Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy



Abstract
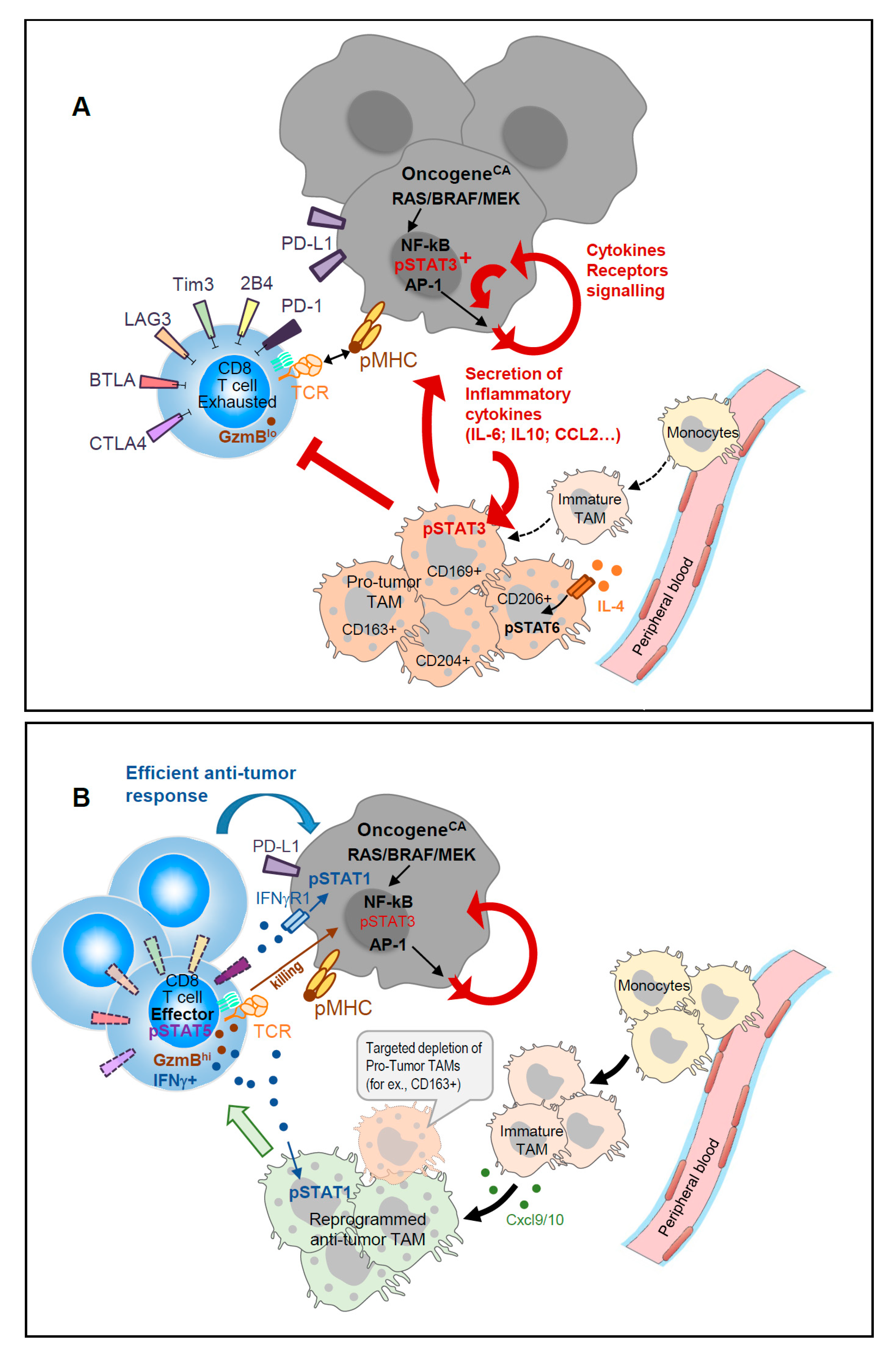
1. Introduction
2. Tumor-Induced Inflammation Drives Accumulation of Tumor-Infiltrating Myeloid Cells
3. Re-Educating TAM to Restore Anti-Tumor T Cell Functions.
4. STAT5 versus STAT3 in Adaptive CD8 T Cell Responses to Cancer
4.1. STAT5 in the Adaptive T Cell Responses
Role of STAT5 in the Generation of Memory CD8 T Cells and Maintenance of Effector Functions
4.2. Role of STAT3 in T Cells
4.2.1. Role of STAT3 in the Maintenance and Memory Formation of CD8 T Cells
4.2.2. Role of STAT3 in the CD8 T Cell-Mediated Anti-Tumor Responses
5. Adoptive CD8 T Cell Therapy and CAR-T Cell Generation for Cancer Immunotherapy
5.1. Increasing the Frequency of Tumor-Specific CD8 T Cells
5.2. On CD8 T cell Intrinsic Modifications For adoptive T Cell Therapies
5.2.1. Promoting Effector Memory CD8 T Cells Through STAT5
5.2.2. Promoting Central Memory CD8 T Cells Through STAT3
5.2.3. On the STAT5 versus STAT3 Balance in T Cells
6. STAT5 and Resistance to Immunosuppression in the Tumor Microenvironment
6.1. Immunosuppression by PD-1
6.2. Other Immune Suppressive Pathways
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.D.; Giresi, P.G.; Lindahl-Allen, M.; Krall, E.B.; Lieb, J.D.; Struhl, K. STAT3 acts through pre-existing nucleosome-depleted regions bound by FOS during an epigenetic switch linking inflammation to cancer. Epigenetics Chromatin 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef]
- Soudja, S.M.; Wehbe, M.; Mas, A.; Chasson, L.; De Tenbossche, C.P.; Huijbers, I.; Van den Eynde, B.; Schmitt-Verhulst, A.M. Tumor-initiated inflammation overrides protective adaptive immunity in an induced melanoma model in mice. Cancer Res. 2010, 70, 3515–3525. [Google Scholar] [CrossRef]
- Wehbe, M.; Soudja, S.M.; Mas, A.; Chasson, L.; Guinamard, R.; Powis de Tenbossche, C.; Verdeil, G.; Van den Eynde, B.; Schmitt-Verhulst, A.M. Epithelial-Mesenchymal-Transition-like and TGFβ pathways associated with autochthonous inflammatory melanoma development in mice. PLoS ONE 2012, 7, e49419. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602. [Google Scholar] [CrossRef]
- Bercovici, N.; Guerin, M.V.; Trautmann, A.; Donnadieu, E. The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front. Immunol. 2019, 10, 1563. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163(+) TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765. [Google Scholar] [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749. [Google Scholar] [CrossRef]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1014–1030. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Croxford, A.L.; Lanzinger, M.; Hartmann, F.J.; Schreiner, B.; Mair, F.; Pelczar, P.; Clausen, B.E.; Jung, S.; Greter, M.; Becher, B. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 2015, 43, 502–514. [Google Scholar] [CrossRef]
- Huen, S.C.; Huynh, L.; Marlier, A.; Lee, Y.; Moeckel, G.W.; Cantley, L.G. GM-CSF Promotes Macrophage Alternative Activation after Renal Ischemia/Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1334–1345. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [PubMed]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jain, A.; Syed, S.N.; Snodgrass, R.G.; Pfluger-Muller, B.; Leisegang, M.S.; Weigert, A.; Brandes, R.P.; Ebersberger, I.; Brune, B.; et al. IL-6 augments IL-4-induced polarization of primary human macrophages through synergy of STAT3, STAT6 and BATF transcription factors. Oncoimmunology 2018, 7, e1494110. [Google Scholar] [CrossRef] [PubMed]
