Abstract
In this review we concentrate on the recent findings describing the oncogenic potential of the protein tyrosine kinase 2 (TYK2). The overview on the current understanding of TYK2 functions in cytokine responses and carcinogenesis focusses on the activation of the signal transducers and activators of transcription (STAT) 3 and 5. Insight gained from loss-of-function (LOF) gene-modified mice and human patients homozygous for Tyk2/TYK2-mutated alleles established the central role in immunological and inflammatory responses. For the description of physiological TYK2 structure/function relationships in cytokine signaling and of overarching molecular and pathologic properties in carcinogenesis, we mainly refer to the most recent reviews. Dysregulated TYK2 activation, aberrant TYK2 protein levels, and gain-of-function (GOF) TYK2 mutations are found in various cancers. We discuss the molecular consequences thereof and briefly describe the molecular means to counteract TYK2 activity under (patho-)physiological conditions by cellular effectors and by pharmacological intervention. For the role of TYK2 in tumor immune-surveillance we refer to the recent Special Issue of Cancers “JAK-STAT Signaling Pathway in Cancer”.
1. TYK2-Mediated Cytokine Signaling and Activation of STAT3 and STAT5
TYK2 was the first identified member of a family of non-receptor kinases later termed Janus kinases (JAK), which additionally comprises JAK1-3 [1,2]. JAKs are associated with cytokine and growth factor receptors and activate STAT (STAT1-4, STAT5A, STAT5B, STAT6) family members [2,3]. JAKs share four functional domains (from N- to C-terminal): (i) a four-point-one, ezrin, radixin, moesin (FERM) homology domain; (ii) an atypical Src-homology 2 (SH2) domain, both facilitating protein-protein interactions (PPIs); (iii) a kinase-like or pseudokinase (JAK homology (JH) 2) domain negatively regulating the kinase activity; and (iv) a tyrosine kinase (JH1) domain which, upon conformational changes at ligand bound receptors, increases its catalytic activity by trans-/autophosphorylation of its activation loop [2,4].
To date, the requirement for TYK2 in signaling has been shown for numerous cytokines, including distinct interleukin (ILs) and interferons (IFNs), which comprise several subtypes (i.e., type I and III IFNs). The heterodimeric cytokine receptor complexes are composed of four distinct TYK2-associated receptor chains (IFNAR1, IL-12Rβ1, IL-10R2, and IL-13Rα1) and a respective second receptor chain associated either with JAK1 or JAK2, which serves as the signal transducing chain harboring STAT docking sites. Usually, these sites contain critical tyrosine residues that are phosphorylated by JAKs upon receptor complex activation (Figure 1). TYK2 also associates with the gp130 receptor chain, yet there is no evidence that gp130-utilizing cytokines rely on TYK2 for signal transduction [5,6]. Note that comprehensive reviews [2,7] provide lists of various other receptors utilizing TYK2-STAT signaling; however, TYK2-STAT activation/utilization is frequently only biochemically assessed by phosphorylation of critical tyrosine residues and cannot be put on a level with dissected downstream cellular activities. Here we review the cytokines which clearly transduce the TYK2 phosphorylation events into downstream physiological changes (Figure 1).
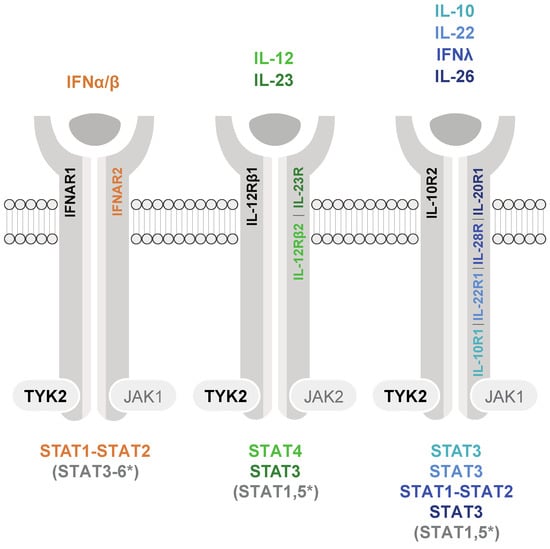
Figure 1.
Cytokine receptor families signaling with the participation of TYK2 and JAK1 or JAK2. Cytokines are depicted only upon appearance in humans and mice and proof of TYK2 dependency. The color codes indicate the major STAT(s) activated by the respective cytokines. STAT1-STAT2 heterodimers combine with IFN regulatory factor (IRF) 9 and form the interferon-stimulated gene factor 3 (ISGF3) complex; * STAT activation is dependent on cell type or of less clear biological relevance.
The biological relevance for TYK2-dependent cytokines activating STAT3 is best established for the IL-10R2 utilizing IL-22 [8,9] and the IL-12Rβ1-utilizing IL-12 and IL-23 [10,11,12]. IL-22 is a central cytokine in tissue-barrier function, wound healing, and epithelial homeostasis and repair. Cancer promoting, as well as restraining, functions were described [13,14]. IL-23 is a key mediator of inflammation, bridges innate and adaptive immune responses, and is known to support tumorigenesis and metastasis [15,16]. IL-12 is central in promoting cell-mediated immunity to infection and cancer [12]. However, this anti-carcinogenic function can be counteracted by IL-12-STAT3-promoted production of pro-carcinogenic IL-23 [17]. While STAT3 is activated by type I and III IFN stimulation in various cell types, its biological functions in the IFN responses are less clear. Growing evidence suggests that STAT3 is a negative regulator of type I IFN activities, thereby providing a pro-viral and pro-survival cellular program [18]; there is, however, also a report on an opposite, i.e., anti-viral activity of STAT3 [19]. The role of TYK2 in IL-10 signaling through STAT3 is not entirely clear and may be cell type- or context-dependent [6]. The double-edged role of IL-10 in immunity and cancer is reviewed elsewhere [9,20]. IL-19, IL-20, IL-24, and IL-26 (absent in mice) constitute a subfamily within the IL-10 cytokine family and signal primarily through activation of STAT3 [9]. Activation of TYK2 at the respective receptors has not been formally shown but can be inferred from the receptor-chain composition. As this subfamily constitutes relatively recently discovered cytokines, cellular responses are still poorly defined, and we refer to recent publications and reviews for a potential cancer connection [21,22,23]. Lastly, without specification of the cytokines involved, TYK2 via STAT3 was reported to be crucial for the mediation of cell death in an auto-inflammatory context [24].
STAT5, in contrast, is not among the primarily activated STATs downstream of TYK2 (Figure 1) and occurs dependently on cell type and differentiation stage, in response to type I and III IFNs [25,26], IL-10R2-, and IL-12Rβ1-receptor family cytokines [9,12]. Neither a cytokine-TYK2-STAT5 axis nor its significance have been established under physiological conditions.
2. Aberrant Expression and/or Activity of TYK2 in Cancers
The JAK-STAT pathway is recognized as a core cancer pathway [27] and directly contributes to all hallmarks of cancer [28]. Oncogenic JAK activity can originate from aberrant JAK expression, deregulated upstream signals, GOF mutations, or generation of fusion proteins, as well as loss of negative feedback regulation [2,29,30,31]. Initially, cancer research focused on JAK1-3, while the TYK2 impact on disease was predominantly studied in inflammatory and (auto-)immune diseases [32,33]. Table 1 summarizes the literature on constitutive or hyperactivated TYK2, as well as GOF-mutated TYK2 and the resulting activation of STATs in cancers.

Table 1.
(Hyper-)active TYK2, GOF-, or LOF-mutated TYK2 and STAT activation in various cancers and cancer cell lines.
