Cancer Cell Membrane-Coated Nanoparticles for Cancer Management


Abstract
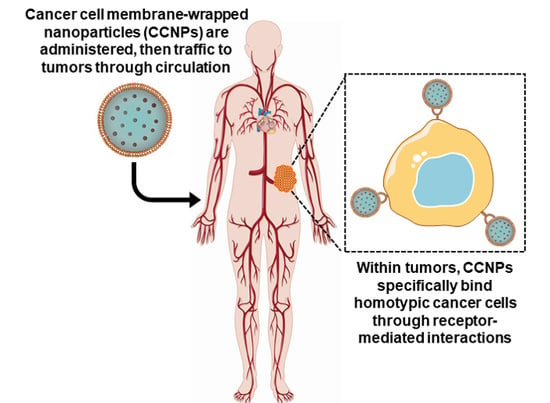
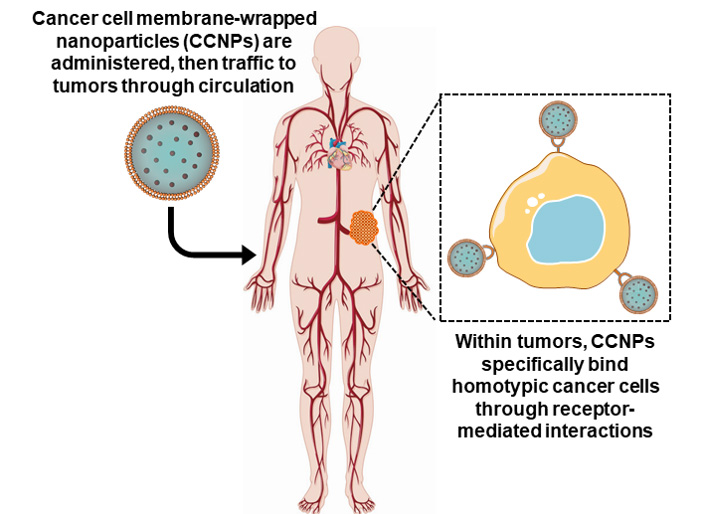
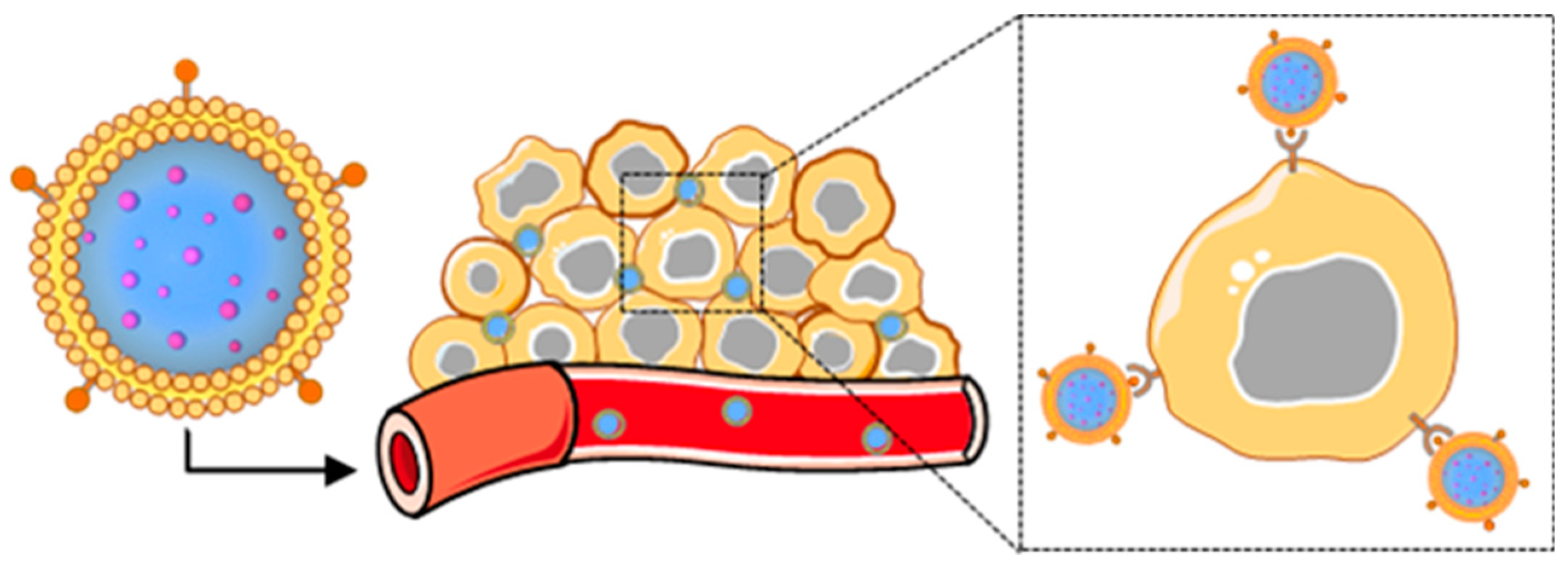
:
1. Introduction to Cancer and Nanomedicine
2. Cancer Cell Membrane-Wrapped Nanovehicles
2.1. Multi-step Synthesis of Cell Membrane-Wrapped Nanovehicles
2.1.1. Membrane Extraction
2.1.2. Selection of Nanoparticle Core
2.1.3. Fusion of Membrane Vesicles with Nanoparticle Cores
2.2. Characterization of Membrane-Coated Nanoparticles
3. Applications of Membrane-Wrapped Nanoparticles in Cancer
3.1. Drug Delivery
3.2. Photothermal and Photodynamic Therapy
3.2.1. Combination Photothermal Therapy and Chemotherapy
3.2.2. Photodynamic Therapy Combined with Chemotherapy or Starvation Therapy
3.3. Tumor Imaging
3.4. Immune Stimulation
4. Challenges and Path Forward
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer Statistics, 2019. CA. Cancer J. Clin. 2019, 57, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Yin, Q.; Li, Y. Progress of Cell-Derived Biomimetic Drug Delivery Systems for Cancer Therapy. Adv. Ther. 2018, 1, 1800053. [Google Scholar] [CrossRef]
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 12320–12364. [Google Scholar]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng. 2012. [Google Scholar] [CrossRef]
- Fan, W.; Yung, B.; Huang, P.; Chen, X. Nanotechnology for Multimodal Synergistic Cancer Therapy. Chem. Rev. 2017, 117, 13566–13638. [Google Scholar] [CrossRef]
- Kim, K.Y. Nanotechnology platforms and physiological challenges for cancer therapeutics. Nanomed. Nanotechnol. Biol. Med. 2007, 3, 103–110. [Google Scholar] [CrossRef]
- Vijayan, V.; Uthaman, S.; Park, I.K. Cell membrane-camouflaged nanoparticles: A promising biomimetic strategy for cancer theragnostics. Polymers (Basel) 2018, 10, 983. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar]
- Thanuja, M.Y.; Anupama, C.; Ranganath, S.H. Bioengineered cellular and cell membrane-derived vehicles for actively targeted drug delivery: So near and yet so far. Adv. Drug Deliv. Rev. 2018, 132, 57–80. [Google Scholar] [CrossRef]
- Matsumara, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accum. Cancer Res. 1987, 46, 6387–6392. [Google Scholar]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Frakhzad, O.C. Cancer Nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Gerlowski, L.E.; Jain, R.K. Microvascular permeability of normal and neoplastic tissues. Microvasc. Res. 1986, 31, 288–305. [Google Scholar] [CrossRef]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Rao, L.; Bu, L.L.; Xu, J.H.; Cai, B.; Yu, G.T.; Yu, X.; He, Z.; Huang, Q.; Li, A.; Guo, S.S.; et al. Red Blood Cell Membrane as a Biomimetic Nanocoating for Prolonged Circulation Time and Reduced Accelerated Blood Clearance. Small 2015, 11, 6225–6236. [Google Scholar] [CrossRef]
