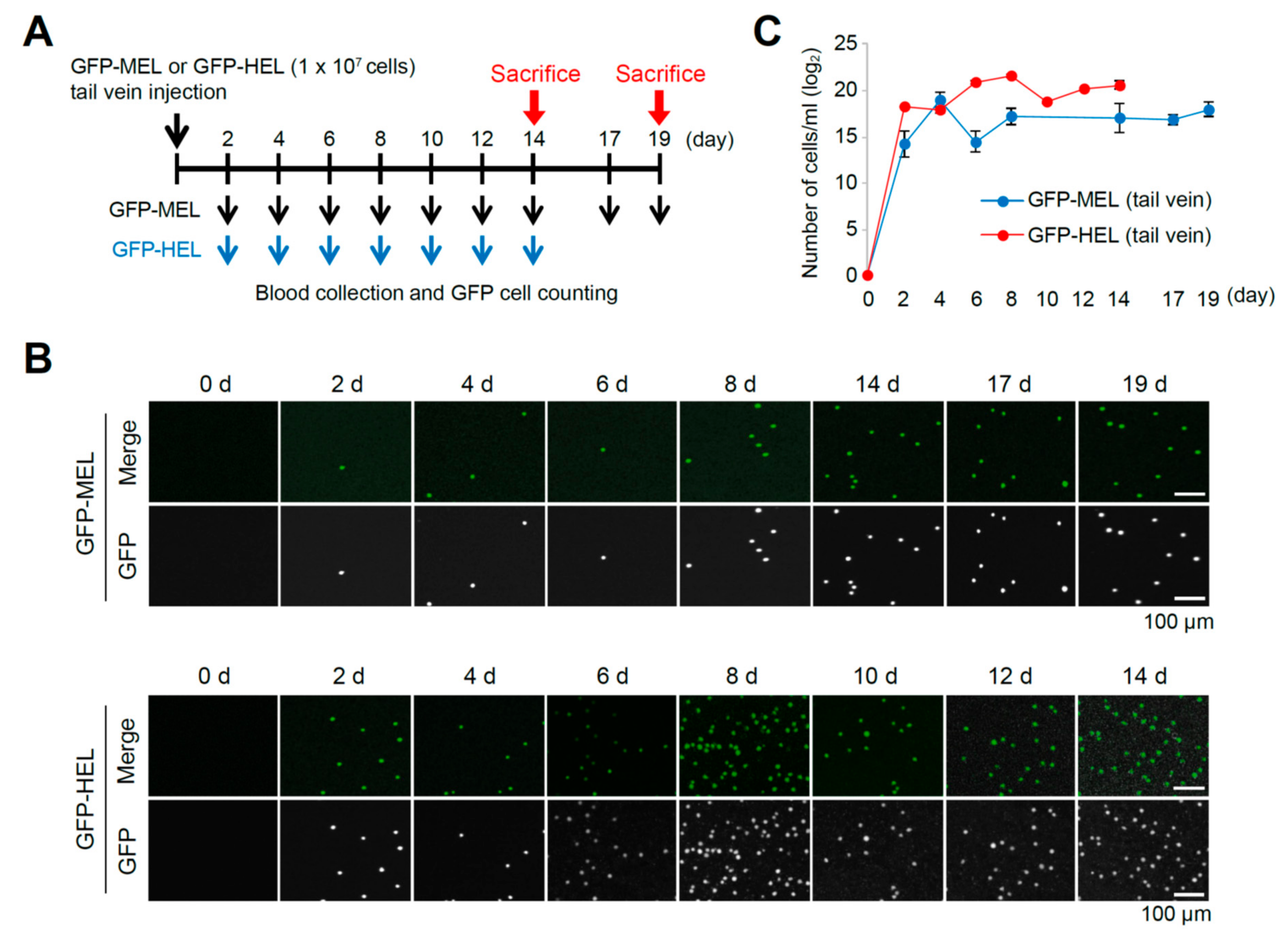
2.2. Erythroleukemia Cell-Transfused Mice as a Diagnostic Model to Evaluate Tumorigenicity Level and Metastatic Potentials in Cancer Researches
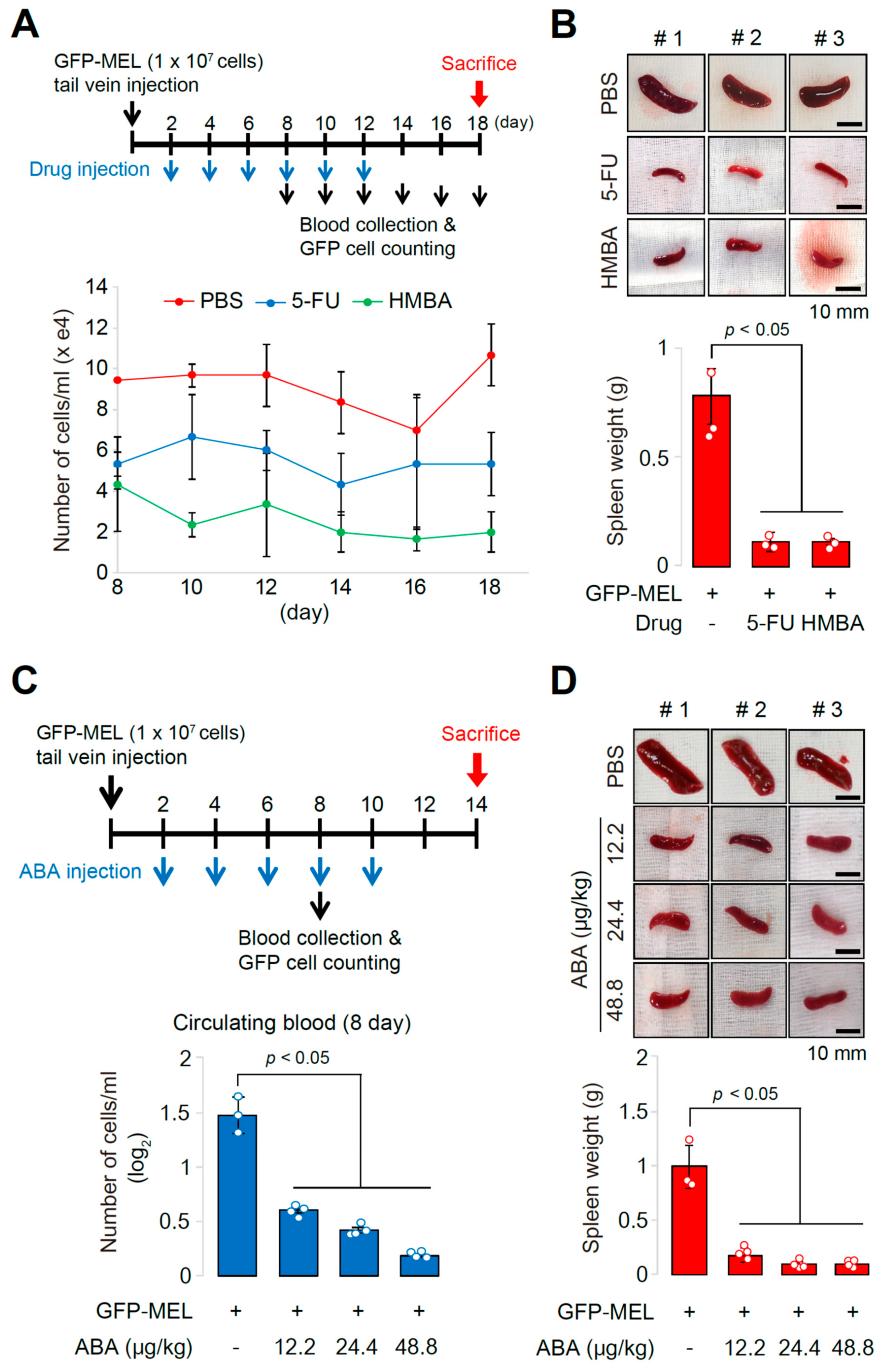
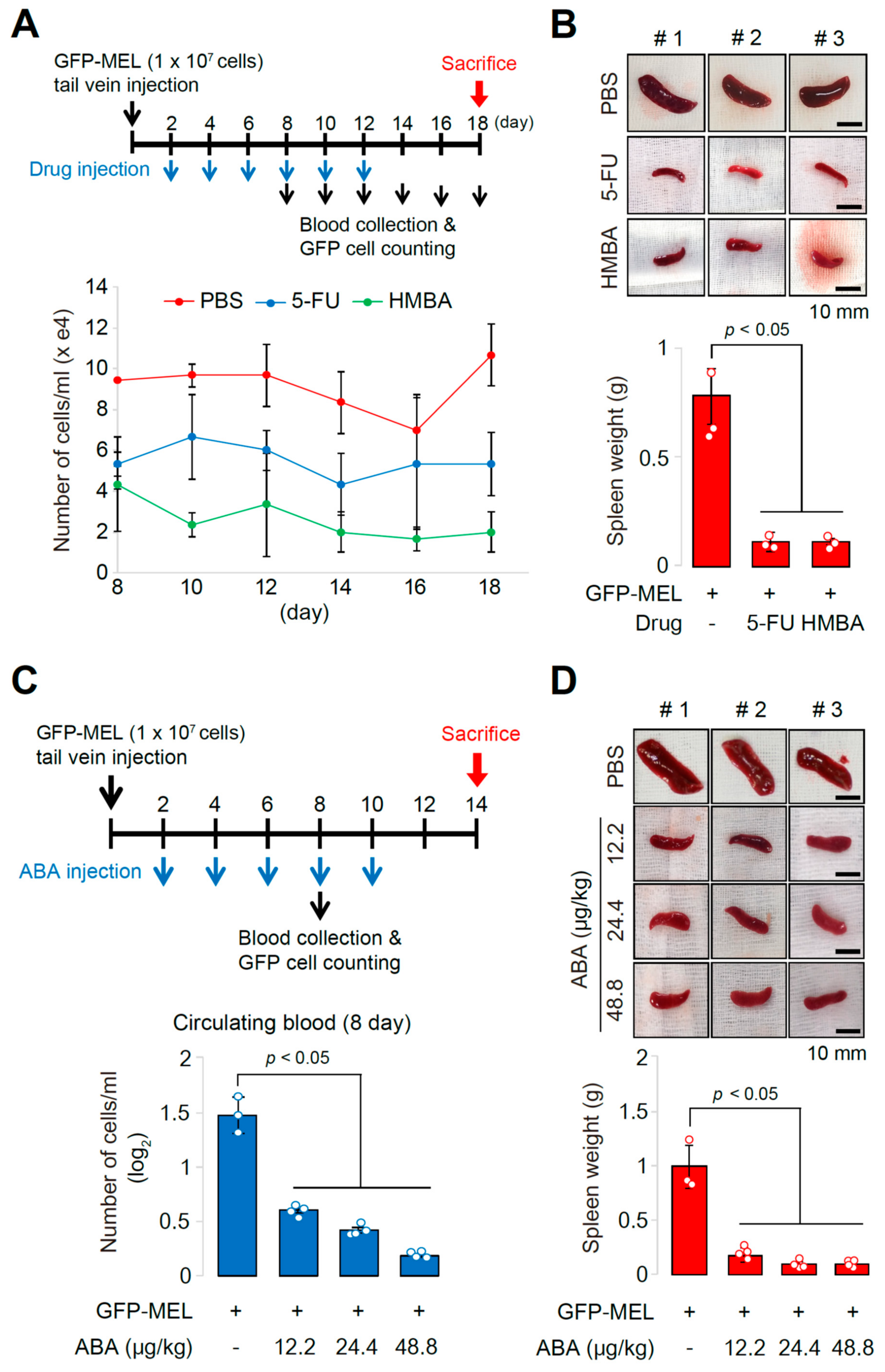
2.3. The Allograft Model for Anti-Cancer Drug Screening and Validation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmid Constructs
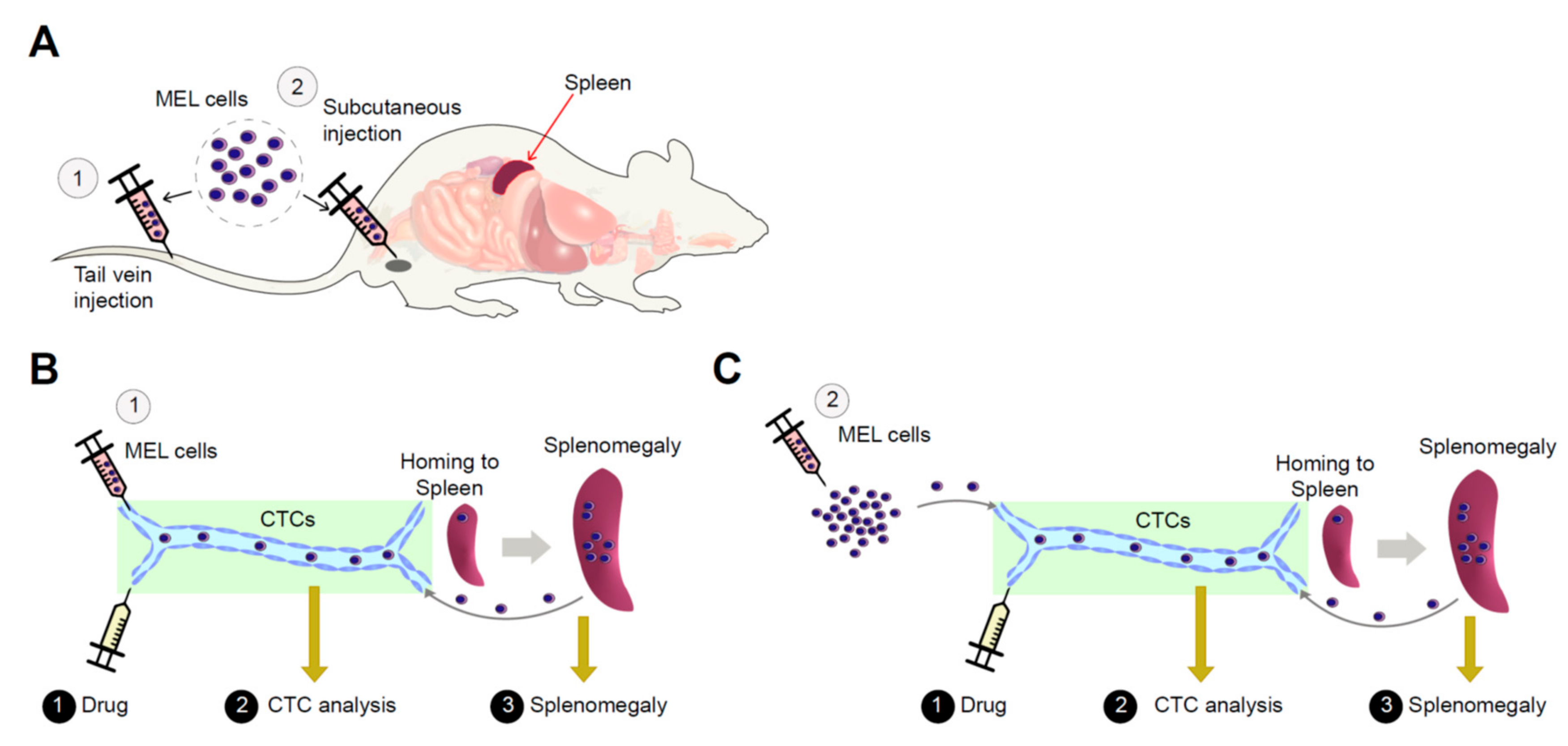
4.3. Animal Studies
4.4. Cell Cycle Analysis
4.5. Quantification and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Santos, F.P.; Bueso-Ramos, C.E.; Ravandi, F. Acute erythroleukemia: Diagnosis and management. Expert Rev. Hematol. 2010, 3, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Boddu, P.; Benton, C.B.; Wang, W.; Borthakur, G.; Khoury, J.D.; Pemmaraju, N. Erythroleukemia-historical perspectives and recent advances in diagnosis and management. Blood Rev. 2018, 32, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.A.; Medeiros, L.J.; Khoury, J.D. Pure erythroid leukemia. Am. J. Hematol. 2017, 92, 292–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.A.; Sheffery, M.; Ramsay, R.; Ikeda, K.; Rifkind, R.A. Induction of transformed cells to terminal differentiation. Ann. N. Y. Acad. Sci. 1987, 511, 246–255. [Google Scholar] [CrossRef]
- Siegel, B.V.; Weaver, W.J.; Koler, R.D. Mouse Erythroleukemia of Viral Etiology. Nature 1964, 201, 1042–1043. [Google Scholar] [CrossRef]
- Kim, M.Y.; Kim, J.S.; Son, S.H.; Lim, C.S.; Eum, H.Y.; Ha, D.H.; Park, M.A.; Baek, E.J.; Ryu, B.Y.; Kang, H.C.; et al. Mbd2-CP2c loop drives adult-type globin gene expression and definitive erythropoiesis. Nucleic Acids Res. 2018, 46, 4933–4949. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, K.M.; Kim, C.G.; Banerji, S.S.; Sheffery, M. Identification and characterization of multiple erythroid cell proteins that interact with the promoter of the murine alpha-globin gene. Mol. Cell. Biol. 1988, 8, 3215–3226. [Google Scholar] [CrossRef]
- Kim, C.G.; Barnhart, K.M.; Sheffery, M. Purification of multiple erythroid cell proteins that bind the promoter of the alpha-globin gene. Mol. Cell. Biol. 1988, 8, 4270–4281. [Google Scholar] [CrossRef]
- Kim, C.G.; Swendeman, S.L.; Barnhart, K.M.; Sheffery, M. Promoter elements and erythroid cell nuclear factors that regulate alpha-globin gene transcription in vitro. Mol. Cell. Biol. 1990, 10, 5958–5966. [Google Scholar] [CrossRef]
- Chae, J.H.; Kim, C.G. CP2 binding to the promoter is essential for the enhanced transcription of globin genes in erythroid cells. Mol. Cells 2003, 15, 40–47. [Google Scholar] [PubMed]
- Chae, J.H.; Lee, Y.H.; Kim, C.G. Transcription factor CP2 is crucial in hemoglobin synthesis during erythroid terminal differentiation in vitro. Biochem. Biophys. Res. Commun. 1999, 263, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Chae, J.H.; Jeon, J.; Kim, W.; Ha, D.H.; Shin, J.H.; Kim, C.G.; Kim, C.G. PIAS1 regulates CP2c localization and active promoter complex formation in erythroid cell-specific alpha-globin expression. Nucleic Acids Res. 2010, 38, 5456–5471. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Chae, J.H.; Lee, Y.H.; Park, M.A.; Shin, J.H.; Kim, S.H.; Ye, S.K.; Cho, Y.S.; Fiering, S.; Kim, C.G. Erythroid cell-specific alpha-globin gene regulation by the CP2 transcription factor family. Mol. Cell. Biol. 2005, 25, 6005–6020. [Google Scholar] [CrossRef] [PubMed]
