Emodin Inhibits EBV Reactivation and Represses NPC Tumorigenesis


 ,
,  and
and 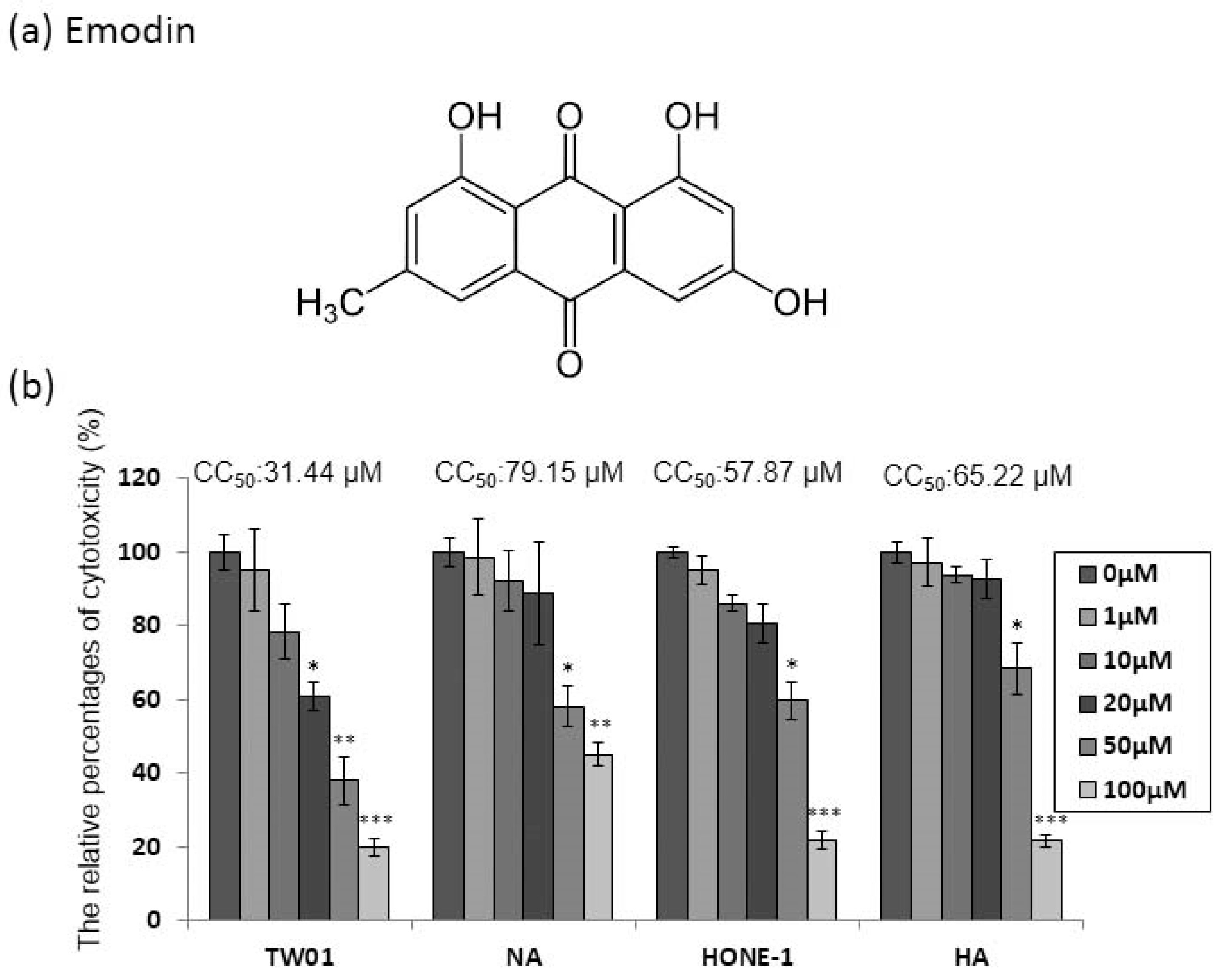{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Cytotoxicity of Emodin to the NPC Cell Lines
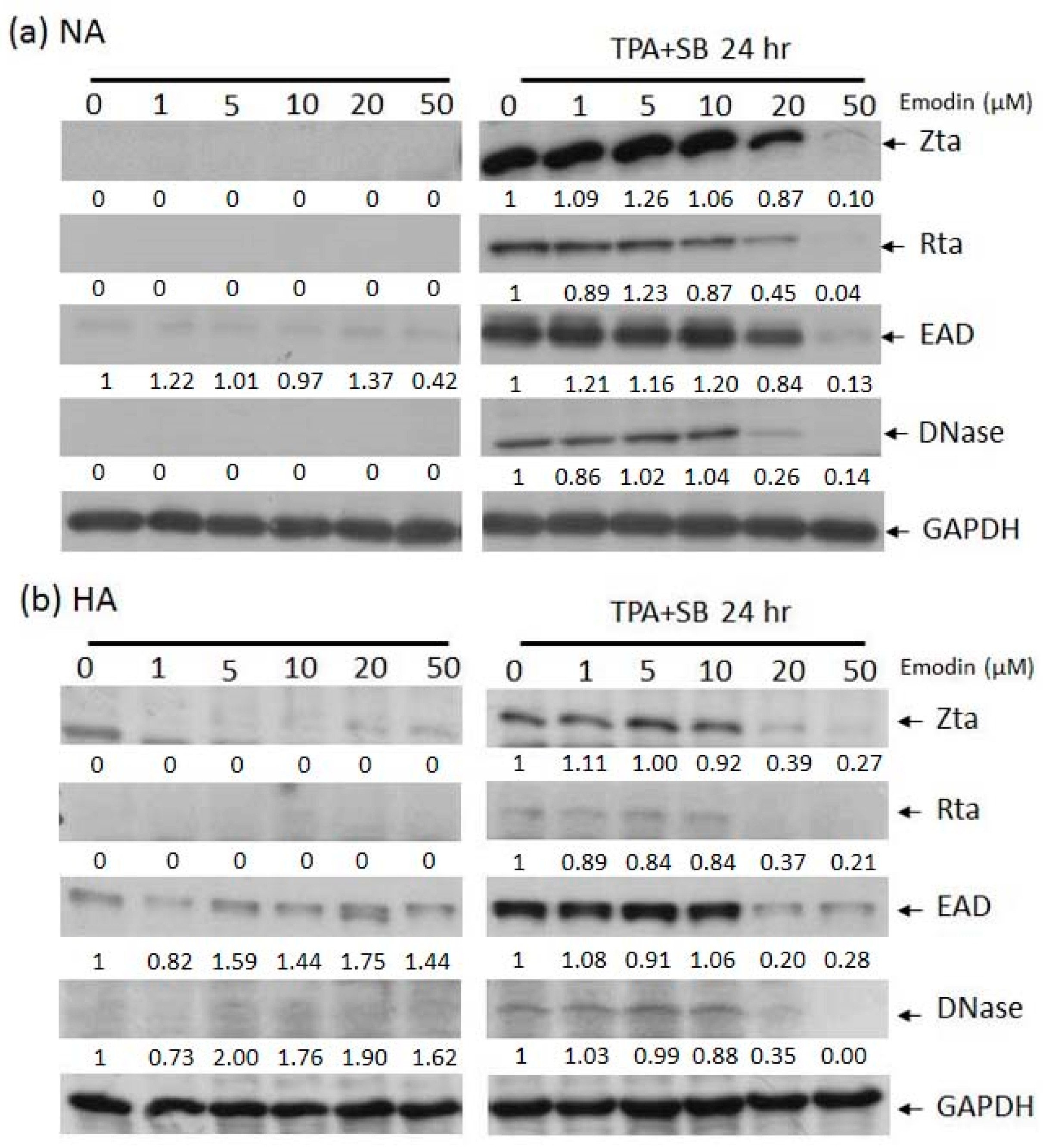
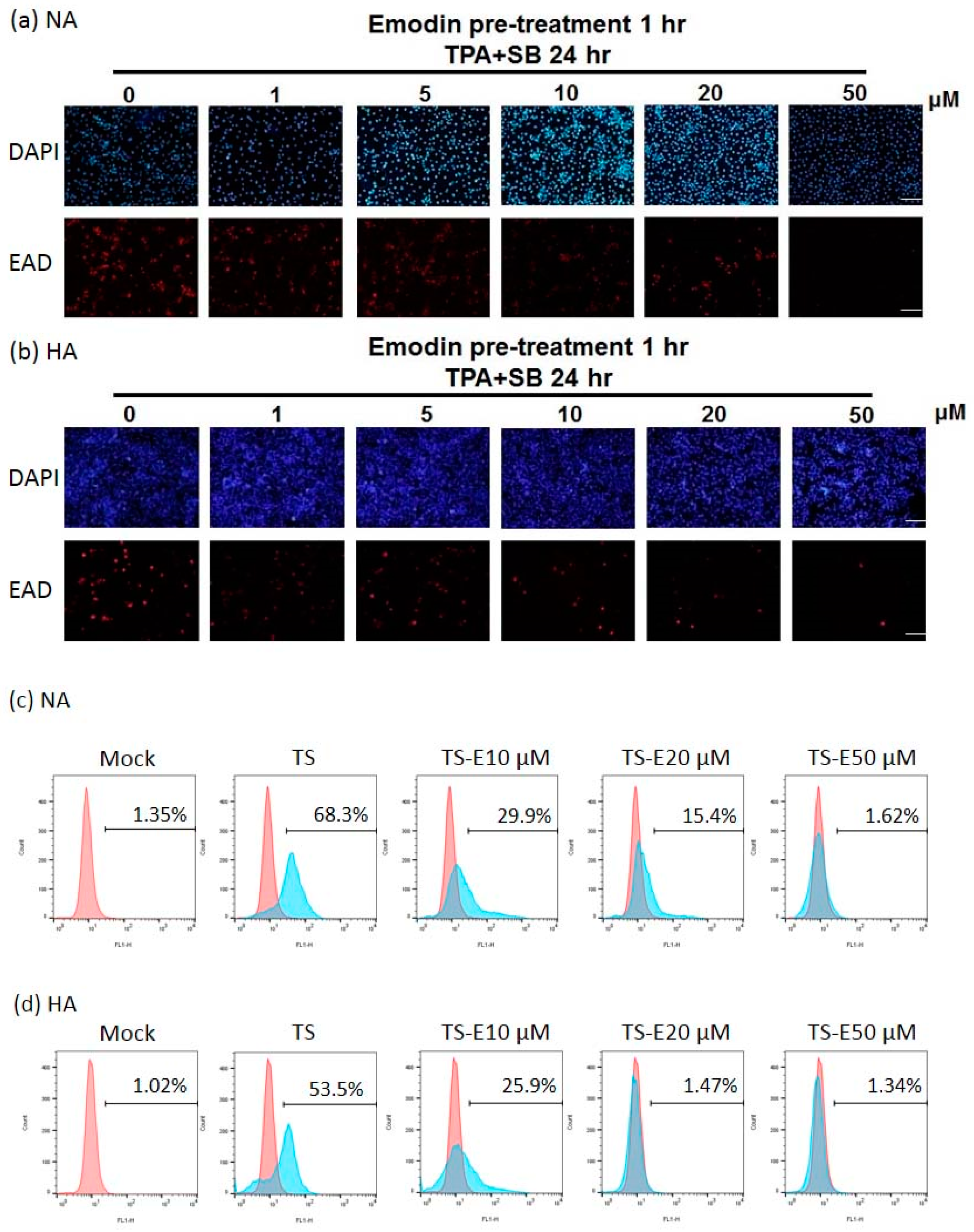
2.2. Emodin Inhibits EBV Lytic Protein Expression in NPC Cells
2.3. The Inhibition of Virion Production by Emodin
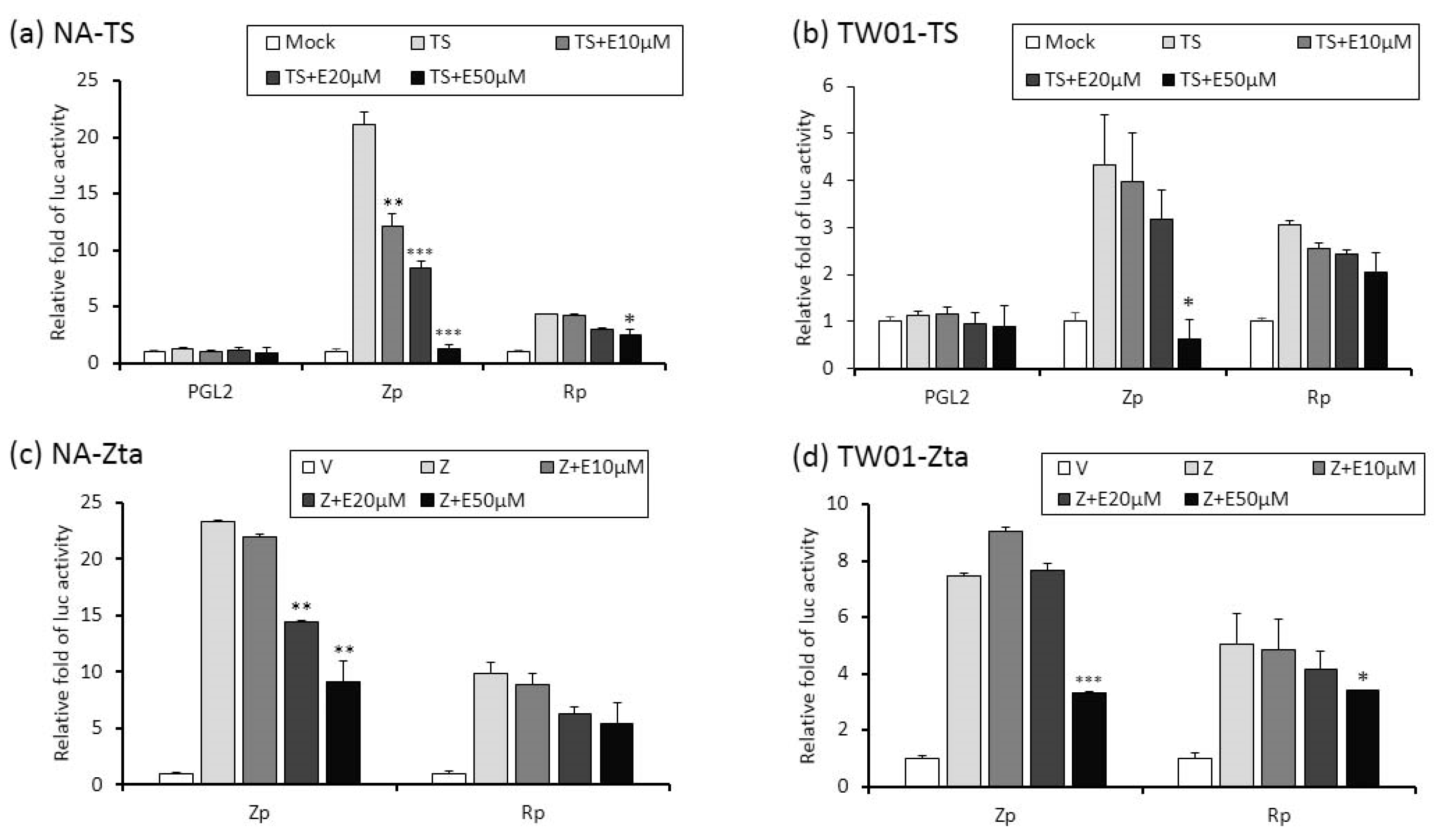
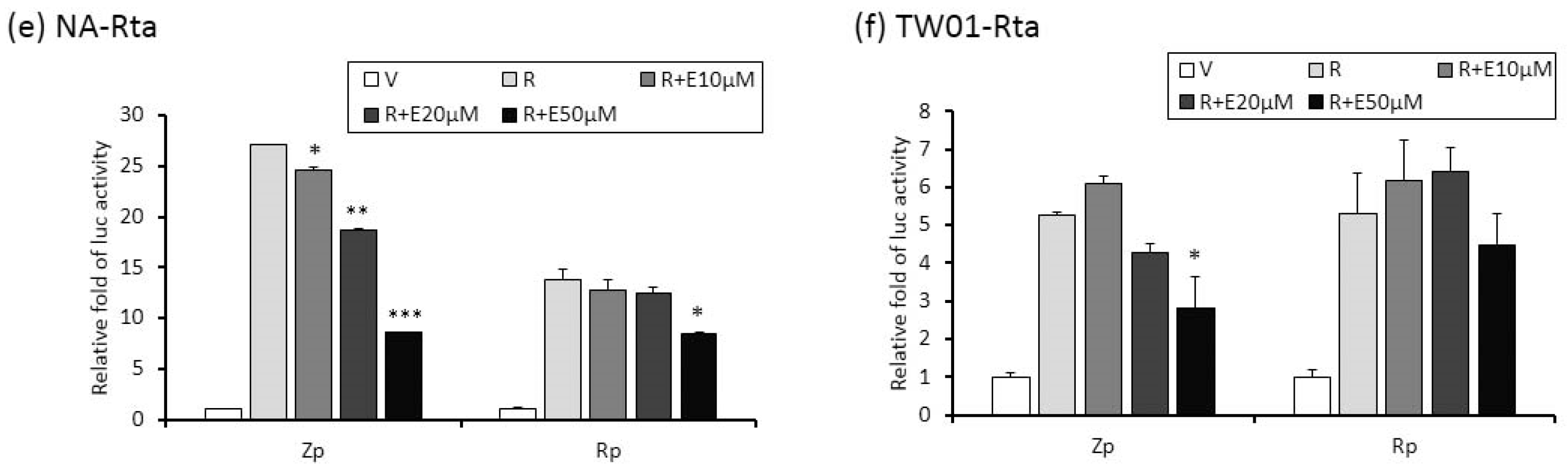
2.4. The Repression of Zta Promoter (Zp) and Rta Promoter (Rp) Transcriptional Activities by Emodin
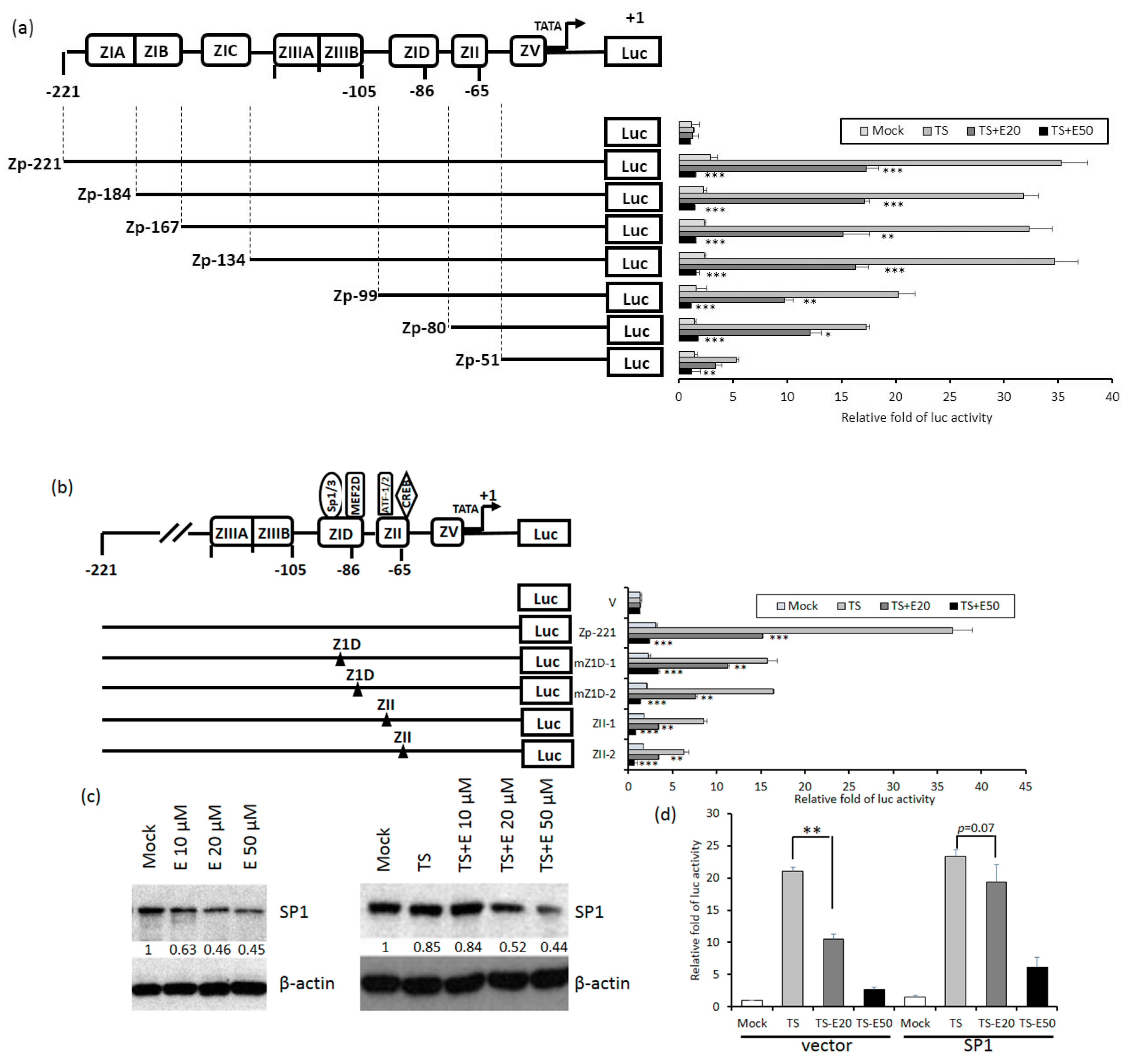
2.5. Identification of Emodin Responsive Element in Zp
2.6. Emodin Attenuates the Reactivation-Induced Tumorigenic Properties of NPC Cells
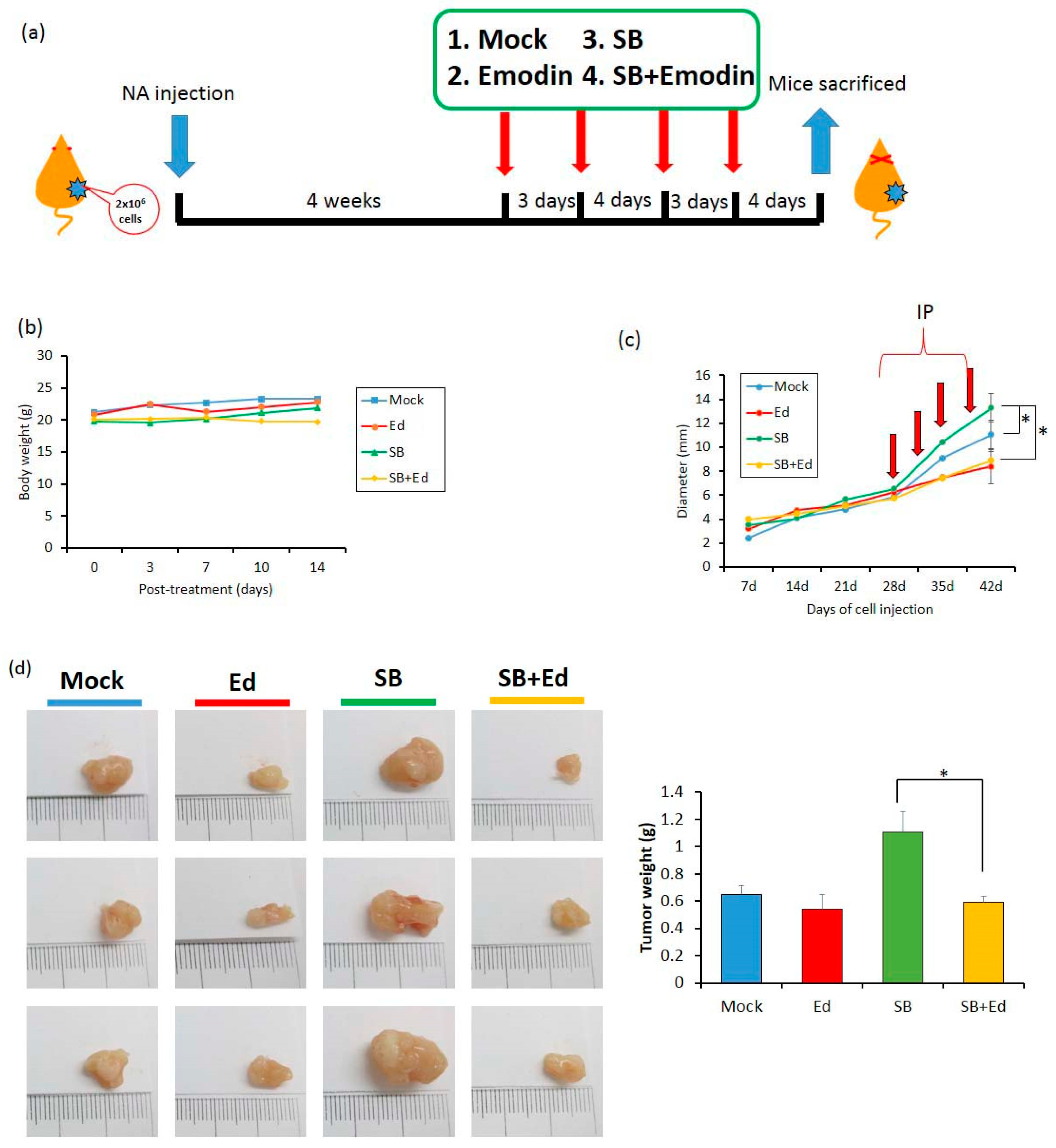
2.7. Inhibition of EBV Reactivation by Emodin Decreases Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines
4.3. EBV Induction and Emodin Treatment
4.4. Evaluation of the Cytotoxicity of Emodin
4.5. Western Blotting Analysis
4.6. Immunofluorecence and Flow Cytometry Analysis
4.7. Determination of the Copy Number of the EBV Genome
4.8. Transfection and Analysis of Luciferase Reporter Activity
4.9. Determination of MN Formation
4.10. Cell Migration Assay
4.11. Cell Invasive Assay
4.12. In Vivo Tumorigenesis Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraf, M.; LeBlanc, M.; Giri, P.G.; Fu, K.K.; Cooper, J.; Vuong, T.; Forastiere, A.A.; Adams, G.; Sakr, W.A.; Schuller, D.E.; et al. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: Phase III randomized intergroup study 0099. J. Clin. Oncol. 1998, 16, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.W.; Foo, W.; Law, S.C.; Peters, L.J.; Poon, Y.F.; Chappell, R.; Sze, W.M.; Tung, S.Y.; Lau, W.H.; Ho, J.H. Total biological effect on late reactive tissues following reirradiation for recurrent nasopharyngeal carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 865–872. [Google Scholar] [CrossRef]