- Verhoef, J.J.F.; Anchordoquy, T.J. Questioning the use of PEGylation for drug delivery. Drug Deliv. Transl. Res. 2013, 3, 499–503. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surfaces B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Vasir, J.K.; Labhasetwar, V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv. Drug Deliv. Rev. 2008, 59, 718–728. [Google Scholar] [CrossRef]
- Astete, C.E.; Sabilov, C.M. Synthesis and characterization of PLGA nanoparticles. J. Mater. Sci. Mater. Electron. 2011, 22, 1761–1765. [Google Scholar] [CrossRef]
- Valcourt, D.M.; Harris, J.; Riley, R.S.; Dang, M.; Wang, J.; Day, E.S. Advances in targeted nanotherapeutics: From bioconjugation to biomimicry. Nano Res. 2018, 11, 4999–5016. [Google Scholar] [CrossRef]
- Fang, R.H.; Kroll, A.V.; Gao, W.; Zhang, L. Cell Membrane Coating Nanotechnology. Adv. Mater. 2018, 30, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Jiang, Y.; Fang, J.C.; Zhang, L. Cell membrane-derived nanomaterials for biomedical applications. Biomaterials 2018, 128, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Luk, B.T.; Zhang, L. Cell Membrane-Camouflaged Nanoparticles for Drug Delivery. J. Control. Release 2016, 25, 289–313. [Google Scholar] [CrossRef] [PubMed]
- Kroll, A.V.; Fang, R.H.; Zhang, L. Biointerfacing and applications of cell membrane-coated nanoparticles. Bioconjug. Chem. 2017, 28, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; He, Y.; Zhang, S.; Qin, J.; Wang, J. Cell membrane-based nanoparticles: A new biomimetic platform for tumor diagnosis and treatment. Acta Pharm. Sin. B 2018, 8, 14–22. [Google Scholar] [CrossRef]
- Hu, C.-M.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef]
- Aryal, S.; Hu, C.-M.J.; Fang, R.H.; Dehaini, D.; Carpenter, C.; Zhang, D.-E.; Zhang, L. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine 2013, 8, 1271–1280. [Google Scholar] [CrossRef]
- Piao, J.G.; Wang, L.; Gao, F.; You, Y.Z.; Xiong, Y.; Yang, L. Erythrocyte membrane is an alternative coating to polyethylene glycol for prolonging the circulation lifetime of gold nanocages for photothermal therapy. ACS Nano 2014, 8, 10414–10425. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, D.; Song, Q.; Wu, T.; Zhuang, X.; Bao, Y.; Kong, M.; Qi, Y.; Tan, S.; Zhang, Z. Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity against Melanoma. ACS Nano 2015, 9, 6918–6933. [Google Scholar] [CrossRef]
- Su, J.; Sun, H.; Meng, Q.; Zhang, P.; Yin, Q.; Li, Y. Enhanced blood suspensibility and laser-Activated tumor-specific drug release of theranostic mesoporous silica nanoparticles by functionalizing with erythrocyte membranes. Theranostics 2017, 7, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zheng, R.; Fang, X.; Wang, X.; Zhang, X.; Yang, W.; Sha, X. Red blood cell membrane camouflaged magnetic nanoclusters for imaging-guided photothermal therapy. Biomaterials 2016, 92, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Quattrocchi, N.; Van De Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Sun, W.; Qian, C.; Wang, C.; Bomba, H.N.; Gu, Z. Anticancer platelet-mimicking nanovehicles. Adv. Mater. 2016, 27, 7043–7050. [Google Scholar] [CrossRef] [PubMed]