- Rebe, C.; Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef]
- Andersen, M.N.; Etzerodt, A.; Graversen, J.H.; Holthof, L.C.; Moestrup, S.K.; Hokland, M.; Moller, H.J. STAT3 inhibition specifically in human monocytes and macrophages by CD163-targeted corosolic acid-containing liposomes. Cancer Immunol. Immunother. 2019, 68, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Deng, J.; Xin, H.; Liu, Y.; Pardoll, D.; Yu, H. A requirement of STAT3 DNA binding precludes Th-1 immunostimulatory gene expression by NF-kappaB in tumors. Cancer Res. 2011, 71, 3772–3780. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Wang, H.W.; Bowman, R.L.; Joyce, J.A. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1alpha Activation. Cell Rep. 2016, 16, 2914–2927. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Deng, J.; Liu, Y.; Lee, H.; Herrmann, A.; Zhang, W.; Zhang, C.; Shen, S.; Priceman, S.J.; Kujawski, M.; Pal, S.K.; et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 2012, 21, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Broz, M.L.; Ranger, J.J.; Ozcelik, J.; Ahn, R.; Zuo, D.; Ursini-Siegel, J.; Hallett, M.T.; Krummel, M.; Muller, W.J. STAT3 Establishes an Immunosuppressive Microenvironment during the Early Stages of Breast Carcinogenesis to Promote Tumor Growth and Metastasis. Cancer Res. 2016, 76, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhang, D.; Gong, B.; Wang, P.; Liu, F. CD163 as a novel target gene of STAT3 is a potential therapeutic target for gastric cancer. Oncotarget 2017, 8, 87244–87262. [Google Scholar] [CrossRef] [PubMed]
- Trovato, R.; Fiore, A.; Sartori, S.; Cane, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef] [PubMed]
- Thorn, M.; Guha, P.; Cunetta, M.; Espat, N.J.; Miller, G.; Junghans, R.P.; Katz, S.C. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene. 2016, 23, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Lamano, J.B.; Lamano, J.B.; Li, Y.D.; DiDomenico, J.D.; Choy, W.; Veliceasa, D.; Oyon, D.E.; Fakurnejad, S.; Ampie, L.; Kesavabhotla, K.; et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin. Cancer Res. 2019, 25, 3643–3657. [Google Scholar] [CrossRef]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wei, S.; Hurt, E.M.; Green, M.D.; Zhao, L.; Vatan, L.; Szeliga, W.; Herbst, R.; Harms, P.W.; Fecher, L.A.; et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liang, Y.; Anders, R.A.; Taube, J.M.; Qiu, X.; Mulgaonkar, A.; Liu, X.; Harrington, S.M.; Guo, J.; Xin, Y.; et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; LaFleur, M.W.; Verschoor, V.L.; Dongre, A.; Zhang, Y.; Nguyen, T.H.; Kolifrath, S.; Aref, A.R.; Lau, C.J.; Paweletz, C.P.; et al. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 16971–16980. [Google Scholar] [CrossRef]
- Wei, Y.; Zhao, Q.; Gao, Z.; Lao, X.M.; Lin, W.M.; Chen, D.P.; Mu, M.; Huang, C.X.; Liu, Z.Y.; Li, B.; et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J. Clin. Investig. 2019, 129, 3347–3360. [Google Scholar] [CrossRef]
- Leonard, W.J.; Lin, J.X.; O’Shea, J.J. The gammac Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 2019, 50, 832–850. [Google Scholar] [CrossRef]
- Raeber, M.E.; Zurbuchen, Y.; Impellizzieri, D.; Boyman, O. The role of cytokines in T-cell memory in health and disease. Immunol Rev. 2018, 283, 176–193. [Google Scholar] [CrossRef]
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659. [Google Scholar] [CrossRef]
- Hand, T.W.; Cui, W.; Jung, Y.W.; Sefik, E.; Joshi, N.S.; Chandele, A.; Liu, Y.; Kaech, S.M. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. USA 2010, 107, 16601–16606. [Google Scholar] [CrossRef]