- Lin, J.C.; Jan, J.S.; Hsu, C.Y.; Jiang, R.S.; Wang, W.Y. Outpatient weekly neoadjuvant chemotherapy followed by radiotherapy for advanced nasopharyngeal carcinoma: High complete response and low toxicity rates. Br. J. Cancer 2003, 88, 187–194. [Google Scholar] [CrossRef]
- Su, S.F.; Han, F.; Zhao, C.; Huang, Y.; Chen, C.Y.; Xiao, W.W.; Li, J.X.; Lu, T.X. Treatment outcomes for different subgroups of nasopharyngeal carcinoma patients treated with intensity-modulated radiation therapy. Chin. J. Cancer 2011, 30, 565–573. [Google Scholar] [CrossRef]
- Rickinson, A.B.; Kieff, E. Epstein-Barr virus. In Field’s Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 75, pp. 2575–2627. [Google Scholar]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef]
- Cao, S.M.; Liu, Z.; Jia, W.H.; Huang, Q.H.; Liu, Q.; Guo, X.; Huang, T.B.; Ye, W.; Hong, M.H. Fluctuations of Epstein-Barr virus serological antibodies and risk for nasopharyngeal carcinoma: A prospective screening study with a 20-year follow-up. PLoS ONE 2011, 6, e19100. [Google Scholar] [CrossRef]
- Chen, J.Y.; Chen, C.J.; Liu, M.Y.; Cho, S.M.; Hsu, M.M.; Lynn, T.C.; Shieh, T.; Tu, S.M.; Beasley, R.P.; Hwang, L.Y.; et al. Antibody to Epstein-Barr virus-specific DNase as a marker for field survey of patients with nasopharyngeal carcinoma in Taiwan. J. Med. Virol. 1989, 27, 269–273. [Google Scholar] [CrossRef]
- Chen, J.Y.; Hwang, L.Y.; Beasley, R.P.; Chien, C.S.; Yang, C.S. Antibody response to Epstein-Barr-virus-specific DNase in 13 patients with nasopharyngeal carcinoma in Taiwan: A retrospective study. J. Med. Virol. 1985, 16, 99–105. [Google Scholar] [CrossRef]
- Chien, Y.C.; Chen, J.Y.; Liu, M.Y.; Yang, H.I.; Hsu, M.M.; Chen, C.J.; Yang, C.S. Serologic markers of Epstein-Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. N. Engl. J. Med. 2001, 345, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.H. Epstein-Barr virus. In Principles and Practice of Clinical Virology, 5th ed.; Zuckerman, A.J., Banatvala, J.E., Pattison, J.R., Griffiths, P., Schoub, B., Eds.; John Wiley and Sons: West Succex, UK, 2004; pp. 123–146. [Google Scholar]