2.1. Aberrant TYK2 Levels
In vitro studies with overexpressed JAKs revealed that aberrant TYK2 levels lead to cellular transformation with constitutive phosphorylation of STAT3 [34]. An unusually high expression of TYK2 associated with or causative for carcinogenesis (reviewed [35]) was described for various cancer cell lines and samples from patients suffering from prostate [36,37], ovarian [38], cervical [39], and breast cancer [40,41], as well as malignant peripheral nerve sheath tumors (MPNST) [42,43]. Conflictingly, lowered TYK2 levels in tumor samples and sections (tumor cells and stroma) are generally considered to be an unfavorable prognostic marker (e.g., [44], www.proteinatlas.org). This is supported by a recently published meta-analysis of JAKs and STATs in hepatocellular carcinoma (HCC) patients, where normal or higher TYK2 levels correlated with longer survival and were found in healthy tissue [45]. The underlying reason for these conflicting reports may be attributed to the anti-proliferative/pro-apoptotic and/or tumor surveillance properties of TYK2 [5], as well as the undetermined tumor cell intrinsic and extrinsic state of TYK2. The important role of TYK2 in immune-surveillance is also in line with findings in patients who carry mutated TYK2 alleles which lead to loss of TYK2, lowered TYK2 levels [46], or expression of kinase-inactive TYK2 [47,48,49], and that show primarily immunodeficiencies. Nonetheless, proteomics suggested that low TYK2 facilitates local metastasis in breast cancer [50], and a comprehensive screen for protein tyrosine kinase variants in numerous cancer cell lines identified splice variants that render TYK2 inactive [51]. On a molecular mechanistic level, the cell intrinsic tumor-promoting consequences of low TYK2 or LOF of TYK2 currently remain elusive.
2.2. Aberrant Activation of TYK2
A comprehensive list of receptors (over-)expressed in various cancer types which allows us to deduce putative upstream signals involved in hyperactivation of TYK2 was compiled recently [7]. Primary hematological neoplasm (ALCL, anaplastic large cell lymphoma; T-ALL, T cell acute lymphoblastic leukemia) patient samples and cell lines were shown to be dependent on TYK2 activated by upstream IL-10 and/or IL-22 signals and established an upregulation of anti-apoptotic BCL2 family members via STAT1 and/or STAT3 [52,53]. A similar high TYK2-STAT1/3-BCL2 axis was found in MPNST [43]. Cytotoxic T-lymphocyte-associated antigen 4 (CTLA4, CD152) is mainly expressed on T cells and is a well-established immune checkpoint. CTLA4 signaling is initiated through binding to CD80 (B7-1) or CD86 (B7-2) on the surface of antigen-presenting cells (APCs). Ectopic expression of CTLA4 was found on diverse B-cell lymphoma. Mechanistically, it was established that CD86-CTLA4 engagement resulted in recruitment/activation of TYK2, which, in turn, led to a STAT3-driven tumor-promoting transcriptional program [54]. A STAT-independent involvement of activated TYK2 in fibroblast growth factor 2 (FGF-2) mediated escape from drug-induced death was reported for a sarcoma cell line [55].
2.3. TYK2 Mutations
Oncogenic JAK2 with the prominent JAK2V617F mutation found in over 50% of myeloproliferative neoplasia (MPN) patients [56] is the paradigm for the understanding of structure/function relations of JAK activity [57,58,59] and for the general alertness of the cancer field for mutated JAK family members as potential oncogenes. TYK2 joined the club of GOF-mutated JAKs causative for patient hematopoietic malignancies only recently: In 2013, the first TYK2 GOF point mutations were found in T-ALL cell lines and characterized to have transforming capacity via STAT1 and a BCL2 family member [53]. With respect to biochemical studies, the first GOF mutation of TYK2 was V678F, which is the homologous mutation to JAK2V617F [60,61]. Until now, this mutation was not found in patients. The only mutation reported in a public cancer genome database (www.stjude.cloud) for this residue is the V678L mutation, albeit with unknown structure/function consequences. Point mutations at the TYK2 locus are distributed throughout the whole gene body, with GOF mutations—similar to the other JAKs—primarily accumulating in the JH1 and JH2 domains ([2,5] and see public databases, e.g., Genomic Data Commons of the National Cancer Institute [62,63], Catalogue of Somatic Mutations in Cancer (COSMIC [64], and cBioPortal for Cancer Genomics [65,66]).
In addition to the somatic cancer cell mutations, two GOF TYK2 germline mutations (P760L and G761V) were found in pediatric patients developing several de novo leukemias. These mutations are located in the JH2 pseudokinase domain of TYK2 and are predicted to attenuate the negative regulation on the JH1 kinase domain, leading to constitutively activated TYK2 [67].
A prominent germline TYK2 mutation is P1104A/V, which was first found to be associated with solid and hematopoietic cancers [68,69] and later with immunological and inflammatory disorders (reviewed in [5]). While analyzing MPNST tumor samples, it was proposed that TYK2P1104A is an unfavorable prognostic marker for the disease [42]. Notably, this study solely genotyped the somatic cancer cells and overlooked that this mutation impairs TYK2 catalytic activity; cellular signaling, however, is not completely abrogated, and the detected induction of BCL2 expression might favor an anti-apoptotic program [69,70]. Recent studies show that TYK2P1104A is a LOF mutation, because patients homozygous for this allele are either susceptible to microbial infection or protected from autoimmune disease [47,49,71]. These mechanistic and phenotypic features of TYK2P1104A were confirmed in independent mouse models [48,71].
2.4. TYK2 Fusion Proteins
Chromosomal rearrangements account for a number of driver kinase fusion genes in cancer [72,73,74]. The first fusion kinase involving a JAK was TEL-JAK2, consisting of a 3′ portion of JAK2 and a 5′ region of TEL, a member of the ETS transcription factor family [75]. This chromosomal translocation is found in T-ALL in patients [75] and transgenic mice expressing TEL-JAK2 develop T-cell leukemia [76]. In vitro studies with a TEL-TYK2 fusion showed constitutive activation of STAT1/3/5 and transforming capacities [77], albeit respective translocations have not yet been identified in patients. As observed for GOF-mutated JAKs, JAK2 kinase fusions occur most frequently compared to the other JAKs, which suggests that the JAK2 locus is a mutation and rearrangement hotspot [56,78,79]. The first leukemia patients carrying TYK2 fusion genes described were combinations of the TYK2 kinase domain and a part of the pseudokinase domain with 5′ portions of nucleophosmin (NPM) 1, polyadenylate binding protein (PABPC) 4, or the transcription factors MYB or NFκB2 [80,81,82]. Structurally and mechanistically, the TYK2 fusion proteins lack the negatively regulating function of the pseudokinase (JH2) domain leading to a GOF kinase activity and hyperactivity of STAT3 and depending on the cellularity also STAT1 and 5 (reviewed in [5,58,59]).
Subsequent analysis of patient samples and cell lines [83,84,85,86,87] and screening of cancer data sets revealed more than 50 chromosomal TYK2 rearrangements found mostly in hematological, but also in solid cancers [88]. For the fusions, it is currently not known if they contribute as driver oncogenes to early tumorigenesis or are rather the result of genomic instability at later tumor stages [89]. Recently, chromothripsis was identified as a new type of chromosomal rearrangement during carcinogenesis. Based on a single chromosome-shattering event and DNA repair complex, intra- and interchromosomal rearrangements, such as fusion genes, are produced within a few cell cycles. If the fusion event(s) allow for growth or survival advantages, a cancer driver gene might be generated [90,91]. Chromothripsis was assigned to genomic alterations in childhood cancer [92], and mechanistically it is caused by defects in the nuclear envelope composition or formation and failures during mitosis [93]. It is tempting to speculate that the remarkably high number of described TYK2 fusions were—at least in part—generated through chromothripsis and thus might act as driver mutations.