- Tripathi, P.; Kurtulus, S.; Wojciechowski, S.; Sholl, A.; Hoebe, K.; Morris, S.C.; Finkelman, F.D.; Grimes, H.L.; Hildeman, D.A. STAT5 is critical to maintain effector CD8+ T cell responses. J. Immunol. 2010, 185, 2116–2124. [Google Scholar] [CrossRef]
- O’Gorman, W.E.; Dooms, H.; Thorne, S.H.; Kuswanto, W.F.; Simonds, E.F.; Krutzik, P.O.; Nolan, G.P.; Abbas, A.K. The initial phase of an immune response functions to activate regulatory T cells. J. Immunol. 2009, 183, 332–339. [Google Scholar] [CrossRef]
- Verdeil, G.; Chaix, J.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Temporal cross-talk between TCR and STAT signals for CD8 T cell effector differentiation. Eur. J. Immunol. 2006, 36, 3090–3100. [Google Scholar] [CrossRef]
- Verdeil, G.; Puthier, D.; Nguyen, C.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. STAT5-mediated signals sustain a TCR-initiated gene expression program toward differentiation of CD8 T cell effectors. J. Immunol. 2006, 176, 4834–4842. [Google Scholar] [CrossRef]
- Altan-Bonnet, G.; Germain, R.N. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005, 3, e356. [Google Scholar] [CrossRef]
- Auphan-Anezin, N.; Mazza, C.; Guimezanes, A.; Barrett-Wilt, G.A.; Montero-Julian, F.; Roussel, A.; Hunt, D.F.; Malissen, B.; Schmitt-Verhulst, A.M. Distinct orientation of the alloreactive monoclonal CD8 T cell activation program by three different peptide/MHC complexes. Eur. J. Immunol. 2006, 36, 1856–1866. [Google Scholar] [CrossRef]
- Chau, L.A.; Bluestone, J.A.; Madrenas, J. Dissociation of intracellular signaling pathways in response to partial agonist ligands of the T cell receptor. J. Exp. Med. 1998, 187, 1699–1709. [Google Scholar] [CrossRef]
- Rollings, C.M.; Sinclair, L.V.; Brady, H.J.M.; Cantrell, D.A.; Ross, S.H. Interleukin-2 shapes the cytotoxic T cell proteome and immune environment-sensing programs. Sci. Signal 2018, 11, eaap8112. [Google Scholar] [CrossRef]
- Ross, S.H.; Rollings, C.; Anderson, K.E.; Hawkins, P.T.; Stephens, L.R.; Cantrell, D.A. Phosphoproteomic Analyses of Interleukin 2 Signaling Reveal Integrated JAK Kinase-Dependent and -Independent Networks in CD8(+) T Cells. Immunity 2016, 45, 685–700. [Google Scholar] [CrossRef]
- Cote-Sierra, J.; Foucras, G.; Guo, L.; Chiodetti, L.; Young, H.A.; Hu-Li, J.; Zhu, J.; Paul, W.E. Interleukin 2 plays a central role in Th2 differentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 3880–3885. [Google Scholar] [CrossRef]
- Grange, M.; Verdeil, G.; Arnoux, F.; Griffon, A.; Spicuglia, S.; Maurizio, J.; Buferne, M.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Active STAT5 regulates T-bet and eomesodermin expression in CD8 T cells and imprints a T-bet-dependent Tc1 program with repressed IL-6/TGF-beta1 signaling. J. Immunol. 2013, 191, 3712–3724. [Google Scholar] [CrossRef]
- Shi, M.; Lin, T.H.; Appell, K.C.; Berg, L.J. Janus-kinase-3-dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity 2008, 28, 763–773. [Google Scholar] [CrossRef]
- Scheeren, F.A.; Naspetti, M.; Diehl, S.; Schotte, R.; Nagasawa, M.; Wijnands, E.; Gimeno, R.; Vyth-Dreese, F.A.; Blom, B.; Spits, H. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nat. Immunol. 2005, 6, 303–313. [Google Scholar] [CrossRef]
- Sheng, W.; Yang, F.; Zhou, Y.; Yang, H.; Low, P.Y.; Kemeny, D.M.; Tan, P.; Moh, A.; Kaplan, M.H.; Zhang, Y.; et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014, 24, 1387–1402. [Google Scholar] [CrossRef]