- de-The, G. Epidemiology of Epstein-Barr virus and associated disease in man. In The Herpesviruses I; Roizman, B., Ed.; Plenum: New York, NY, USA; London, UK, 1982; pp. 25–103. [Google Scholar]
- Liu, M.Y.; Chou, W.H.; Nutter, L.; Hsu, M.M.; Chen, J.Y.; Yang, C.S. Antibody against Epstein-Barr virus DNA polymerase activity in sera of patients with nasopharyngeal carcinoma. J. Med. Virol. 1989, 28, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Cabras, G.; Decaussin, G.; Zeng, Y.; Djennaoui, D.; Melouli, H.; Broully, P.; Bouguermouh, A.M.; Ooka, T. Epstein-Barr virus encoded BALF1 gene is transcribed in Burkitt’s lymphoma cell lines and in nasopharyngeal carcinoma’s biopsies. J. Clin. Virol. 2005, 34, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 1995, 76 Pt 6, 1401–1408. [Google Scholar] [CrossRef]
- Sbih-Lammali, F.; Berger, F.; Busson, P.; Ooka, T. Expression of the DNase encoded by the BGLF5 gene of Epstein-Barr virus in nasopharyngeal carcinoma epithelial cells. Virology 1996, 222, 64–74. [Google Scholar] [CrossRef]
- Chang, Y.H.; Lee, C.P.; Su, M.T.; Wang, J.T.; Chen, J.Y.; Lin, S.F.; Tsai, C.H.; Hsieh, M.J.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase retards cellular S-phase progression and induces chromosomal abnormality. PLoS ONE 2012, 7, e39217. [Google Scholar] [CrossRef]
- Chiu, S.H.; Wu, C.C.; Fang, C.Y.; Yu, S.L.; Hsu, H.Y.; Chow, Y.H.; Chen, J.Y. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583–8601. [Google Scholar] [CrossRef]
- Li, L.Y.; Shih, H.M.; Liu, M.Y.; Chen, J.Y. The cellular protein PRA1 modulates the anti-apoptotic activity of Epstein-Barr virus BHRF1, a homologue of Bcl-2, through direct interaction. J. Biol. Chem. 2001, 276, 27354–27362. [Google Scholar] [CrossRef]
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W.; et al. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef]
- Fang, C.Y.; Huang, S.Y.; Wu, C.C.; Hsu, H.Y.; Chou, S.P.; Tsai, C.H.; Chang, Y.; Takada, K.; Chen, J.Y. The synergistic effect of chemical carcinogens enhances Epstein-Barr virus reactivation and tumor progression of nasopharyngeal carcinoma cells. PLoS ONE 2012, 7, e44810. [Google Scholar] [CrossRef]