- Xuan, M.; Shao, J.; Dai, L.; He, Q.; Li, J. Macrophage Cell Membrane Camouflaged Mesoporous Silica Nanocapsules for In Vivo Cancer Therapy. Adv. Healthc. Mater. 2015, 4, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Fang, R.; Wang, K.; Luk, B.T.; Thamphiwatana, S.; Dehaini, D.; Nguyen, P.; Angsantikul, P.; Wen, C.H.; Kroll, A.V.; et al. Nanoparticle biointerfacing via platelet membrane cloaking. Nature 2016, 526, 118–121. [Google Scholar] [CrossRef]
- Zhang, Q.; Dehaini, D.; Zhang, Y.; Zhou, J.; Chen, X.; Zhang, L.; Fang, R.H.; Gao, W.; Zhang, L. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 2018, 13, 1182–1190. [Google Scholar] [CrossRef]
- Rao, L.; Bu, L.L.; Ma, L.; Wang, W.; Liu, H.; Wan, D.; Liu, J.F.; Li, A.; Guo, S.S.; Zhang, L.; et al. Platelet-Facilitated Photothermal Therapy of Head and Neck Squamous Cell Carcinoma. Angew. Chem. Int. Ed. 2018, 57, 986–991. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Qian, C.; Bomba, H.N.; Xin, H.; Gu, Z. Relay Drug Delivery for Amplifying Targeting Signal and Enhancing Anticancer Efficacy. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef]
- Sun, H.; Su, J.; Meng, Q.; Yin, Q.; Chen, L.; Gu, W.; Zhang, P.; Zhang, Z.; Yu, H.; Wang, S.; et al. Cancer-Cell-Biomimetic Nanoparticles for Targeted Therapy of Homotypic Tumors. Adv. Mater. 2016, 28, 9581–9588. [Google Scholar] [CrossRef]
- Dehaini, D.; Fang, R.H.; Zhang, L. Biomimetic strategies for targeted nanoparticle delivery. Bioeng. Transl. Med. 2016, 1, 30–46. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Su, J.; Meng, Q.; Yin, Q.; Chen, L.; Gu, W.; Zhang, Z.; Yu, H.; Zhang, P.; Wang, S.; et al. Cancer Cell Membrane-Coated Gold Nanocages with Hyperthermia-Triggered Drug Release and Homotypic Target Inhibit Growth and Metastasis of Breast Cancer. Adv. Funct. Mater. 2017, 27. [Google Scholar] [CrossRef]
- Jin, J.; Krishnamachary, B.; Barnett, J.D.; Chatterjee, S.; Chang, D.; Mironchik, Y.; Wildes, F.; Jaffee, E.M.; Nimmagadda, S.; Bhujwalla, Z.M. Human Cancer Cell Membrane-Coated Biomimetic Nanoparticles Reduce Fibroblast-Mediated Invasion and Metastasis and Induce T-Cells. ACS Appl. Mater. Interfaces 2019, 11, 7850–7861. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Zheng, D.W.; Zhang, M.K.; Yu, W.Y.; Qiu, W.X.; Hu, J.J.; Feng, J.; Zhang, X.Z. Preferential Cancer Cell Self-Recognition and Tumor Self-Targeting by Coating Nanoparticles with Homotypic Cancer Cell Membranes. Nano Lett. 2016, 16, 5895–5901. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Hu, C.M.J.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, M.; He, J. Cancer cell membrane cloaking nanoparticles for targeted co-delivery of doxorubicin and PD-L1 siRNA. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Luk, B.T.; Zhang, L. Cell membrane-camouflaged nanoparticles for drug delivery. J. Control. Release 2015, 220, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Su, J.; Ran, W.; Zhang, P.; Yin, Q.; Zhang, Z.; Yu, H.; Li, Y. Preparation and application of cell membrane-camouflaged nanoparticles for cancer therapy. Theranostics 2017, 7, 2575–2592. [Google Scholar] [CrossRef]