- Olson, M.R.; Verdan, F.F.; Hufford, M.M.; Dent, A.L.; Kaplan, M.H. STAT3 Impairs STAT5 Activation in the Development of IL-9-Secreting T Cells. J. Immunol. 2016, 196, 3297–3304. [Google Scholar] [CrossRef]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef]
- Yao, Z.; Kanno, Y.; Kerenyi, M.; Stephens, G.; Durant, L.; Watford, W.T.; Laurence, A.; Robinson, G.W.; Shevach, E.M.; Moriggl, R.; et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 2007, 109, 4368–4375. [Google Scholar] [CrossRef]
- Yang, X.P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.W.; Wei, L.; Vahedi, G.; et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef]
- Nurieva, R.I.; Podd, A.; Chen, Y.; Alekseev, A.M.; Yu, M.; Qi, X.; Huang, H.; Wen, R.; Wang, J.; Li, H.S.; et al. STAT5 protein negatively regulates T follicular helper (Tfh) cell generation and function. J. Biol. Chem. 2012, 287, 11234–11239. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Chen, Z.; Larjo, A.; Kanduri, K.; Nousiainen, K.; Aijo, T.; Ricano-Ponce, I.; Hrdlickova, B.; Tuomela, S.; Laajala, E.; et al. Genome-wide Analysis of STAT3-Mediated Transcription during Early Human Th17 Cell Differentiation. Cell Rep. 2017, 19, 1888–1901. [Google Scholar] [CrossRef]
- Poholek, A.C.; Jankovic, D.; Villarino, A.V.; Petermann, F.; Hettinga, A.; Shouval, D.S.; Snapper, S.B.; Kaech, S.M.; Brooks, S.R.; Vahedi, G.; et al. IL-10 induces a STAT3-dependent autoregulatory loop in TH2 cells that promotes Blimp-1 restriction of cell expansion via antagonism of STAT5 target genes. Sci. Immunol. 2016, 1, eaaf8612. [Google Scholar]
- Li, P.; Mitra, S.; Spolski, R.; Oh, J.; Liao, W.; Tang, Z.; Mo, F.; Li, X.; West, E.E.; Gromer, D.; et al. STAT5-mediated chromatin interactions in superenhancers activate IL-2 highly inducible genes: Functional dissection of the Il2ra gene locus. Proc. Natl. Acad. Sci. USA 2017, 114, 12111–12119. [Google Scholar] [CrossRef]
- Onishi, M.; Nosaka, T.; Misawa, K.; Mui, A.L.; Gorman, D.; McMahon, M.; Miyajima, A.; Kitamura, T. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol. Cell. Biol. 1998, 18, 3871–3879. [Google Scholar] [CrossRef]
- Gotthardt, D.; Putz, E.M.; Grundschober, E.; Prchal-Murphy, M.; Straka, E.; Kudweis, P.; Heller, G.; Bago-Horvath, Z.; Witalisz-Siepracka, A.; Cumaraswamy, A.A.; et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 2016, 6, 414–429. [Google Scholar] [CrossRef]
- Cayrol, F.; Praditsuktavorn, P.; Fernando, T.M.; Kwiatkowski, N.; Marullo, R.; Calvo-Vidal, M.N.; Phillip, J.; Pera, B.; Yang, S.N.; Takpradit, K.; et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun. 2017, 8, 14290. [Google Scholar] [CrossRef]
- Nivarthi, H.; Prchal-Murphy, M.; Swoboda, A.; Hager, M.; Schlederer, M.; Kenner, L.; Tuckermann, J.; Sexl, V.; Moriggl, R.; Ermakova, O. Stat5 gene dosage in T cells modulates CD8+ T-cell homeostasis and attenuates contact hypersensitivity response in mice. Allergy 2015, 70, 67–79. [Google Scholar] [CrossRef]
- Liao, W.; Spolski, R.; Li, P.; Du, N.; West, E.E.; Ren, M.; Mitra, S.; Leonard, W.J. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc. Natl. Acad. Sci. USA 2014, 111, 3508–3513. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Mohn, S.E.; Weinmann, A.S. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat. Immunol. 2012, 13, 405–411. [Google Scholar] [CrossRef]
- Son, H.J.; Lee, S.H.; Lee, S.Y.; Kim, E.K.; Yang, E.J.; Kim, J.K.; Seo, H.B.; Park, S.H.; Cho, M.L. Oncostatin M Suppresses Activation of IL-17/Th17 via SOCS3 Regulation in CD4+ T Cells. J. Immunol. 2017, 198, 1484–1491. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Muller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Moriggl, R.; Sexl, V.; Kenner, L.; Duntsch, C.; Stangl, K.; Gingras, S.; Hoffmeyer, A.; Bauer, A.; Piekorz, R.; Wang, D.; et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell 2005, 7, 87–99. [Google Scholar] [CrossRef]