- Kloet, S.L.; Baymaz, H.I.; Makowski, M.; Groenewold, V.; Jansen, P.W.; Berendsen, M.; Niazi, H.; Kops, G.J.; Vermeulen, M. Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J. 2015, 282, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.A.; Webb, H.D.; Sinanan, L.M.; Scarsdale, J.N.; Walavalkar, N.M.; Ginder, G.D.; Williams, D.C., Jr. An intrinsically disordered region of methyl-CpG binding domain protein 2 (MBD2) recruits the histone deacetylase core of the NuRD complex. Nucleic Acids Res. 2015, 43, 3100–3113. [Google Scholar] [CrossRef]
- Gnanapragasam, M.N.; Scarsdale, J.N.; Amaya, M.L.; Webb, H.D.; Desai, M.A.; Walavalkar, N.M.; Wang, S.Z.; Zu Zhu, S.; Ginder, G.D.; Williams, D.C., Jr. p66Alpha-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc. Natl. Acad. Sci. USA 2011, 108, 7487–7492. [Google Scholar] [CrossRef]
- Kim, M.Y.; Na, I.; Kim, J.S.; Son, S.H.; Choi, S.; Lee, S.E.; Kim, J.H.; Jang, K.; Alterovitz, G.; Chen, Y.; et al. Rational discovery of anti-metastatic agents targeting the intrinsically disordered region of MBD2. Sci. Adv. 2019, in press. [Google Scholar]
- Baldwin, J.G.; Wagner, F.; Martine, L.C.; Holzapfel, B.M.; Theodoropoulos, C.; Bas, O.; Savi, F.M.; Werner, C.; De-Juan-Pardo, E.M.; Hutmacher, D.W. Periosteum tissue engineering in an orthotopic in vivo platform. Biomaterials 2017, 121, 193–204. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Hesami, P.; Holzapfel, B.M.; Taubenberger, A.; Roudier, M.; Fazli, L.; Sieh, S.; Thibaudeau, L.; Gregory, L.S.; Hutmacher, D.W.; Clements, J.A. A humanized tissue-engineered in vivo model to dissect interactions between human prostate cancer cells and human bone. Clin. Exp. Metastasis 2014, 31, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, B.M.; Hutmacher, D.W.; Nowlan, B.; Barbier, V.; Thibaudeau, L.; Theodoropoulos, C.; Hooper, J.D.; Loessner, D.; Clements, J.A.; Russell, P.J.; et al. Tissue engineered humanized bone supports human hematopoiesis in vivo. Biomaterials 2015, 61, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Krijgsman, O.; Cornelissen-Steijger, P.; Shahrabi, A.; Weeber, F.; Song, J.Y.; Kuilman, T.; Vis, D.J.; Wessels, L.F.; Voest, E.E.; et al. Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts. EMBO Mol. Med. 2015, 7, 1104–1118. [Google Scholar] [CrossRef]
- Kostenuik, P.J.; Nguyen, H.Q.; McCabe, J.; Warmington, K.S.; Kurahara, C.; Sun, N.; Chen, C.; Li, L.; Cattley, R.C.; Van, G.; et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J. Bone Miner. Res. 2009, 24, 182–195. [Google Scholar] [CrossRef]
- Krepler, C.; Xiao, M.; Sproesser, K.; Brafford, P.A.; Shannan, B.; Beqiri, M.; Liu, Q.; Xu, W.; Garman, B.; Nathanson, K.L.; et al. Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin. Cancer Res. 2016, 22, 1592–1602. [Google Scholar] [CrossRef]
- Suri, A.; Sheng, X.; Schuler, K.M.; Zhong, Y.; Han, X.; Jones, H.M.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. The effect of celecoxib on tumor growth in ovarian cancer cells and a genetically engineered mouse model of serous ovarian cancer. Oncotarget 2016, 7, 39582–39594. [Google Scholar] [CrossRef]
- Wagner, F.; Holzapfel, B.M.; Thibaudeau, L.; Straub, M.; Ling, M.T.; Grifka, J.; Loessner, D.; Levesque, J.P.; Hutmacher, D.W. A Validated Preclinical Animal Model for Primary Bone Tumor Research. J. Bone Joint Surg. Am. 2016, 98, 916–925. [Google Scholar] [CrossRef]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M.; Reardon, D.A. Cancer: The nuances of therapy. Nature 2009, 458, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, M.; McGovern, J.A.; Friedl, P.; Hutmacher, D.W. Rational Design of Mouse Models for Cancer Research. Trends Biotechnol. 2018, 36, 242–251. [Google Scholar] [CrossRef] [PubMed]