- Fang, C.Y.; Lee, C.H.; Wu, C.C.; Chang, Y.T.; Yu, S.L.; Chou, S.P.; Huang, P.T.; Chen, C.L.; Hou, J.W.; Chang, Y.; et al. Recurrent chemical reactivations of EBV promotes genome instability and enhances tumor progression of nasopharyngeal carcinoma cells. Int. J. Cancer 2009, 124, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Fang, C.Y.; Hsu, H.Y.; Chen, Y.J.; Chou, S.P.; Huang, S.Y.; Cheng, Y.J.; Lin, S.F.; Chang, Y.; Tsai, C.H.; et al. Luteolin inhibits Epstein-Barr virus lytic reactivation by repressing the promoter activities of immediate-early genes. Antivir. Res. 2016, 132, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Fang, C.Y.; Hsu, H.Y.; Chuang, H.Y.; Chen, Y.J.; Chou, S.P.; Huang, S.Y.; Cheng, Y.J.; Lin, S.F.; Chang, Y.; et al. EBV reactivation as a target of luteolin to repress NPC tumorigenesis. Oncotarget 2016, 7, 18999–19017. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Fang, C.Y.; Cheng, Y.J.; Hsu, H.Y.; Chou, S.P.; Huang, S.Y.; Tsai, C.H.; Chen, J.Y. Inhibition of Epstein-Barr virus reactivation by the flavonoid apigenin. J. Biomed. Sci. 2017, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Fu, J.; Yin, X.; Cao, S.; Li, X.; Lin, L.; Huyiligeqi, N.J. Emodin: A Review of its Pharmacology, Toxicity and Pharmacokinetics. Phytother Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.Y.; Wu, S.L.; Chen, J.C.; Li, C.C.; Hsiang, C.Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2007, 74, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Shuangsuo, D.; Zhengguo, Z.; Yunru, C.; Xin, Z.; Baofeng, W.; Lichao, Y.; Yan’an, C. Inhibition of the replication of hepatitis B virus in vitro by emodin. Med. Sci. Monit. 2006, 12, BR302–BR306. [Google Scholar]
- Liu, Z.; Wei, F.; Chen, L.J.; Xiong, H.R.; Liu, Y.Y.; Luo, F.; Hou, W.; Xiao, H.; Yang, Z.Q. In vitro and in vivo studies of the inhibitory effects of emodin isolated from Polygonum cuspidatum on Coxsakievirus B(4). Molecules 2013, 18, 11842–11858. [Google Scholar] [CrossRef]