- Grange, M.; Giordano, M.; Mas, A.; Roncagalli, R.; Firaguay, G.; Nunes, J.A.; Ghysdael, J.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Control of CD8 T cell proliferation and terminal differentiation by active STAT5 and CDKN2A/CDKN2B. Immunology 2015, 145, 543–557. [Google Scholar] [CrossRef]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132–2142. [Google Scholar] [CrossRef]
- Grange, M.; Buferne, M.; Verdeil, G.; Leserman, L.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Activated STAT5 promotes long-lived cytotoxic CD8+ T cells that induce regression of autochthonous melanoma. Cancer Res. 2012, 72, 76–87. [Google Scholar] [CrossRef]
- Maurer, B.; Nivarthi, H.; Wingelhofer, B.; Pham, H.T.T.; Schlederer, M.; Suske, T.; Grausenburger, R.; Schiefer, A.I.; Prchal-Murphy, M.; Chen, D.; et al. High activation of STAT5A drives peripheral T-cell lymphoma and leukemia. Haematologica 2019. [Google Scholar] [CrossRef]
- Burchill, M.A.; Goetz, C.A.; Prlic, M.; O’Neil, J.J.; Harmon, I.R.; Bensinger, S.J.; Turka, L.A.; Brennan, P.; Jameson, S.C.; Farrar, M.A. Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: Development of CD4+CD25+ regulatory T cells versus CD8+ memory T cells. J. Immunol. 2003, 171, 5853–5864. [Google Scholar] [CrossRef]
- Heltemes-Harris, L.M.; Larson, J.D.; Starr, T.K.; Hubbard, G.K.; Sarver, A.L.; Largaespada, D.A.; Farrar, M.A. Sleeping Beauty transposon screen identifies signaling modules that cooperate with STAT5 activation to induce B-cell acute lymphoblastic leukemia. Oncogene 2016, 35, 3454–3464. [Google Scholar] [CrossRef]
- Katerndahl, C.D.S.; Heltemes-Harris, L.M.; Willette, M.J.L.; Henzler, C.M.; Frietze, S.; Yang, R.; Schjerven, H.; Silverstein, K.A.T.; Ramsey, L.B.; Hubbard, G.; et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 2017, 18, 694–704. [Google Scholar] [CrossRef]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef]
- De Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P.; et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef]
- Jones, L.L.; Alli, R.; Li, B.; Geiger, T.L. Differential T Cell Cytokine Receptivity and Not Signal Quality Distinguishes IL-6 and IL-10 Signaling during Th17 Differentiation. J. Immunol. 2016, 196, 2973–2985. [Google Scholar] [CrossRef]
- Ashouri, J.F.; Hsu, L.Y.; Yu, S.; Rychkov, D.; Chen, Y.; Cheng, D.A.; Sirota, M.; Hansen, E.; Lattanza, L.; Zikherman, J.; et al. Reporters of TCR signaling identify arthritogenic T cells in murine and human autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2019, 116, 18517–18527. [Google Scholar] [CrossRef]
- Logotheti, S.; Putzer, B.M. STAT3 and STAT5 Targeting for Simultaneous Management of Melanoma and Autoimmune Diseases. Cancers 2019, 11, 1448. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef]
- Cui, W.; Liu, Y.; Weinstein, J.S.; Craft, J.; Kaech, S.M. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity 2011, 35, 792–805. [Google Scholar] [CrossRef]
- Siegel, A.M.; Heimall, J.; Freeman, A.F.; Hsu, A.P.; Brittain, E.; Brenchley, J.M.; Douek, D.C.; Fahle, G.H.; Cohen, J.I.; Holland, S.M.; et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity 2011, 35, 806–818. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef]
- Kujawski, M.; Zhang, C.; Herrmann, A.; Reckamp, K.; Scuto, A.; Jensen, M.; Deng, J.; Forman, S.; Figlin, R.; Yu, H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010, 70, 9599–9610. [Google Scholar] [CrossRef]