3. Tumor-Promoting Activities of (Hyper-)Active TYK2
The molecular contribution of TYK2 signaling and known protein–protein interactions to the hallmarks of cancer were reviewed previously [5,28]. Here, we highlight the latest findings on the consequences of TYK2 hyperactivity in cancer cells.
3.1. TYK2 Activation of (Oncogenic) STAT Signaling
As shown in Figure 1, the heterodimeric cytokine receptors with engagement of TYK2 are capable of activating all STATs. Hyperactive, GOF-mutated TYK2 or TYK2 fusions in oncogenic settings preferentially lead to aberrant activation of STAT1, STAT3, and STAT5. The oncogenic potential of STAT3 and STAT5 was recognized early on and is well documented [94,95]. STAT1 was initially considered to exert tumor suppressor functions, and its oncogenic potential emerged more recently [96,97,98].
STAT1/3/5 were found hyperactivated in patient-tailored cell lines with activated TYK2 [53], as well as carrying somatic or germline TYK2 GOF mutations [53,67] or TYK2-NPM1 and -NFkB2 fusions [80,82]. In other tumor samples or experimental tissue culture settings, STAT3 only, or other dual combinations of activated STAT1/3/5, are described (see Table 1).
Interestingly, TYK2 does not only phosphorylate the major phosphorylation site Y705 in STAT3, but also Y640, which represses STAT3 activation [99]. This phosphorylation site in STAT3 is often mutated in cancers [100,101]. Neither the general (patho-)physiological impact nor the contribution to malignancies of this phosphorylation event are currently known.
3.2. TYK2 Stimulation of Tumor Cell Invasion
The families of tight junction proteins claudins (CLDNs) and of matrix metalloproteinases (MMPs) are central for the invasion of tumor cells and, in consequence, metastasis formation [106,107]. Recent studies show that, in liver and lung carcinoma, high levels of CLDN9/12/17 caused activation of TYK2 and STAT1/3 and promoted metastasis [102,104,105]. The promoters of various MMP genes harbor STAT binding sites, and many MMPs are transcriptionally activated through TYK2-associated cytokine receptors [108,109]. Gene-targeted mice revealed that TYK2 and STAT1 are required for expression of MMP2/9/14 under inflammatory conditions [110]. Biochemical studies showed that, dependent on context and inflammatory conditions, MMP1/3 induction involves STAT1 alone [108] or also STAT3 [111]. In a hematopoietic tumor TYK2-STAT3 induced MMP9 and tumor cell invasiveness [54] and in a solid tumor TYK2-STAT3 signaling induced MMP1 expression [103].
The urokinase-type plasminogen activator (uPA)/receptor (uPAR) system is central for a cascade of proteolytic events, including activation of MMPs, which allow for tumor cell migration and metastasis [112]. Signaling via uPAR involves TYK2 and PI3K [113], and, at the post-transcriptional level, TYK2 inhibits the accumulation of plasminogen activator inhibitor (PAI) 2 [114]. In prostate cancer, high levels of TYK2 correlate with invasion and metastasis [36,37]. In an ovarian cancer cell line pY-STAT3 co-localizes with TYK2 and JAK2 at focal adhesions, and hyperactive STAT3 was shown to promote cancer cell motility [38]. Without providing molecular details, a mouse model for aggressive lymphoma showed reduced tumor cell invasiveness upon loss of TYK2 [115]. In addition, without providing molecular insights, a siRNA screen assessing the role of the tyrosine kinome in metastasis formation identified TYK2 as a promoter of invadopodia, which are cellular structures characteristic for tumor cell migration [116,117]. Connexin43 (Cx43) is the most widely expressed member of a large family of transmembrane proteins involved in gap junction formation. Cx43 can be both pro- and anti-tumorigenic, e.g., by promoting invasion and metastasis and by acting as a tumor suppressor [118,119]. TYK2 was found to play a dual role in regulation of Cx43: On the one hand, TYK2 is capable of directly phosphorylating Cx43, thereby decreasing its stability; on the other hand, angiotensin II-activated TYK2 increased Cx43 levels in a STAT3-dependent manner [120]. This regulatory loop has not yet been studied in the context of carcinogenesis. Furthermore, knockdown of TYK2 reduced migration of breast cancer cell lines [50].
3.3. TYK2 Prevention of Apoptosis
IFNs in general are capable of promoting apoptosis of cancer cells [121]; hence, provided that IFN stimulus and responsiveness in the tumor is given, TYK2 acts tumor suppressive. Tumor cells are able to resist cell death by upregulation of anti-apoptotic BCL-2 family members [122,123]. TYK2 was shown to drive either in a STAT1- and/or a STAT3-dependent manner or in a STAT-independent but ERK1/2-dependent manner high expression of BCL-2 [43,53,55] or its family members BCL-2L1 [54] and MCL1 [52,55]. In contrast, an in vitro study demonstrated that TYK2 physically interacts with SIVA-1 and promotes SIVA-1 mediated apoptosis, as well as inhibits BCL-2 [124].
3.4. TYK2 Crosstalk to Oncogenes and Proto-Oncogenic Pathways
In a mouse model of ALCL, as well as in patient cells, TYK2 showed co-operativity with the oncogenic fusion kinase NPM-ALK [52]. In contrast, no co-operation of TYK2 with mutated FLT3-ITD or JAK2V617F in MPN mouse models was found [125,126]. The latter is consistent with the observation that, in JAK2V617F MPN patients (see below) resistant to pharmacological JAK2 inhibition, only JAK1, and not TYK2, leads to heterodimeric STAT activation, despite both kinases show equal tyrosine phosphorylation at the activating loop [127]. This is to be expected, since, in contrast to the other JAKs, loss of TYK2 at heterodimeric JAK-associated cytokine receptors leads only to a partial impairment in signaling [5,6], and, as experimentally described for the IFNAR receptor, TYK2 is the subordinated JAK at cytokine receptors [128,129].
Early biochemical studies suggest that, upon type I IFN treatment, TYK2 interacts with various proto-oncogenes, including the guanine nucleotide exchange factor 1 VAV, the E3 ubiquitin-protein ligase C-CBL, and the SRC family tyrosine kinases FYN and LYN [130,131,132,133,134]. The importance of these PPIs for tumorigenesis is currently unknown. In cancer samples or cell lines, TYK2 was found to cooperate with other oncogenic effectors and pathways, such as the RAF/ERK [53,55,61], MAPKs [135], PIM1/2 [84], and PI3K/AKT/mTOR pathway [36,53,61]. Reported solely in the context of skin inflammation is the TYK2-STAT3 requirement for expression of IκBζ (encoded by NFKBIZ) [136]; however, emerging reports suggest cell-intrinsic oncogenic, as well as tumor-suppressive, functions of IκBζ [137].
The mapped and predicted PPIs of TYK2 based on proteomics [138,139] and next generation sequencing (NGS) are accessible at various open-source databases (for a review, see [140]). The TYK2 kinase domain and a STAT3-based reporter system were used to establish the first mammalian two hybrid kinase substrate sensor (KISS) screening platform [141,142]. These databases and the screening approaches should be systematically exploited to further define and fine tune the TYK2 interactome in health and disease.
4. Deactivation and Stabilization of TYK2 under (Patho-)Physiological Conditions
JAK activity is counter regulated by molecule-intrinsic events, such as post-translational modifications (PTMs) and the inhibitory function of the pseudokinase domain [143] as well as by extrinsic inhibitory regulators, such as suppressor of cytokine signaling (SOCS) proteins and protein tyrosine phosphatases (PTPs) [144].