- Chen, W.; Zeng, K.; Liu, H.; Ouyang, J.; Wang, L.; Liu, Y.; Wang, H.; Deng, L.; Liu, Y.N. Cell Membrane Camouflaged Hollow Prussian Blue Nanoparticles for Synergistic Photothermal-/Chemotherapy of Cancer. Adv. Funct. Mater. 2017, 27. [Google Scholar] [CrossRef]
- Xia, Q.; Zhang, Y.; Li, Z.; Hou, X.; Feng, N. Red blood cell membrane-camouflaged nanoparticles: A novel drug delivery system for antitumor application. Acta Pharm. Sin. B 2019. [Google Scholar] [CrossRef]
- Rao, L.; Cai, B.; Bu, L.L.; Liao, Q.Q.; Guo, S.S.; Zhao, X.Z.; Dong, W.F.; Liu, W. Microfluidic Electroporation-Facilitated Synthesis of Erythrocyte Membrane-Coated Magnetic Nanoparticles for Enhanced Imaging-Guided Cancer Therapy. ACS Nano 2017, 11, 3496–3505. [Google Scholar] [CrossRef] [PubMed]
- Tsong, T.Y. Electroporation of cell membranes. Biophys. J. 1991, 60, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Zhou, H.; Li, P.Y.; Speer, J.E.; Cheng, H. Structural elucidation of cell membrane-derived nanoparticles using molecular probes. J. Mater. Chem. B 2014, 2, 8231–8238. [Google Scholar] [CrossRef]
- Rao, L.; Bu, L.L.; Cai, B.; Xu, J.H.; Li, A.; Zhang, W.F.; Sun, Z.J.; Guo, S.S.; Liu, W.; Wang, T.H.; et al. Cancer Cell Membrane-Coated Upconversion Nanoprobes for Highly Specific Tumor Imaging. Adv. Mater. 2016, 28, 3460–3466. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Deng, W.W.; Zan, M.; Rao, L.; Yu, G.T.; Zhu, D.M.; Wu, W.T.; Chen, B.; Ji, L.W.; Chen, L.; et al. Cancer Cell Membrane Camouflaged Nanoparticles to Realize Starvation Therapy Together with Checkpoint Blockades for Enhancing Cancer Therapy. ACS Nano 2019, 13, 2849–2857. [Google Scholar] [CrossRef]
- Liu, C.M.; Chen, G.B.; Chen, H.H.; Zhang, J.B.; Li, H.Z.; Sheng, M.X.; Weng, W.B.; Guo, S.M. Cancer cell membrane-cloaked mesoporous silica nanoparticles with a pH-sensitive gatekeeper for cancer treatment. Colloids Surfaces B Biointerfaces 2019, 175, 477–486. [Google Scholar] [CrossRef]
- Wang, D.; Dong, H.; Li, M.; Cao, Y.; Yang, F.; Zhang, K.; Dai, W.; Wang, C.; Zhang, X. Erythrocyte-Cancer Hybrid Membrane Camouflaged Hollow Copper Sulfide Nanoparticles for Prolonged Circulation Life and Homotypic-Targeting Photothermal/Chemotherapy of Melanoma. ACS Nano 2018, 12, 5241–5252. [Google Scholar] [CrossRef]
- Rao, L.; Meng, Q.F.; Huang, Q.; Liu, P.; Bu, L.L.; Kondamareddy, K.K.; Guo, S.S.; Liu, W.; Zhao, X.Z. Photocatalytic Degradation of Cell Membrane Coatings for Controlled Drug Release. Adv. Healthc. Mater. 2016, 5, 1420–1427. [Google Scholar] [CrossRef]
- Copp, J.A.; Fang, R.H.; Luk, B.T.; Hu, C.-M.J.; Gao, W.; Zhang, K.; Zhang, L. Clearance of pathological antibodies using biomimetic nanoparticles. Proc. Natl. Acad. Sci. USA 2014, 111, 13481–13486. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Fan, Z.; Lemons, P.K.; Cheng, H. A facile approach to functionalize cell membrane-coated nanoparticles. Theranostics 2016, 6, 1012–1022. [Google Scholar] [CrossRef]