- McGovern, J.A.; Griffin, M.; Hutmacher, D.W. Animal models for bone tissue engineering and modelling disease. Dis. Model Mech. 2018, 11, dmm033084. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Simile, M.M.; Peitta, G.; Seddaiu, M.A.; Feo, F.; Calvisi, D.F. Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers 2019, 11, 1450. [Google Scholar] [CrossRef] [PubMed]
- Perrin, S. Preclinical research: Make mouse studies work. Nature 2014, 507, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, N.; Brown, S. The mouse ascending: Perspectives for human-disease models. Nat. Cell Biol. 2007, 9, 993–999. [Google Scholar] [CrossRef]
- Hunter, K.W. Mouse models of cancer: Does the strain matter? Nat. Rev. Cancer 2012, 12, 144–149. [Google Scholar] [CrossRef]
- Witte, J.S.; Visscher, P.M.; Wray, N.R. The contribution of genetic variants to disease depends on the ruler. Nat. Rev. Genet. 2014, 15, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Attarwala, H. TGN1412: From Discovery to Disaster. J. Young Pharm. 2010, 2, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, C.H. Why drugs fail: Of mice and men revisited. Clin. Cancer Res. 2001, 7, 229–230. [Google Scholar]
- Zeimet, A.G.; Marth, C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol. 2003, 4, 415–422. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Frau, M.; Tomasi, M.L.; Feo, F.; Pascale, R.M. Deregulation of signalling pathways in prognostic subtypes of hepatocellular carcinoma: Novel insights from interspecies comparison. Biochim. Biophys. Acta 2012, 1826, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Manenti, G.; Galvan, A.; Falvella, F.S.; Pascale, R.M.; Spada, E.; Milani, S.; Gonzalez Neira, A.; Feo, F.; Dragani, T.A. Genetic control of resistance to hepatocarcinogenesis by the mouse Hpcr3 locus. Hepatology 2008, 48, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Mott, R.; Flint, J. Simultaneous detection and fine mapping of quantitative trait loci in mice using heterogeneous stocks. Genetics 2002, 160, 1609–1618. [Google Scholar]
- Andersen, J.B.; Loi, R.; Perra, A.; Factor, V.M.; Ledda-Columbano, G.M.; Columbano, A.; Thorgeirsson, S.S. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology 2010, 51, 1401–1409. [Google Scholar] [CrossRef]
- Frau, M.; Simile, M.M.; Tomasi, M.L.; Demartis, M.I.; Daino, L.; Seddaiu, M.A.; Brozzetti, S.; Feo, C.F.; Massarelli, G.; Solinas, G.; et al. An expression signature of phenotypic resistance to hepatocellular carcinoma identified by cross-species gene expression analysis. Cell. Oncol. (Dordr.) 2012, 35, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef]
- Martine, L.C.; Holzapfel, B.M.; McGovern, J.A.; Wagner, F.; Quent, V.M.; Hesami, P.; Wunner, F.M.; Vaquette, C.; De-Juan-Pardo, E.M.; Brown, T.D.; et al. Engineering a humanized bone organ model in mice to study bone metastases. Nat. Protoc. 2017, 12, 639–663. [Google Scholar] [CrossRef]
- Reinisch, A.; Thomas, D.; Corces, M.R.; Zhang, X.; Gratzinger, D.; Hong, W.J.; Schallmoser, K.; Strunk, D.; Majeti, R. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat. Med. 2016, 22, 812–821. [Google Scholar] [CrossRef]