- Batista, M.N.; Braga, A.C.S.; Campos, G.R.F.; Souza, M.M.; Matos, R.P.A.; Lopes, T.Z.; Candido, N.M.; Lima, M.L.D.; Machado, F.C.; Andrade, S.T.Q.; et al. Natural Products Isolated from Oriental Medicinal Herbs Inactivate Zika Virus. Viruses 2019, 11, 49. [Google Scholar] [CrossRef]
- Dai, J.P.; Wang, Q.W.; Su, Y.; Gu, L.M.; Zhao, Y.; Chen, X.X.; Chen, C.; Li, W.Z.; Wang, G.F.; Li, K.S. Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways. Molecules 2017, 22, 1754. [Google Scholar] [CrossRef]
- Hsiang, C.Y.; Ho, T.Y. Emodin is a novel alkaline nuclease inhibitor that suppresses herpes simplex virus type 1 yields in cell cultures. Br. J. Pharmacol. 2008, 155, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.R.; Luo, J.; Hou, W.; Xiao, H.; Yang, Z.Q. The effect of emodin, an anthraquinone derivative extracted from the roots of Rheum tanguticum, against herpes simplex virus in vitro and in vivo. J. Ethnopharmacol. 2011, 133, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Yiu, C.Y.; Chen, S.Y.; Yang, T.H.; Chang, C.J.; Yeh, D.B.; Chen, Y.J.; Lin, T.P. Inhibition of Epstein-Barr virus lytic cycle by an ethyl acetate subfraction separated from Polygonum cuspidatum root and its major component, emodin. Molecules 2014, 19, 1258–1272. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Wu, C.C.; Cheng, Y.J.; Chou, S.P.; Jiang, Y.J.; Chu, K.C.; Tsai, C.H.; Lin, S.F.; Chen, J.Y. Epstein-Barr virus BRLF1 induces genomic instability and progressive malignancy in nasopharyngeal carcinoma cells. Oncotarget 2017, 8, 78948–78964. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hu, J.; Luo, X.; Bode, A.M.; Dong, Z.; Cao, Y. Therapies based on targeting Epstein-Barr virus lytic replication for EBV-associated malignancies. Cancer Sci. 2018, 109, 2101–2108. [Google Scholar] [CrossRef]
- Chang, L.K.; Wei, T.T.; Chiu, Y.F.; Tung, C.P.; Chuang, J.Y.; Hung, S.K.; Li, C.; Liu, S.T. Inhibition of Epstein-Barr virus lytic cycle by (-)-epigallocatechin gallate. Biochem. Biophys. Res. Commun. 2003, 301, 1062–1068. [Google Scholar] [CrossRef]