Databases [145,146] provide curated PTMs, but with the exception of the well described activating phosphotyrosines, there is still a lack of information on the properties of JAKs that are defined by PTMs. For TYK2, ubiquitination and phosphorylation are detected at multiple residues and discussed in the context of stability/decay (PhosphoSitePlus®, [146]), albeit the (patho-) physiological relevance is unknown.
SOCS proteins are encoded by STAT target genes and are negative feedback inhibitors of JAK signaling. SOCS1 and 3 are the most potent JAK inhibitors because, in addition to recruitment of JAKs to E3 ubiquitination/degradation mediated by all SOCS family members, they also harbor a kinase inhibitory region (KIR), which efficiently shuts down JAK activity by binding to the JH1 domain [147]. Activated JAKs and cytokine receptor chains are dephosphorylated by multiple PTPs [148]. The current literature regarding deactivation of TYK2 by SOCS1/3, the PTPs PTB1B and SHP1, as well as the global impact of SOCS and PTP family members in cancer are reviewed elsewhere [5,149,150,151].
In vitro studies showed that in hematopoietic tumor cells the PTP SHP1 suppresses growth via accelerating the TYK2 protein degradation [152]. In lung cancer cells, overexpression of the E3 ubiquitin ligase seven-in-absentia-2 (SIAH2) accelerates the proteasomal degradation of TYK2, thereby attenuating STAT3 signaling [103].
HSP90 is a chaperone supporting folding, stability, and function of many client proteins, including JAKs and STATs [153,154,155]. Cancer cells frequently use HSP90 to stabilize and/or increase the function of numerous oncogenes, and HSP90 inhibitors have been studied as anticancer drugs for more than two decades [156,157]. Physical interaction of HSP90 with TYK2 was demonstrated in cancer cell lines and confirmed in a proteome-wide assessment of the HSP90 interactome [158,159]. HSP90 inhibitor treatments in various tumor settings showed beneficial effects by reducing the activity of TYK2 or its fusion proteins [158,160,161].
An emerging field is the involvement of noncoding RNAs in the regulation of the JAK-STAT pathway in carcinogenesis [162,163,164]. Recently, the long noncoding RNA (lncRNA) MEG3 in concert with a microRNA (miR-147) was reported to modulate JAK-STAT signaling in chronic myeloid leukemia (CML). Interestingly, the lncRNA was found to physically interact with TYK2, JAK2, and STAT3, thereby diminishing the activity level of STAT3 (and STAT5) [165].
5. Pharmaceutical TYK2 Inhibition
The first selective JAK inhibitor (JAKinib) to be tested in humans was tofacitinib, which potently inhibits JAK3 and JAK1, and, to a lesser extent JAK2, and has little effect on TYK2 [166]. Historically, JAKinibs were developed as immunosuppressive drugs for the clinical use in organ transplants and autoimmune diseases [167]. The success story of ruxolitinib, a JAK2 and JAK1 inhibitor which was the first JAKinib approved for treatment of a hematopoietic malignancy, pushed the perception of JAKinibs as anticancer drugs [168,169]. For insight in development and clinical use, as well as side effects of JAKinibs, we refer to the most recent reviews [170,171,172,173].
TYK2inibs are mainly envisaged as therapeutics for treatment of autoimmune and inflammatory diseases [33,174], in which JAKinib selectivity is currently considered not to be of utmost importance [175]. As for the other JAKinibs, the first generation TYK2inibs are directed to the JH1 domain and compete with ATP in binding to the enzymatic pocket. These inhibitors are potent in inhibiting wildtype (overexpressed) TYK2, mutated (hyperactive) TYK2, and TYK2 fusion proteins harboring the JH1 domain. Since the JAKs show high homology in the JH1 domain, it is hard to develop ATP-competing inhibitors with high selectivity for one particular JAK family member [170,172]. A next-generation inhibitor of TYK2 is directed against the JH2 domain and recently passed the phase II clinical trial for psoriasis treatment [176]. A comprehensive report on the high selectivity and the biological effects of this TYK2inib in mouse models, as well as its efficacy in human cells collected from autoimmune patients, was recently published [177]. JH2-specific TYK2inibs are currently further improved, and additional compounds are being developed [178,179,180,181]. The only TYK2inib reported and successfully tested to block TYK2 activity in an oncogenic setting is a JH1-specific TYK2inib [135]. Notably, JH2 domain inhibitors might not be working for treatment of diseases driven by TYK2 fusion genes missing parts of the JH2 domain.
6. Conclusions and Future Perspectives
Since the discovery of TYK2 and the JAK-STAT signaling paradigm in the early 1990s, enormous progress has been made in the structural and functional understanding of the linear JAK-STAT axis and the crosstalk of JAKs or STATs to other signaling hubs, as well as the cell type-specific contributions of JAKs and STATs in health and disease. The striking phenotypical similarities between mouse models deficient for TYK2 or engineered to express kinase-inactive TYK2 and human patients carrying the respective germline mutations established TYK2 as a fundamental component in both innate and adaptive immunity. The (patho-)physiological and molecular pathway similarities of TYK2 in human and mice allow for highly informative comparative biomedical studies and efficient translation of basic molecular insights into clinical applications. The use of TYK2inibs in the treatment of immunological and inflammatory diseases is within reach [182] and is also attractive for malignancies with the involvement of hyperactivated TYK2. The role of TYK2 and GOF-mutated TYK2 upstream of oncogenic STAT3—and, less frequently, STAT1—is established, while, up to now, no mechanistic evidence for an oncogenic TYK2-STAT5 axis is given. Mouse models as genetic mimics of kinase-inhibited TYK2 exist [48,71,183,184] and are currently exploited to further dissect the kinase-dependent from the scaffolding functions of TYK2.
In a short-term perspective, work should concentrate on the use of refined TYK2 mouse models that allow studying the kinase-independent and cell type-specific functions, in order to fully in vivo assess TYK2inibs with respect to their benefits and unwanted side effects. Mouse models to study the consequences of aberrant high TYK2 and GOF-mutated TYK2 are underway (K. Wöss, T. Rülicke et al., unpublished). For pharmacological intervention with oncogenic TYK2, TYK2inibs with the highest possible selectivity are required, and efforts should focus on the further development and in vivo testing of these next-generation TYK2inibs.
In a long-term perspective, the further understanding of the TYK2 function requires the in-depth elucidation of the PTMs and the interactome of TYK2 under spatiotemporal conditions. Additionally, computational modelling and structure predictions (e.g., [185]) should complement the attempts to determine the holo-crystal structure of TYK2 and to use high-resolution imaging (e.g., [186]) to gain insight into the structural features of full-length wildtype and mutated TYK2, as well as its conformation bound to various cytokine receptors.
Author Contributions
K.W., S.M.-M., and M.M. designed the draft of the review and performed the literature search. K.W. and S.M.-M. compiled the table, and N.S. designed the figure. S.M.-M., M.M., N.S., and B.S. provided the final version of the manuscript.