- Yue, C.; Shen, S.; Deng, J.; Priceman, S.J.; Li, W.; Huang, A.; Yu, H. STAT3 in CD8+ T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis. Cancer Immunol. Res. 2015, 3, 864–870. [Google Scholar] [CrossRef]
- Triplett, T.A.; Tucker, C.G.; Triplett, K.C.; Alderman, Z.; Sun, L.; Ling, L.E.; Akporiaye, E.T.; Weinberg, A.D. STAT3 Signaling Is Required for Optimal Regression of Large Established Tumors in Mice Treated with Anti-OX40 and TGFbeta Receptor Blockade. Cancer Immunol. Res. 2015, 3, 526–535. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Krieg, C.; Letourneau, S.; Pantaleo, G.; Boyman, O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11906–11911. [Google Scholar] [CrossRef]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef]
- Verdeil, G.; Marquardt, K.; Surh, C.D.; Sherman, L.A. Adjuvants targeting innate and adaptive immunity synergize to enhance tumor immunotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 16683–16688. [Google Scholar] [CrossRef]
- Boyman, O.; Arenas-Ramirez, N. Development of a novel class of interleukin-2 immunotherapies for metastatic cancer. Swiss Med. Wkly. 2019, 149, w14697. [Google Scholar] [CrossRef]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211. [Google Scholar] [CrossRef]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J., Jr.; Rosenberg, S.A. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef]
- Yang, S.; Ji, Y.; Gattinoni, L.; Zhang, L.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol. Immunother. 2013, 62, 727–736. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Milner, J.D.; Vogel, T.P.; Forbes, L.; Ma, C.A.; Stray-Pedersen, A.; Niemela, J.E.; Lyons, J.J.; Engelhardt, K.R.; Zhang, Y.; Topcagic, N.; et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015, 125, 591–599. [Google Scholar] [CrossRef]
- Sadreev, I.I.; Chen, M.Z.Q.; Umezawa, Y.; Biktashev, V.N.; Kemper, C.; Salakhieva, D.V.; Welsh, G.I.; Kotov, N.V. The competitive nature of signal transducer and activator of transcription complex formation drives phenotype switching of T cells. Immunology 2018, 153, 488–501. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Giordano, M.; Henin, C.; Maurizio, J.; Imbratta, C.; Bourdely, P.; Buferne, M.; Baitsch, L.; Vanhille, L.; Sieweke, M.H.; Speiser, D.E.; et al. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J. 2015, 34, 2042–2058. [Google Scholar] [CrossRef]
- Speiser, D.E.; Ho, P.C.; Verdeil, G. Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol. 2016, 16, 599–611. [Google Scholar] [CrossRef]
- Ladak, K.; Bass, A.R. Checkpoint inhibitor-associated autoimmunity. Best Pract. Res. Clin. Rheumatol. 2018, 32, 781–802. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Celis-Gutierrez, J.; Blattmann, P.; Zhai, Y.; Jarmuzynski, N.; Ruminski, K.; Gregoire, C.; Ounoughene, Y.; Fiore, F.; Aebersold, R.; Roncagalli, R.; et al. Quantitative Interactomics in Primary T Cells Provides a Rationale for Concomitant PD-1 and BTLA Coinhibitor Blockade in Cancer Immunotherapy. Cell Rep. 2019, 27, 3315–3330. [Google Scholar] [CrossRef]
- Franceschini, D.; Paroli, M.; Francavilla, V.; Videtta, M.; Morrone, S.; Labbadia, G.; Cerino, A.; Mondelli, M.U.; Barnaba, V. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. J. Clin. Investig. 2009, 119, 551–564. [Google Scholar] [CrossRef]