- Li, J.; Zhen, X.; Lyu, Y.; Jiang, Y.; Huang, J.; Pu, K. Cell Membrane Coated Semiconducting Polymer Nanoparticles for Enhanced Multimodal Cancer Phototheranostics. ACS Nano 2018, 12, 8520–8530. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wu, S.; Wang, J. Cancer cell membrane–coated nanocarriers for homologous target inhibiting the growth of hepatocellular carcinoma. J. Bioact. Compat. Polym. 2019, 34, 58–71. [Google Scholar] [CrossRef]
- Tian, H.; Luo, Z.; Liu, L.; Zheng, M.; Chen, Z.; Ma, A.; Liang, R.; Han, Z.; Lu, C.; Cai, L. Cancer Cell Membrane-Biomimetic Oxygen Nanocarrier for Breaking Hypoxia-Induced Chemoresistance. Adv. Funct. Mater. 2017, 27, 1–7. [Google Scholar] [CrossRef]
- Yang, R.; Xu, J.; Xu, L.; Sun, X.; Chen, Q.; Zhao, Y.; Peng, R.; Liu, Z. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano 2018, 12, 5121–5129. [Google Scholar] [CrossRef]
- Yu, G.T.; Rao, L.; Wu, H.; Yang, L.L.; Bu, L.L.; Deng, W.W.; Wu, L.; Nan, X.; Zhang, W.F.; Zhao, X.Z.; et al. Myeloid-Derived Suppressor Cell Membrane-Coated Magnetic Nanoparticles for Cancer Theranostics by Inducing Macrophage Polarization and Synergizing Immunogenic Cell Death. Adv. Funct. Mater. 2018, 28, 1–9. [Google Scholar] [CrossRef]
- Song, Q.; Yin, Y.; Shang, L.; Wu, T.; Zhang, D.; Kong, M.; Zhao, Y.; He, Y.; Tan, S.; Guo, Y.; et al. Tumor Microenvironment Responsive Nanogel for the Combinatorial Antitumor Effect of Chemotherapy and Immunotherapy. Nano Lett. 2017, 17, 6366–6375. [Google Scholar] [CrossRef]
- Pan, W.; Ge, Y.; Yu, Z.; Zhou, P.; Cui, B.; Li, N.; Tang, B. A cancer cell membrane-encapsulated MnO2 nanoreactor for combined photodynamic-starvation therapy. Chem. Commun. 2019, 55, 5115–5118. [Google Scholar] [CrossRef]
- Jiang, Q.; Liu, Y.; Guo, R.; Yao, X.; Sung, S.; Pang, Z.; Yang, W. Erythrocyte-cancer hybrid membrane-camouflaged melanin nanoparticles for enhancing photothermal therapy efficacy in tumors. Biomaterials 2019, 192, 292–308. [Google Scholar] [CrossRef]
- Gao, L.; Han, L.; Ding, X.; Xu, J.; Wang, J.; Zhu, J.; Lu, W.; Sun, J.; Yu, L.; Yan, Z.; et al. An effective intracellular delivery system of monoclonal antibody for treatment of tumors: Erythrocyte membrane-coated self-associated antibody nanoparticles. Nanotechnology 2017, 28. [Google Scholar] [CrossRef]
- Gao, W.; Hu, C.M.J.; Fang, R.H.; Luk, B.T.; Su, J.; Zhang, L. Surface Functionalization of Gold Nanoparticles with Red Blood Cell Membranes. Adv. Mater. 2014, 25, 3549–3553. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, L.R.; Stafford, R.J.; Bankson, J.A.; Sershen, S.R.; Rivera, B.; Price, R.E.; Hazle, J.D.; Halas, N.J.; West, J.L. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc. Natl. Acad. Sci. USA 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, X.; Zheng, D.; Lin, X.; Wei, Z.; Zhang, D.; Li, Z.; Zhang, Y.; Wu, M.; Liu, X. Cancer cell membrane-coated magnetic nanoparticles for MR/NIR fluorescence dual-modal imaging and photodynamic therapy. Biomater. Sci. 2018, 6, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Cheng, H.; Qiu, W.X.; Zhang, L.; Wan, S.S.; Zeng, J.Y.; Zhang, X.Z. Cancer