Funding
This work was funded by the Austrian Science Fund FWF DK W1212, SFB F6101 and F6106, and DocFund DOC32-B28. We are thankful to Tanja Bulat for critically reading the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Krolewski, J.J.; Lee, R.; Eddy, R.; Shows, T.B.; Dalla-Favera, R. Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene 1990, 5, 277–282. [Google Scholar] [PubMed]
- Hammaren, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E. The JAK-STAT Pathway at Twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Leitner, N.R.; Witalisz-Siepracka, A.; Strobl, B.; Muller, M. Tyrosine kinase 2-Surveillant of tumours and bona fide oncogene. Cytokine 2017, 89, 209–218. [Google Scholar] [CrossRef]
- Strobl, B.; Stoiber, D.; Sexl, V.; Mueller, M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front. Biosci. 2011, 16, 3214–3232. [Google Scholar] [CrossRef]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and biology of IL-23 and IL-27: Related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef]
- Rutz, S.; Wang, X.; Ouyang, W. The IL-20 subfamily of cytokines--from host defence to tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Gronke, K.; Diefenbach, A. A catch-22: Interleukin-22 and cancer. Eur. J. Immunol. 2018, 48, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Savan, R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev. 2014, 25, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Yan, J.M.; Smyth, M.J.; Teng, M.W.L. Interleukin (IL)-12 and IL-23 and Their Conflicting Roles in Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a028530. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Tsai, M.H.; Pai, L.M.; Lee, C.K. Fine-Tuning of Type I Interferon Response by STAT3. Front. Immunol. 2019, 10, 1448. [Google Scholar] [CrossRef]
- Mahony, R.; Gargan, S.; Roberts, K.L.; Bourke, N.; Keating, S.E.; Bowie, A.G.; O’Farrelly, C.; Stevenson, N.J. A novel anti-viral role for STAT3 in IFN-alpha signalling responses. Cell. Mol. Life Sci. 2017, 74, 1755–1764. [Google Scholar] [CrossRef]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Li, C.F.; Yeh, C.H.; Chang, M.S.; Hsing, C.H. Interleukin-19 in breast cancer. Clin. Dev. Immunol. 2013, 2013, 294320. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Hruz, P.; Kaymak, T. The Interleukin-20 Cytokines in Intestinal Diseases. Front. Immunol. 2018, 9, 1373. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Tang, Q.; Zhang, C.; Wu, J.; Gu, C.; Wu, Z.; Li, X. IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PLoS ONE 2013, 8, e63588. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Fu, A.K.; Ip, F.C.; Ng, H.K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V. IFN-lambdas. Curr. Opin. Immunol. 2011, 23, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G. The molecular basis for differential type I interferon signaling. J. Biol. Chem. 2017, 292, 7285–7294. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, E.; Staudt, L.M.; Green, A.R. Janus kinase deregulation in leukemia and lymphoma. Immunity 2012, 36, 529–541. [Google Scholar] [CrossRef]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Knoops, L.; Hornakova, T.; Royer, Y.; Constantinescu, S.N.; Renauld, J.C. JAK kinases overexpression promotes in vitro cell transformation. Oncogene 2008, 27, 1511–1519. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ubel, C.; Mousset, S.; Trufa, D.; Sirbu, H.; Finotto, S. Establishing the role of tyrosine kinase 2 in cancer. Oncoimmunology 2013, 2, e22840. [Google Scholar] [CrossRef]
- Ide, H.; Nakagawa, T.; Terado, Y.; Kamiyama, Y.; Muto, S.; Horie, S. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem. Biophys. Res. Commun. 2008, 369, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Mesquita, D.; Barros-Silva, J.D.; Jeronimo, C.; Henrique, R.; Morais, A.; Paulo, P.; Teixeira, M.R. Uncovering potential downstream targets of oncogenic GRPR overexpression in prostate carcinomas harboring ETS rearrangements. Oncoscience 2015, 2, 497–507. [Google Scholar] [CrossRef][Green Version]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (STAT) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lv, J.; Yu, L.; Zhu, X.; Wu, J.; Zou, S.; Jiang, S. Proteomic identification of differentially-expressed proteins in squamous cervical cancer. Gynecol. Oncol. 2009, 112, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Christy, J.; Priyadharshini, L. Differential expression analysis of JAK/STAT pathway related genes in breast cancer. Meta Gene 2018, 16, 122–129. [Google Scholar] [CrossRef]
- Song, X.C.; Fu, G.; Yang, X.; Jiang, Z.; Wang, Y.; Zhou, G.W. Protein expression profiling of breast cancer cells by dissociable antibody microarray (DAMA) staining. Mol. Cell. Proteom. 2008, 7, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Kaushal, M.; Sharma, M.K.; Dahiya, S.; Pekmezci, M.; Perry, A.; Gutmann, D.H. Clinical genomic profiling identifies TYK2 mutation and overexpression in patients with neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Cancer 2017, 123, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.J.; Godec, A.; Zhang, X.C.; Zhu, C.G.; Shao, J.Y.; Tao, Y.; Bu, X.Z.; Hirlbe, A.C. TYK2 promotes malignant peripheral nerve sheath tumor progression through inhibition of cell death. Cancer Med. 2019, 8, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Wang, X.; Liao, X.; Yu, T.; Gong, Y.; Zhang, L.; Huang, J.; Yang, C.; Han, C.; Yu, L.; Zhu, G.; et al. Analysis of clinical significance and prospective molecular mechanism of main elements of the JAK/STAT pathway in hepatocellular carcinoma. Int. J. Oncol. 2019, 55, 805–822. [Google Scholar] [CrossRef]
- Nemoto, M.; Hattori, H.; Maeda, N.; Akita, N.; Muramatsu, H.; Moritani, S.; Kawasaki, T.; Maejima, M.; Ode, H.; Hachiya, A.; et al. Compound heterozygous TYK2 mutations underlie primary immunodeficiency with T-cell lymphopenia. Sci. Rep. 2018, 8, 6956. [Google Scholar] [CrossRef]
- Boisson-Dupuis, S.; Ramirez-Alejo, N.; Li, Z.; Patin, E.; Rao, G.; Kerner, G.; Lim, C.K.; Krementsov, D.N.; Hernandez, N.; Ma, C.S.; et al. Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci. Immunol. 2018, 3, eaau8714. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149. [Google Scholar] [CrossRef]
- Kerner, G.; Ramirez-Alejo, N.; Seeleuthner, Y.; Yang, R.; Ogishi, M.; Cobat, A.; Patin, E.; Quintana-Murci, L.; Boisson-Dupuis, S.; Casanova, J.L.; et al. Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc. Natl. Acad. Sci. USA 2019, 116, 10430–10434. [Google Scholar] [CrossRef]
- Sang, Q.X.; Man, Y.G.; Sung, Y.M.; Khamis, Z.I.; Zhang, L.; Lee, M.H.; Byers, S.W.; Sahab, Z.J. Non-receptor tyrosine kinase 2 reaches its lowest expression levels in human breast cancer during regional nodal metastasis. Clin. Exp. Metastasis 2012, 29, 143–153. [Google Scholar] [CrossRef]
- Ruhe, J.E.; Streit, S.; Hart, S.; Wong, C.H.; Specht, K.; Knyazev, P.; Knyazeva, T.; Tay, L.S.; Loo, H.L.; Foo, P.; et al. Genetic alterations in the tyrosine kinase transcriptome of human cancer cell lines. Cancer Res. 2007, 67, 11368–11376. [Google Scholar] [CrossRef]