- Hergenhahn, M.; Soto, U.; Weninger, A.; Polack, A.; Hsu, C.H.; Cheng, A.L.; Rosl, F. The chemopreventive compound curcumin is an efficient inhibitor of Epstein-Barr virus BZLF1 transcription in Raji DR-LUC cells. Mol. Carcinog. 2002, 33, 137–145. [Google Scholar] [CrossRef]
- De Leo, A.; Arena, G.; Lacanna, E.; Oliviero, G.; Colavita, F.; Mattia, E. Resveratrol inhibits Epstein Barr Virus lytic cycle in Burkitt’s lymphoma cells by affecting multiple molecular targets. Antivir. Res. 2012, 96, 196–202. [Google Scholar] [CrossRef]
- Wu, C.C.; Chuang, H.Y.; Lin, C.Y.; Chen, Y.J.; Tsai, W.H.; Fang, C.Y.; Huang, S.Y.; Chuang, F.Y.; Lin, S.F.; Chang, Y. Inhibition of Epstein-Barr virus reactivation in nasopharyngeal carcinoma cells by dietary sulforaphane. Mol. Carcinog. 2013, 52, 946–958. [Google Scholar] [CrossRef]
- Daibata, M.; Mellinghoff, I.; Takagi, S.; Humphreys, R.E.; Sairenji, T. Effect of genistein, a tyrosine kinase inhibitor, on latent EBV activation induced by cross-linkage of membrane IgG in Akata B cells. J. Immunol. 1991, 147, 292–297. [Google Scholar]
- Tung, C.P.; Chang, F.R.; Wu, Y.C.; Chuang, D.W.; Hunyadi, A.; Liu, S.T. Inhibition of the Epstein-Barr virus lytic cycle by protoapigenone. J. Gen. Virol. 2011, 92, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.R.; Hsieh, Y.C.; Chang, Y.F.; Lee, K.H.; Wu, Y.C.; Chang, L.K. Inhibition of the Epstein-Barr virus lytic cycle by moronic acid. Antivir. Res. 2010, 85, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.P.; Chen, S.Y.; Duh, P.D.; Chang, L.K.; Liu, Y.N. Inhibition of the epstein-barr virus lytic cycle by andrographolide. Biol. Pharm. Bull. 2008, 31, 2018–2023. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Wu, J.; Zheng, F.; Hann, S.S.; Chen, Y. Emodin Increases Expression of Insulin-Like Growth Factor Binding Protein 1 through Activation of MEK/ERK/AMPKalpha and Interaction of PPARgamma and Sp1 in Lung Cancer. Cell. Physiol. Biochem. 2017, 41, 339–357. [Google Scholar] [CrossRef]