cell membrane-coated biomimetic platform for tumor targeted photodynamic therapy and hypoxia-amplified bioreductive therapy. Biomaterials 2017, 142, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Luo, Z.; Zheng, M.; Tian, H.; Gong, P.; Gao, G.; Pan, H.; Liu, L.; Ma, A.; et al. Cancer Cell Membrane-Biomimetic Nanoparticles for Homologous-Targeting Dual-Modal Imaging and Photothermal Therapy. ACS Nano 2016, 10, 10049–10057. [Google Scholar] [CrossRef] [PubMed]
- Kroll, A.V.; Fang, R.H.; Jiang, Y.; Zhou, J.; Wei, X.; Yu, C.L.; Gao, J.; Luk, B.T.; Dehaini, D.; Gao, W.; et al. Nanoparticulate Delivery of Cancer Cell Membrane Elicits Multiantigenic Antitumor Immunity. Adv. Mater. 2017, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, M.; Sun, X.; Jia, H.; Liu, W. NIR-responsive cancer cytomembrane-cloaked carrier-free nanosystems for highly efficient and self-targeted tumor drug delivery. Biomaterials 2018, 159, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Astete, C.E.; Sabliov, C.M. Synthesis and characterization of PLGA nanoparticles. J. Biomater. Sci. Polym. Ed. 2006, 17, 247–289. [Google Scholar] [CrossRef]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Giaccone, G.; Schiller, J.H.; Natale, R.B.; Miller, V.; Manegold, C.; Scagliotti, G.; Rosell, R.; Oliff, I.; Reeves, J.A.; et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: A phase III trial—INTACT 2. J. Clin. Oncol. 2004, 22, 785–794. [Google Scholar] [CrossRef]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef]
- Dezube, B.J.; Pantanowitz, L.; Aboulafia, D.M. Management of AIDS-Related Kaposi Sarcoma: Advances in Target Discovery and Treatment. AIDS Read. 2004, 1–8. [Google Scholar]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A.; Mackey, D. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Fehrenbacher, L.; Novotny, W.F.; Herbst, R.S.; Nemunaitis, J.J.; Jablons, D.M.; Langer, C.J.; DeVore, R.F.; Gaudreault, J.; Damico, L.A.; et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- du Bois, A.; Luck, H.-J.; Meier, W.; Adams, H.-P.; Mobus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schroder, W.; et al. A Randomized Clinical Trial of Cisplatin/Paclitaxel Versus Carboplatin/Paclitaxel as First-Line Treatment of Ovarian Cancer. J. Natl. Cancer Inst. 2003, 95, 1320–1329. [Google Scholar] [CrossRef]
- Sparano, J.A.; Wang, M.; Martino, S.; Jones, V.; Perez, E.A.; Sapher, T.; Wolff, A.; Sledge, G.W.; Wood, W.C.; Davidson, N.E. Weekly Paclitaxel in the Adjuvant Treatment of Breast Cancer. N. Engl. J. Med. 2008, 358, 1663–1671. [Google Scholar] [CrossRef]
- Fay, B.L.; Melamed, J.R.; Day, E.S. Nanoshell-mediated photothermal therapy can enhance chemotherapy in inflammatory breast cancer cells. Int. J. Nanomed. 2015, 10, 6931–6941. [Google Scholar]
- Valcourt, D.M.; Dang, M.N.; Day, E.S. IR820-loaded PLGA nanoparticles for photothermal therapy of triple-negative breast cancer. J. Biomed. Mater. Res 2019, 107, 1702–1712. [Google Scholar] [CrossRef]