- Taylor, D.K.; Walsh, P.T.; LaRosa, D.F.; Zhang, J.; Burchill, M.A.; Farrar, M.A.; Turka, L.A. Constitutive activation of STAT5 supersedes the requirement for cytokine and TCR engagement of CD4+ T cells in steady-state homeostasis. J. Immunol. 2006, 177, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Buferne, M.; Chasson, L.; Grange, M.; Mas, A.; Arnoux, F.; Bertuzzi, M.; Naquet, P.; Leserman, L.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. IFNgamma producing CD8+ T cells modified to resist major immune checkpoints induce regression of MHC class I-deficient melanomas. Oncoimmunology 2015, 4, e974959. [Google Scholar] [CrossRef] [PubMed]
- Auphan-Anezin, N.; Verdeil, G.; Grange, M.; Soudja, S.M.; Wehbe, M.; Buferne, M.; Mas, A.; Schmitt-Verhulst, A.M. Immunosuppression in inflammatory melanoma: Can it be resisted by adoptively transferred T cells? Pigment Cell Melanoma Res. 2013, 26, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Verdeil, G.; Fuertes Marraco, S.A.; Murray, T.; Speiser, D.E. From T cell “exhaustion” to anti-cancer immunity. Biochim. Biophys. Acta 2016, 1865, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fu, B.; Gao, Y.; Liao, X.; Sun, R.; Tian, Z.; Wei, H. TGF-beta1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. 2012, 8, e1002594. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Benahmed, M.; Meresse, B.; Arnulf, B.; Barbe, U.; Mention, J.J.; Verkarre, V.; Allez, M.; Cellier, C.; Hermine, O.; Cerf-Bensussan, N. Inhibition of TGF-beta signaling by IL-15: A new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 2007, 132, 994–1008. [Google Scholar] [CrossRef]
- Xiao, Q.; Wu, J.; Wang, W.J.; Chen, S.; Zheng, Y.; Yu, X.; Meeth, K.; Sahraei, M.; Bothwell, A.L.M.; Chen, L.; et al. DKK2 imparts tumor immunity evasion through beta-catenin-independent suppression of cytotoxic immune-cell activation. Nat Med. 2018, 24, 262–270. [Google Scholar] [CrossRef]
- Mardiana, S.; Lai, J.; House, I.G.; Beavis, P.A.; Darcy, P.K. Switching on the green light for chimeric antigen receptor T-cell therapy. Clin. Transl. Immunol. 2019, 8, e1046. [Google Scholar] [CrossRef]
- Zhang, J.; Endres, S.; Kobold, S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotherapy 2019, 11, 201–213. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Horton, B.L.; Williams, J.B.; Cabanov, A.; Spranger, S.; Gajewski, T.F. Intratumoral CD8(+) T-cell Apoptosis Is a Major Component of T-cell Dysfunction and Impedes Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.S.; Huang, J.T.; Lu, X.; Simons, D.M.; Park, C.; Chang, A.; Tamaki, W.; Liu, E.; Roybal, K.T.; Seagal, J.; et al. Clonal Deletion of Tumor-Specific T Cells by Interferon-gamma Confers Therapeutic Resistance to Combination Immune Checkpoint Blockade. Immunity 2019, 50, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Powis de Tenbossche, C.G.; Cane, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.M.; Liljestrom, P.; Uyttenhove, C.; Van den Eynde, B.J. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Commun. 2017, 8, 1404. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target Genes | STAT3 Input | Cancer Type | TAM Phenotype |
|---|---|---|---|
| Cytokines/Cytokine receptors | |||
| Il10 * | positive | m-melanoma | CD11b+ [28] |
| m-PDAC | CD68+, IL-10Ra+ [29] | ||
| Il23a * | positive | m-melanoma | CD11b+ CD11c− [30] |
| Il10ra, Il4ra | positive | m-PDAC | CD68+, IL-10Ra+ [29] |
| Tgfb1 * | positive | m-melanoma | CD11b+ CD11c− [30] |
| Il12a | negative | m-melanoma | CD11b+ [28]; CD11b+ CD11c− [30] |
| Ifng | negative | m-melanoma | CD11b+ [28] |
| Chemokines/Chemokine Receptors | |||
| Ccl5, Cxcl9-10-11 | Negative | m-melanoma | CD11b+ [28] |
| Cxcl2, Cxcl12 | positive | m-melanoma | BMDM+Tumor conditioned media [31] |