- Tang, Q.; Zhao, S.; Wu, J.; Zheng, F.; Yang, L.; Hu, J.; Hann, S.S. Inhibition of integrin-linked kinase expression by emodin through crosstalk of AMPKalpha and ERK1/2 signaling and reciprocal interplay of Sp1 and c-Jun. Cell. Signal. 2015, 27, 1469–1477. [Google Scholar] [CrossRef]
- Alam, Z.; Al-Mahdi, Z.; Zhu, Y.; McKee, Z.; Parris, D.S.; Parikh, H.I.; Kellogg, G.E.; Kuchta, A.; McVoy, M.A. Anti-cytomegalovirus activity of the anthraquinone atanyl blue PRL. Antivir. Res. 2015, 114, 86–95. [Google Scholar] [CrossRef]
- Nicolas, M.; Noe, V.; Ciudad, C.J. Transcriptional regulation of the human Sp1 gene promoter by the specificity protein (Sp) family members nuclear factor Y (NF-Y) and E2F. Biochem. J. 2003, 371, 265–275. [Google Scholar] [CrossRef]
- Xu, K.; Al-Ani, M.K.; Wang, C.; Qiu, X.; Chi, Q.; Zhu, P.; Dong, N. Emodin as a selective proliferative inhibitor of vascular smooth muscle cells versus endothelial cells suppress arterial intima formation. Life Sci. 2018, 207, 9–14. [Google Scholar] [CrossRef]
- Tapias, A.; Ciudad, C.J.; Roninson, I.B.; Noe, V. Regulation of Sp1 by cell cycle related proteins. Cell Cycle 2008, 7, 2856–2867. [Google Scholar] [CrossRef]
- Dong, X.; Ni, B.; Fu, J.; Yin, X.; You, L.; Leng, X.; Liang, X.; Ni, J. Emodin induces apoptosis in human hepatocellular carcinoma HepaRG cells via the mitochondrial caspasedependent pathway. Oncol. Rep. 2018, 40, 1985–1993. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Zaman, K.; Kubilis, J.; Ferrante, R.J.; Ross, B.D.; Neve, R.; Ratan, R.R. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J. Neurosci. 2003, 23, 3597–3606. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Liu, M.T.; Chen, M.R.; Lu, J.; Yang, H.L.; Chen, J.Y.; Yang, C.S. Characterization of monoclonal antibodies to the Zta and DNase proteins of Epstein-Barr virus. J. Biomed. Sci. 1997, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Williams, M.V.; Glaser, R. Characterization of two monoclonal antibodies to Epstein-Barr virus diffuse early antigen which react to two different epitopes and have different biological function. J. Virol. Methods 1991, 33, 47–52. [Google Scholar] [CrossRef]