- Prutsch, N.; Gurnhofer, E.; Suske, T.; Liang, H.C.; Schlederer, M.; Roos, S.; Wu, L.C.; Simonitsch-Klupp, I.; Alvarez-Hernandez, A.; Kornauth, C.; et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia 2019, 33, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Tyner, J.W.; Gutierrez, A.; Ngo, V.N.; Glover, J.; Chang, B.H.; Yost, A.; Ma, W.; Fleischman, A.G.; Zhou, W.; et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013, 3, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Lahtz, C.; Nagao, T.; Song, J.Y.; Chan, W.C.; Lee, H.; Yue, C.; Look, T.; Mulfarth, R.; Li, W.; et al. CTLA4 Promotes Tyk2-STAT3-Dependent B-cell Oncogenicity. Cancer Res. 2017, 77, 5118–5128. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.R.; Lyons-Lewis, J.; Seckl, M.J.; Costa-Pereira, A.P. A Novel Requirement for Janus Kinases as Mediators of Drug Resistance Induced by Fibroblast Growth Factor-2 in Human Cancer Cells. PLoS ONE 2011, 6, e19861. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Hubbard, S.R. Mechanistic Insights into Regulation of JAK2 Tyrosine Kinase. Front. Endocrinol. 2017, 8, 361. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Silvennoinen, O.; Hubbard, S.R. Molecular insights into regulation of JAK2 in myeloproliferative neoplasms. Blood 2015, 125, 3388–3392. [Google Scholar] [CrossRef]
- Gakovic, M.; Ragimbeau, J.; Francois, V.; Constantinescu, S.N.; Pellegrini, S. The Stat3-activating Tyk2 V678F mutant does not up-regulate signaling through the type I interferon receptor but confers ligand hypersensitivity to a homodimeric receptor. J. Biol. Chem. 2008, 283, 18522–18529. [Google Scholar] [CrossRef]
- Staerk, J.; Kallin, A.; Demoulin, J.B.; Vainchenker, W.; Constantinescu, S.N. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: Cross-talk with IGF1 receptor. J. Biol. Chem. 2005, 280, 41893–41899. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.A.; Ferretti, V.; Grossman, R.L.; Staudt, L.M. The NCI Genomic Data Commons as an engine for precision medicine. Blood 2017, 130, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Waanders, E.; Scheijen, B.; Jongmans, M.C.; Venselaar, H.; van Reijmersdal, S.V.; van Dijk, A.H.; Pastorczak, A.; Weren, R.D.; van der Schoot, C.E.; van de Vorst, M.; et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia 2017, 31, 821–828. [Google Scholar] [CrossRef]
- Kaminker, J.S.; Zhang, Y.; Waugh, A.; Haverty, P.M.; Peters, B.; Sebisanovic, D.; Stinson, J.; Forrest, W.F.; Bazan, J.F.; Seshagiri, S.; et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007, 67, 465–473. [Google Scholar] [CrossRef]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.E.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef]
- Li, Z.; Gakovic, M.; Ragimbeau, J.; Eloranta, M.L.; Ronnblom, L.; Michel, F.; Pellegrini, S. Two rare disease-associated Tyk2 variants are catalytically impaired but signaling competent. J. Immunol. 2013, 190, 2335–2344. [Google Scholar] [CrossRef]
- Gorman, J.A.; Hundhausen, C.; Kinsman, M.; Arkatkar, T.; Allenspach, E.J.; Clough, C.; West, S.E.; Thomas, K.; Eken, A.; Khim, S.; et al. The TYK2-P1104A Autoimmune Protective Variant Limits Coordinate Signals Required to Generate Specialized T Cell Subsets. Front. Immunol. 2019, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.S.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.J.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Medves, S.; Demoulin, J.B. Tyrosine kinase gene fusions in cancer: Translating mechanisms into targeted therapies. J. Cell. Mol. Med. 2012, 16, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffe, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef]
- Carron, C.; Cormier, F.; Janin, A.; Lacronique, V.; Giovannini, M.; Daniel, M.T.; Bernard, O.; Ghysdael, J. TEL-JAK2 transgenic mice develop T-cell leukemia. Blood 2000, 95, 3891–3899. [Google Scholar] [CrossRef]
- Lacronique, V.; Boureux, A.; Monni, R.; Dumon, S.; Mauchauffe, M.; Mayeux, P.; Gouilleux, F.; Berger, R.; Gisselbrecht, S.; Ghysdael, J.; et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood 2000, 95, 2076–2083. [Google Scholar] [CrossRef]
- Ho, K.; Valdez, F.; Garcia, R.; Tirado, C.A. JAK2 Translocations in hematological malignancies: Review of the literature. J. Assoc. Genet. Technol. 2010, 36, 107–109. [Google Scholar]
- Levavi, H.; Tripodi, J.; Marcellino, B.; Mascarenhas, J.; Jones, A.V.; Cross, N.C.P.; Gruenstein, D.; Najfeld, V. A Novel t(1;9)(p36;p24.1) JAK2 Translocation and Review of the Literature. Acta Haematol. 2019, 142, 105–112. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Keeton, E.K.; Ye, M.; Casas-Selves, M.; Chen, H.; Dillman, K.S.; Gale, R.E.; Stengel, C.; Zinda, M.; Linch, D.C.; et al. Next-generation sequencing identifies a novel ELAVL1-TYK2 fusion gene in MOLM-16, an AML cell line highly sensitive to the PIM kinase inhibitor AZD1208. Leuk. Lymphoma 2016, 57, 2927–2929. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef]
- Roberts, K.G.; Yang, Y.L.; Payne-Turner, D.; Lin, W.; Files, J.K.; Dickerson, K.; Gu, Z.; Taunton, J.; Janke, L.J.; Chen, T.; et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. 2017, 1, 1657–1671. [Google Scholar] [CrossRef]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; Garcia-Muret, M.P.; et al. Identification of gene mutations and fusion genes in patients with Sezary Syndrome. J. Investig. Dermatol. 2016, 136, 1490–1499. [Google Scholar] [CrossRef]
- Kim, P.; Zhou, X. FusionGDB: Fusion gene annotation DataBase. Nucleic Acids Res. 2019, 47, D994–D1004. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Muller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012, 1, 65–72. [Google Scholar] [CrossRef]
- Meissl, K.; Macho-Maschler, S.; Muller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z. STAT1 in cancer: Friend or foe? Discov. Med. 2017, 24, 19–29. [Google Scholar]
- Mori, R.; Wauman, J.; Icardi, L.; Van der Heyden, J.; De Cauwer, L.; Peelman, F.; De Bosscher, K.; Tavernier, J. TYK2-induced phosphorylation of Y640 suppresses STAT3 transcriptional activity. Sci. Rep. 2017, 7, 15919. [Google Scholar] [CrossRef]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagstrom, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.M.; Feng, L.S.; Cui, J.W. Increased expression of claudin-17 promotes a malignant phenotype in hepatocyte via Tyk2/Stat3 signaling and is associated with poor prognosis in patients with hepatocellular carcinoma. Diagn. Pathol. 2018, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Chen, Y.; Ginter, T.; Schafer, C.; Buchwald, M.; Schmitz, L.M.; Klitzsch, J.; Schutz, A.; Haitel, A.; Schmid, K.; et al. SIAH2 antagonizes TYK2-STAT3 signaling in lung carcinoma cells. Oncotarget 2014, 5, 3184–3196. [Google Scholar] [CrossRef]