| MDSC: CD11b+ GR1+ CD11c− [22] | |||
| Scavenger receptors/Endocytosis | |||
| Mrc1 (CD206) | positive | m-breast | CD11b+ Ly-6Clo F4/80hi CD24lo MHC-IIlo [32] |
| CD163 | positive | h-gastric | CD163+ CD209a+ [33] |
| h-SCC | ERK5+ CD163+ [23] | ||
| m-PDAC | CD68+, IL-10Ra+ [29] | ||
| Cd209a | positive | m-PDAC | CD68+, IL-10Ra+ [29] |
| Immune suppression | |||
| Arg1 | positive | m-PDAC; h-PDAC | CD68+, IL-10Ra+ [29]; blood CD14+ [34] |
| Cox2 | positive | m-melanoma | BMDM + Tumor conditioned media [31] |
| Ido1 | positive | m-liver metastasis | liver-MDSC: CD11b+ Ly-6Cint/hi Ly-6G+ [35] |
| Pdl1 (CD274) | positive | h- & m-glioma | h-CD68+; m-CD11b+ CD115+ [36] |
| h-breast | CD163+ [37] | ||
| m-liver metastasis | liver-MDSC: CD11b+ Ly-6Cint/hi Ly-6G+ [35] | ||
| Extra-cellular Matrix/Angiogenesis | |||
| Mmp2 | positive | m-melanoma | BMDM + Tumor conditioned media [31] |
| Vegf | positive | m-melanoma | CD11b+ [28]; MDSC: CD11b+ GR1+ CD11c- [22] |
| Cathepsin (B, L) | positive | m-PDAC | CD68+, IL-10Ra+ [29] |
| Cell cycle/TFs | |||
| Ccnd1 | positive | m-melanoma | BMDM + Tumor conditioned media [31] |
| ATF6, sXBP1 | positive | m-PDAC | CD68+, IL-10Ra+ [29] |
| STAT3 and STAT5-Regulated Genes in T and NK cells | ||||
|---|---|---|---|---|
| Target Genes | STAT3 Input | Cell Subsets | STAT5 Input | Cell Subsets |
| Cytokines/Cytokine receptors | ||||
| Il17 | positive | hCD4 Th-17 [69] | ||
| Il10 | positive | mCD4 Th-2 [70] | ||
| Il2ra (CD25) | positive | mCD4 Th-21 [71] | positive | mCD4 Th-1 [71], mCD8 [60], mNK [72] |
| Il7ra (CD127) | negative | mCD8 [60] | ||
| Il6st | negative | mCD8 [60] | ||
| Tgfb2r | negative | mCD8 [60] | ||
| Homing | ||||
| CCR7 | negative | mCD8 [60] | ||
| Sell (CD62L) | negative | mCD8 [60] | ||
| TFs | ||||
| Prdm1 (Blimp) | positive | mCD4 Th-2 [70] | ||
| Eomes | positive | mCD8 [60] | ||
| Tbx21 (Tbet) | positive | mCD8 [60] | ||
| Cytotoxicity | ||||
| Gzmb | positive | mCD8 [60], mNK [73] | ||
| Prf1 | positive | mCD8 [60], mNK [73] | ||
| Ifng | positive | mCD8 [60], mNK [73] | ||
| Fasl | positive | mCD8 [60] | ||
| Cell cycle/Cell survival | ||||
| Myc | positive | hT lymphoma [74] | positive | mCD8 [60] |
| Pim1 | positive | hT lymphoma [74] | positive | mCD8 [60,75] |
| Bcl2 | positive | hT lymphoma [74] | positive | mCD8 [49,60]; NK [73] |
| Competitive gene regulation by STAT3 and STAT5 in T cells * | ||||
| Target Genes | STAT3 Input | STAT5 Input | T Cell Subsets | |
| Il17 | positive | competitor | mCD4 Th-17 [67] | |
| Il9 | competitor | positive | mCD4 Th-9 [64,76] | |
| Bcl6 | positive | competitor | mCD4 Tfh; mCD4 Th-1 [68,77] | |
| Socs3 | positive | positive | mCD4 Th-17/Treg balance [78] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdeil, G.; Lawrence, T.; Schmitt-Verhulst, A.-M.; Auphan-Anezin, N. Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy. Cancers 2019, 11, 1832. https://doi.org/10.3390/cancers11121832
Verdeil G, Lawrence T, Schmitt-Verhulst A-M, Auphan-Anezin N. Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy. Cancers. 2019; 11(12):1832. https://doi.org/10.3390/cancers11121832
Chicago/Turabian StyleVerdeil, Grégory, Toby Lawrence, Anne-Marie Schmitt-Verhulst, and Nathalie Auphan-Anezin. 2019. "Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy" Cancers 11, no. 12: 1832. https://doi.org/10.3390/cancers11121832
APA StyleVerdeil, G., Lawrence, T., Schmitt-Verhulst, A.-M., & Auphan-Anezin, N. (2019). Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy. Cancers, 11(12), 1832. https://doi.org/10.3390/cancers11121832