- Lin, C.T.; Wong, C.I.; Chan, W.Y.; Tzung, K.W.; Ho, J.K.; Hsu, M.M.; Chuang, S.M. Establishment and characterization of two nasopharyngeal carcinoma cell lines. Labor. Investig. J. Tech. Methods Pathol. 1990, 62, 713–724. [Google Scholar]
- Glaser, R.; Zhang, H.Y.; Yao, K.T.; Zhu, H.C.; Wang, F.X.; Li, G.Y.; Wen, D.S.; Li, Y.P. Two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein-Barr virus that were derived from nasopharyngeal carcinomas. Proc. Natl. Acad. Sci. USA 1989, 86, 9524–9528. [Google Scholar] [CrossRef]
- Chang, Y.; Tung, C.H.; Huang, Y.T.; Lu, J.; Chen, J.Y.; Tsai, C.H. Requirement for cell-to-cell contact in Epstein-Barr virus infection of nasopharyngeal carcinoma cells and keratinocytes. J. Virol. 1999, 73, 8857–8866. [Google Scholar]
- Liu, M.T.; Chen, Y.R.; Chen, S.C.; Hu, C.Y.; Lin, C.S.; Chang, Y.T.; Wang, W.B.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 2004, 23, 2531–2539. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-C.; Chen, M.-S.; Cheng, Y.-J.; Ko, Y.-C.; Lin, S.-F.; Chiu, I.-M.; Chen, J.-Y. Emodin Inhibits EBV Reactivation and Represses NPC Tumorigenesis. Cancers 2019, 11, 1795. https://doi.org/10.3390/cancers11111795
Wu C-C, Chen M-S, Cheng Y-J, Ko Y-C, Lin S-F, Chiu I-M, Chen J-Y. Emodin Inhibits EBV Reactivation and Represses NPC Tumorigenesis. Cancers. 2019; 11(11):1795. https://doi.org/10.3390/cancers11111795
Chicago/Turabian StyleWu, Chung-Chun, Mei-Shu Chen, Yu-Jhen Cheng, Ying-Chieh Ko, Su-Fang Lin, Ing-Ming Chiu, and Jen-Yang Chen. 2019. "Emodin Inhibits EBV Reactivation and Represses NPC Tumorigenesis" Cancers 11, no. 11: 1795. https://doi.org/10.3390/cancers11111795
APA StyleWu, C.-C., Chen, M.-S., Cheng, Y.-J., Ko, Y.-C., Lin, S.-F., Chiu, I.-M., & Chen, J.-Y. (2019). Emodin Inhibits EBV Reactivation and Represses NPC Tumorigenesis. Cancers, 11(11), 1795. https://doi.org/10.3390/cancers11111795