- Bankert, R.B.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher, R.J., Jr.; Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS ONE 2011, 6, e24420. [Google Scholar] [CrossRef]
- Chu, Y.; Tang, H.; Guo, Y.; Guo, J.; Huang, B.; Fang, F.; Cai, J.; Wang, Z. Adipose-derived mesenchymal stem cells promote cell proliferation and invasion of epithelial ovarian cancer. Exp. Cell Res. 2015, 337, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Eyre, R.; Alferez, D.G.; Spence, K.; Kamal, M.; Shaw, F.L.; Simoes, B.M.; Santiago-Gomez, A.; Sarmiento-Castro, A.; Bramley, M.; Absar, M.; et al. Patient-derived Mammosphere and Xenograft Tumour Initiation Correlates with Progression to Metastasis. J. Mammary Gland Biol. Neoplasia 2016, 21, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Gil-Bernabe, A.M.; Ferjancic, S.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- House, C.D.; Hernandez, L.; Annunziata, C.M. Recent technological advances in using mouse models to study ovarian cancer. Front. Oncol. 2014, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Kovachev, A.; Wierling, C.; Lange, B.M.H.; Lehrach, H. Models of Models: A Translational Route for Cancer Treatment and Drug Development. Front. Oncol. 2017, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Burghoff, S.; Gong, X.; Viethen, C.; Jacoby, C.; Flogel, U.; Bongardt, S.; Schorr, A.; Hippe, A.; Homey, B.; Schrader, J. Growth and metastasis of B16-F10 melanoma cells is not critically dependent on host CD73 expression in mice. BMC Cancer 2014, 14, 898. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Li, Q.; Kazarov, A.R.; Epardaud, M.; Elpek, K.; Turley, S.J.; Hemler, M.E. Diminished metastasis in tetraspanin CD151-knockout mice. Blood 2011, 118, 464–472. [Google Scholar] [CrossRef]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Gresser, I.; Moss, J.; Woodrow, D.; Le Bousse, C.; Maury, C.; Proietti, E.; Belardelli, F. Influence of the site of tumor growth on the capacity of a low tumorigenic line of Friend erythroleukemia cells to differentiate. Am. J. Pathol. 1991, 138, 1125–1133. [Google Scholar]
- Yoon, Y.; Lee, J.; Ra, M.; Gwon, H.; Lee, S.; Kim, M.Y.; Yoo, K.C.; Sul, O.; Kim, C.G.; Kim, W.Y.; et al. Continuous Separation of Circulating Tumor Cells from Whole Blood Using a Slanted Weir Microfluidic Device. Cancers 2019, 11, 200. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.Y.; Choi, S.; Lee, S.E.; Kim, J.S.; Son, S.H.; Lim, Y.S.; Kim, B.-J.; Ryu, B.-Y.; Uversky, V.N.; Lee, Y.J.; et al. Development of a MEL Cell-Derived Allograft Mouse Model for Cancer Research. Cancers 2019, 11, 1707. https://doi.org/10.3390/cancers11111707
Kim MY, Choi S, Lee SE, Kim JS, Son SH, Lim YS, Kim B-J, Ryu B-Y, Uversky VN, Lee YJ, et al. Development of a MEL Cell-Derived Allograft Mouse Model for Cancer Research. Cancers. 2019; 11(11):1707. https://doi.org/10.3390/cancers11111707
Chicago/Turabian StyleKim, Min Young, Sungwoo Choi, Seol Eui Lee, Ji Sook Kim, Seung Han Son, Young Soo Lim, Bang-Jin Kim, Buom-Yong Ryu, Vladimir N. Uversky, Young Jin Lee, and et al. 2019. "Development of a MEL Cell-Derived Allograft Mouse Model for Cancer Research" Cancers 11, no. 11: 1707. https://doi.org/10.3390/cancers11111707
APA StyleKim, M. Y., Choi, S., Lee, S. E., Kim, J. S., Son, S. H., Lim, Y. S., Kim, B. -J., Ryu, B. -Y., Uversky, V. N., Lee, Y. J., & Kim, C. G. (2019). Development of a MEL Cell-Derived Allograft Mouse Model for Cancer Research. Cancers, 11(11), 1707. https://doi.org/10.3390/cancers11111707