- Liu, H.; Wang, M.; Liang, N.; Guan, L. Claudin-9 enhances the metastatic potential of hepatocytes via Tyk2/Stat3 signaling. Turk. J. Gastroenterol. 2019, 30, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.M.; Feng, L.S.; Cui, J.W. Increased expression of claudin-12 promotes the metastatic phenotype of human bronchial epithelial cells and is associated with poor prognosis in lung squamous cell carcinoma. Exp. Ther. Med. 2019, 17, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Siegel, P.M. The role of claudins in cancer metastasis. Oncogene 2017, 36, 1176–1190. [Google Scholar] [CrossRef]
- Cutler, S.J.; Doecke, J.D.; Ghazawi, I.; Yang, J.; Griffiths, L.R.; Spring, K.J.; Ralph, S.J.; Mellick, A.S. Novel STAT binding elements mediate IL-6 regulation of MMP-1 and MMP-3. Sci. Rep. 2017, 7, 8526. [Google Scholar] [CrossRef]
- Fanjul-Fernandez, M.; Folgueras, A.R.; Cabrera, S.; Lopez-Otin, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 2010, 1803, 3–19. [Google Scholar] [CrossRef]
- Costantino, G.; Egerbacher, M.; Kolbe, T.; Karaghiosoff, M.; Strobl, B.; Vogl, C.; Helmreich, M.; Muller, M. Tyk2 and signal transducer and activator of transcription 1 contribute to intestinal I/R injury. Shock 2008, 29, 238–244. [Google Scholar] [CrossRef]
- Araki, Y.; Tsuzuki Wada, T.; Aizaki, Y.; Sato, K.; Yokota, K.; Fujimoto, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Histone Methylation and STAT-3 Differentially Regulate Interleukin-6-Induced Matrix Metalloproteinase Gene Activation in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol. 2016, 68, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Kusch, A.; Tkachuk, S.; Haller, H.; Dietz, R.; Gulba, D.C.; Lipp, M.; Dumler, I. Urokinase stimulates human vascular smooth muscle cell migration via a phosphatidylinositol 3-kinase-Tyk2 interaction. J. Biol. Chem. 2000, 275, 39466–39473. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.; Miller, I.; Grunert, T.; Marchetti-Deschmann, M.; Vogl, C.; O’Donoghue, N.; Dunn, M.J.; Kolbe, T.; Allmaier, G.; Gemeiner, M.; et al. The impact of tyrosine kinase 2 (Tyk2) on the proteome of murine macrophages and their response to lipopolysaccharide (LPS). Proteomics 2008, 8, 3469–3485. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.; Muller, M.; Freissmuth, M.; Sexl, V.; Stoiber, D. Commentary on H. Ide. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem. Biophys. Res. Commun. 2008, 366, 869–870. [Google Scholar] [CrossRef]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef]
- Revach, O.Y.; Sandler, O.; Samuels, Y.; Geiger, B. Cross-Talk between Receptor Tyrosine Kinases AXL and ERBB3 Regulates Invadopodia Formation in Melanoma Cells. Cancer Res. 2019, 79, 2634–2648. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Bonacquisti, E.E.; Nguyen, J. Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett. 2019, 442, 439–444. [Google Scholar] [CrossRef]
- Li, H.; Spagnol, G.; Zheng, L.; Stauch, K.L.; Sorgen, P.L. Regulation of Connexin43 Function and Expression by Tyrosine Kinase 2. J. Biol. Chem. 2016, 291, 15867–15880. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Gamero, A.M. Interferons as inducers of apoptosis in malignant cells. J. Interferon Cytokine Res. 2013, 33, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, H.K.; Shide, K.; Kameda, T.; Matsunaga, T.; Shimoda, K. Tyrosine kinase 2 interacts with the proapoptotic protein Siva-1 and augments its apoptotic functions. Biochem. Biophys. Res. Commun. 2010, 400, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Shibata, F.; Kumagai, H.; Shimoda, K.; Kitamura, T. Tyk2 is dispensable for induction of myeloproliferative disease by mutant FLT3. Int. J. Hematol. 2006, 84, 54–59. [Google Scholar] [CrossRef]
- Yamaji, T.; Shide, K.; Kameda, T.; Sekine, M.; Kamiunten, A.; Hidaka, T.; Kubuki, Y.; Shimoda, H.; Abe, H.; Miike, T.; et al. Loss of Tyrosine Kinase 2 Does Not Affect the Severity of Jak2V617F-induced Murine Myeloproliferative Neoplasm. Anticancer Res. 2017, 37, 3841–3847. [Google Scholar] [CrossRef]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012, 489, 155–159. [Google Scholar] [CrossRef]
- Kohlhuber, F.; Rogers, N.C.; Watling, D.; Feng, J.; Guschin, D.; Briscoe, J.; Witthuhn, B.A.; Kotenko, S.V.; Pestka, S.; Stark, G.R.; et al. A JAK1/JAK2 chimera can sustain alpha and gamma interferon responses. Mol. Cell. Biol. 1997, 17, 695–706. [Google Scholar] [CrossRef]
- Briscoe, J.; Rogers, N.C.; Witthuhn, B.A.; Watling, D.; Harpur, A.G.; Wilks, A.; Stark, G.R.; Ihle, J.N.; Kerr, I.M. Kinase-negative mutants of JAK1 can sustain interferon-gamma-inducible gene expression but not an antiviral state. EMBO J. 1996, 15, 799–809. [Google Scholar] [CrossRef]
- Adam, L.; Bandyopadhyay, D.; Kumar, R. Interferon-alpha signaling promotes nucleus-to-cytoplasmic redistribution of p95Vav, and formation of a multisubunit complex involving Vav, Ku80, and Tyk2. Biochem. Biophys. Res. Commun. 2000, 267, 692–696. [Google Scholar] [CrossRef]
- Uddin, S.; Gardziola, C.; Dangat, A.; Yi, T.; Platanias, L.C. Interaction of the c-cbl proto-oncogene product with the Tyk-2 protein tyrosine kinase. Biochem. Biophys. Res. Commun. 1996, 225, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Grumbach, I.M.; Yi, T.; Colamonici, O.R.; Platanias, L.C. Interferon alpha activates the tyrosine kinase Lyn in haemopoietic cells. Br. J. Haematol. 1998, 101, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Sher, D.A.; Alsayed, Y.; Pons, S.; Colamonici, O.R.; Fish, E.N.; White, M.F.; Platanias, L.C. Interaction of p59(fyn) with interferon-activated Jak kinases. Biochem. Biophys. Res. Commun. 1997, 235, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Sweet, M.; Colamonici, O.R.; Krolewski, J.J.; Platanias, L.C. The vav proto-oncogene product (p95vav) interacts with the Tyk-2 protein tyrosine kinase. FEBS Lett. 1997, 403, 31–34. [Google Scholar] [CrossRef]
- Akahane, K.; Li, Z.; Etchin, J.; Berezovskaya, A.; Gjini, E.; Masse, C.E.; Miao, W.; Rocnik, J.; Kapeller, R.; Greenwood, J.R.; et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2017, 177, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Tawa, K.; Ohgakiuchi, Y.; Sato, A.; Saino, Y.; Hirashima, K.; Minoguchi, H.; Kitai, Y.; Kashiwakura, J.I.; Shimoda, K.; et al. IkappaB-zeta Expression Requires Both TYK2/STAT3 Activity and IL-17-Regulated mRNA Stabilization. Immunohorizons 2019, 3, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Willems, M.; Dubois, N.; Musumeci, L.; Bours, V.; Robe, P.A. IkappaBzeta: An emerging player in cancer. Oncotarget 2016, 7, 66310–66322. [Google Scholar] [CrossRef]
- Luck, K.; Sheynkman, G.M.; Zhang, I.; Vidal, M. Proteome-Scale Human Interactomics. Trends Biochem. Sci. 2017, 42, 342–354. [Google Scholar] [CrossRef]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Jensen, L.J. Protein-protein interaction databases. Methods Mol. Biol. 2015, 1278, 39–56. [Google Scholar] [CrossRef]
- Lievens, S.; Gerlo, S.; Lemmens, I.; De Clercq, D.J.; Risseeuw, M.D.; Vanderroost, N.; De Smet, A.S.; Ruyssinck, E.; Chevet, E.; Van Calenbergh, S.; et al. Kinase Substrate Sensor (KISS), a mammalian in situ protein interaction sensor. Mol. Cell. Proteom. 2014, 13, 3332–3342. [Google Scholar] [CrossRef] [PubMed]
- Masschaele, D.; Gerlo, S.; Lemmens, I.; Lievens, S.; Tavernier, J. KISS: A Mammalian Two-Hybrid Method for In Situ Analysis of Protein-Protein Interactions. Methods Mol. Biol. 2018, 1794, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, H.; Wu, C.H. Protein Bioinformatics Databases and Resources. Methods Mol. Biol. 2017, 1558, 3–39. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of PhosphoSitePlus(R): Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef]
- Kershaw, N.J.; Murphy, J.M.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. Regulation of Janus kinases by SOCS proteins. Biochem. Soc. Trans. 2013, 41, 1042–1047. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. JAKSTAT 2013, 2, e24053. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Guan, Q.; Wang, Y.; Zhao, Z.J.; Zhou, G.W. SHP-1 suppresses cancer cell growth by promoting degradation of JAK kinases. J. Cell. Biochem. 2003, 90, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, C.E.; Kasembeli, M.M.; Roh, S.H.; Tweardy, D.J. Contribution of chaperones to STAT pathway signaling. JAKSTAT 2014, 3, e970459. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.M.; Whitesell, L. HSP90: Enabler of Cancer Adaptation. Annu. Rev. Cancer Biol. 2019, 3, 275–297. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Caldas-Lopes, E.; Cerchietti, L.; Ahn, J.H.; Clement, C.C.; Robles, A.I.; Rodina, A.; Moulick, K.; Taldone, T.; Gozman, A.; Guo, Y.; et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA 2009, 106, 8368–8373. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Akahane, K.; Sanda, T.; Mansour, M.R.; Radimerski, T.; DeAngelo, D.J.; Weinstock, D.M.; Look, A.T. HSP90 inhibition leads to degradation of the TYK2 kinase and apoptotic cell death in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 219–228. [Google Scholar] [CrossRef]
- Schoof, N.; von Bonin, F.; Trumper, L.; Kube, D. HSP90 is essential for Jak-STAT signaling in classical Hodgkin Lymphoma cells. Cell Commun. Sig. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Witte, S.; Muljo, S.A. Integrating non-coding RNAs in JAK-STAT regulatory networks. JAKSTAT 2014, 3, e28055. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.E.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.; Stevens, J.R.; Wolff, R.K.; Slattery, M.L. MicroRNA-messenger RNA interactions involving JAK-STAT signaling genes in colorectal cancer. Genes Cancer 2018, 9, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Pham, H.T.; Schmoellerl, J.; Javaheri, T.; Schlederer, M.; Culig, Z.; Merkel, O.; Moriggl, R.; Grebien, F.; Kenner, L. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine 2016, 87, 26–36. [Google Scholar] [CrossRef]
- Li, Z.Y.; Yang, L.; Liu, X.J.; Wang, X.Z.; Pan, Y.X.; Luo, J.M. The Long Noncoding RNA MEG3 and its Target miR-147 Regulate JAK/STAT Pathway in Advanced Chronic Myeloid Leukemia. EBioMedicine 2018, 34, 61–75. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef]
- Changelian, P.S.; Flanagan, M.E.; Ball, D.J.; Kent, C.R.; Magnuson, K.S.; Martin, W.H.; Rizzuti, B.J.; Sawyer, P.S.; Perry, B.D.; Brissette, W.H.; et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003, 302, 875–878. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Gadina, M.; Le, M.T.; Schwartz, D.M.; Silvennoinen, O.; Nakayamada, S.; Yamaoka, K.; O’Shea, J.J. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatology 2019, 58, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Leroy, E.; Gilles, L.; Marty, C.; Plo, I.; Constantinescu, S.N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res 2018, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. Biodrugs 2019, 33, 15–32. [Google Scholar] [CrossRef] [PubMed]
- He, X.R.; Chen, X.B.; Zhang, H.C.; Xie, T.; Ye, X.Y. Selective Tyk2 inhibitors as potential therapeutic agents: A patent review (2015-2018). Expert. Opin. Ther. Pat. 2019, 29, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Argollo, M.; Le Berre, C.; Peyrin-Biroulet, L. JAK selectivity for inflammatory bowel disease treatment: Does it clinically matter? Gut 2019, 68, 1893–1899. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Gordon, K.; Thaci, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D.; et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019, 11, eaaw1736. [Google Scholar] [CrossRef]
- Liu, C.; Lin, J.; Moslin, R.; Tokarski, J.S.; Muckelbauer, J.; Chang, C.; Tredup, J.; Xie, D.; Park, H.; Li, P.; et al. Identification of Imidazo[1,2-b]pyridazine Derivatives as Potent, Selective, and Orally Active Tyk2 JH2 Inhibitors. ACS Med. Chem. Lett. 2019, 10, 383–388. [Google Scholar] [CrossRef]
- Moslin, R.; Gardner, D.; Santella, J.; Zhang, Y.; Duncia, J.V.; Liu, C.; Lin, J.; Tokarski, J.S.; Strnad, J.; Pedicord, D.; et al. Identification of imidazo[1,2-b] pyridazine TYK2 pseudokinase ligands as potent and selective allosteric inhibitors of TYK2 signalling. Medchemcomm 2017, 8, 700–712. [Google Scholar] [CrossRef]
- Moslin, R.; Zhang, Y.; Wrobleski, S.T.; Lin, S.; Mertzman, M.; Spergel, S.; Tokarski, J.S.; Strnad, J.; Gillooly, K.; McIntyre, K.W.; et al. Identification of N-Methyl Nicotinamide and N-Methyl Pyridazine-3-Carboxamide Pseudokinase Domain Ligands as Highly Selective Allosteric Inhibitors of Tyrosine Kinase 2 (TYK2). J. Med. Chem. 2019, 62, 8953–8972. [Google Scholar] [CrossRef]
- Wrobleski, S.T.; Moslin, R.; Lin, S.; Zhang, Y.; Spergel, S.; Kempson, J.; Tokarski, J.S.; Strnad, J.; Zupa-Fernandez, A.; Cheng, L.; et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J. Med. Chem. 2019, 62, 8973–8995. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. TYK2 inhibition shows promise. Nat. Rev. Drug Discov. 2019, 18, 668. [Google Scholar] [CrossRef] [PubMed]
- Prchal-Murphy, M.; Semper, C.; Lassnig, C.; Wallner, B.; Gausterer, C.; Teppner-Klymiuk, I.; Kobolak, J.; Muller, S.; Kolbe, T.; Karaghiosoff, M.; et al. TYK2 kinase activity is required for functional type I interferon responses in vivo. PLoS ONE 2012, 7, e39141. [Google Scholar] [CrossRef] [PubMed]
- Raje, V.; Derecka, M.; Cantwell, M.; Meier, J.; Szczepanek, K.; Sisler, J.D.; Strobl, B.; Gamero, A.; Harris, T.E.; Larner, A.C. Kinase Inactive Tyrosine Kinase (Tyk2) Supports Differentiation of Brown Fat Cells. Endocrinology 2017, 158, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lesgidou, N.; Eliopoulos, E.; Goulielmos, G.N.; Vlassi, M. Insights on the alteration of functionality of a tyrosine kinase 2 variant: A molecular dynamics study. Bioinformatics 2018, 34, i781–i786. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Skiniotis, G.; Rice, A.J.; Thomas, C.; Fischer, S.; Walz, T.; Garcia, K.C. Structural snapshots of full-length Jak1, a transmembrane gp130/IL-6/IL-6Ralpha cytokine receptor complex, and the receptor-Jak1 holocomplex. Structure 2011, 19, 45–55. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).