- Riley, R.S.; Day, E.S. Gold nanoparticle-mediated photothermal therapy: Applications and opportunities for multimodal cancer treatment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9. [Google Scholar] [CrossRef]
- Riley, R.S.; O’sullivan, R.K.; Potocny, A.M.; Rosenthal, J.; Day, E.S. Evaluating nanoshells and a potent biladiene photosensitizer for dual photothermal and photodynamic therapy of triple negative breast cancer cells. Nanomaterials 2018, 8, 658. [Google Scholar] [CrossRef] [Green Version]
- Day, E.S.; Thompson, P.A.; Zhang, L.; Lewinski, N.A.; Ahmed, N.; Drezek, R.A.; Blaney, S.M.; West, J.L. Nanoshell-mediated photothermal therapy improves survival in a murine glioma model. J. Neurooncol. 2011, 104, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Day, E.S.; Zhang, L.; Thompson, P.A.; Zawaski, J.A.; Kaffes, C.C.; Gaber, M.W.; Blaney, S.M.; West, J.L. Vascular-targeted photothermal therapy of an orthotopic murine glioma model. Nanomedicine 2012, 7, 1133–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamed, J.R.; Edelstein, R.S.; Day, E.S. Elucidating the fundamental mechanisms of cell death triggered by photothermal therapy. ACS Nano 2015, 9, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-M.J.; Fang, R.H.; Luk, B.T.; Chen, K.N.H.; Carpenter, C.; Gao, W.; Zhang, K.; Zhang, L. ‘Marker-of-self’ functionalization of nanoscale particles through a top-down cellular membrane coating approach. Nanoscale 2013, 5, 2664. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Lin, Z.; Wu, Z.; Lin, X.; He, Q. Stem-Cell-Membrane Camouflaging on Near-Infrared Photoactivated Upconversion Nanoarchitectures for in Vivo Remote-Controlled Photodynamic Therapy. ACS Appl. Mater. Interfaces 2016, 8, 34252–34260. [Google Scholar] [CrossRef]
- Xuan, M.; Shao, J.; Dai, L.; Li, J.; He, Q. Macrophage Cell Membrane Camouflaged Au Nanoshells for in Vivo Prolonged Circulation Life and Enhanced Cancer Photothermal Therapy. ACS Appl. Mater. Interfaces 2016, 8, 9610–9618. [Google Scholar] [CrossRef]
- Liu, W.L.; Liu, T.; Zou, M.Z.; Yu, W.Y.; Li, C.X.; He, Z.Y.; Zhang, M.K.; Liu, M.D.; Li, Z.H.; Feng, J.; et al. Aggressive Man-Made Red Blood Cells for Hypoxia-Resistant Photodynamic Therapy. Adv. Mater. 2018, 30, 1–10. [Google Scholar] [CrossRef]
- Xuan, M.; Shao, J.; Zhao, J.; Li, Q.; Dai, L.; Li, J. Magnetic Mesoporous Silica Nanoparticles Cloaked by Red Blood Cell Membranes: Applications in Cancer Therapy. Angew. Chem. Int. Ed. 2018, 57, 6049–6053. [Google Scholar] [CrossRef]
- Zhen, X.; Cheng, P.; Pu, K. Recent Advances in Cell Membrane–Camouflaged Nanoparticles for Cancer Phototherapy. Small 2019, 15, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Von Maltzahn, G.; Xu, M.J.; Fogal, V.; Kotamraju, V.R.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Cooperative nanomaterial system to sensitize, target, and treat tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.; Son, S.; Ochyl, L.J.; Kuai, R.; Schwendeman, A.; Moon, J.J. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat. Commun. 2018. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ye, Y.; Hu, Q.; Bellotti, A.; Gu, Z. Tailoring Biomaterials for Cancer Immunotherapy: Emerging Trends and Future Outlook. Adv. Mater. 2017, 29, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. Cancer vaccines: Between the idea and the reality. Nat. Rev. Immunol. 2003, 3, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 1–12. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, J.M.; Butterfield, L.H.; Tarhini, A.A.; Zarour, H.; Kalinski, P.; Ferrone, S. Immunotherapy of Cancer in 2012. CA Cancer J. Clin. 2012, 62, 309–335. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [Green Version]
- Dehaini, D.; Wei, X.; Fang, R.H.; Masson, S.; Angsantikul, P.; Luk, B.T.; Zhang, Y.; Ying, M.; Jiang, Y.; Kroll, A.V.; et al. Erythrocyte–Platelet Hybrid Membrane Coating for Enhanced Nanoparticle Functionalization. Adv. Mater. 2017, 29, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane Source | Core NP Material | Cargo Loaded | Particle Purpose (Besides Homotypic Targeting) | Year | Ref. |
|---|---|---|---|---|---|
| 4T1 | poly(caprolactone); Pluronic F-68 | paclitaxel | drug delivery | 2016 | [40] |
| 4T1 | gold nanocages | doxorubicin | PTT; hyperthermia-triggered drug release | 2017 | [42] |
| 4T1 | poly(cyclopentadithiophene-alt-benzothiadiazole) | PTT; PDT; PA imaging | 2018 | [61] | |
| 4T1 | PCN-224 | tirapazamine | PDT; drug delivery | 2017 | [73] |
| MDA-MB-435 | Ln-doped upconversion nanocrystal | FL imaging | 2016 | [54] | |
| MDA-MB-435 | PLGA | DiD fluorophore | FL imaging | 2014 | [45] |
| Luciferase- expressing MDA-MB-231 | PLGA | FL imaging | 2019 | [43] | |
| MCF-7 1 | PLGA | indocyanine green | PTT; PA/FL imaging | 2016 | [74] |
| MCF-7 1 | PLGA | doxorubicin; hemoglobin | PDT; drug delivery | 2017 | [63] |
| MCF-7 2 | melanin | PTT; PA imaging | 2019 | [68] | |
| B16-F10 2 | hollow copper sulfide | doxorubicin | drug delivery | 2018 | [57] |
| B16-F10 | mesoporous silica | glucose oxidase | immunotherapy; starvation therapy | 2019 | [55] |
| B16-F10 | hollow manganese dioxide | chlorin e6; glucose oxidase | PDT; starvation therapy | 2019 | [67] |
| B16-F10 | PLGA | CpG 1826 | Immunotherapy 4 | 2017 | [75] |
| B16-F10 | PLGA | monophosphoryl lipid A | Immunotherapy 4 | 2014 | [45] |
| B16-OVA 3 | PLGA | imiquimod | Immunotherapy 4 | 2018 | [64] |
| HeLa | iron oxide | doxorubicin | drug delivery | 2016 | [44] |
| HeLa | PLGA | doxorubicin; siRNA | drug delivery | 2019 | [46] |
| HeLa | doxorubicin; indocyanine green | PTT; drug delivery (carrier free) | 2018 | [76] | |
| HepG2 | PLGA | doxorubicin | drug delivery | 2019 | [62] |
| H22 | iron oxide | doxorubicin | drug delivery | 2016 | [44] |
| SMMC-7721 | superparamagnetic iron oxide | chlorin e6 | PDT; MR/NIR imaging | 2018 | [72] |
| UM-SCC-7 | iron oxide | doxorubicin | drug delivery | 2016 | [44] |
| CAL 27 | Ln-doped upconversion nanocrystal | FL imaging | 2016 | [54] | |
| LNCaP-Al | mesoporous silica | doxorubicin; calcium carbonate | drug delivery; pH sensitive release | 2019 | [56] |
| DU 145 | Ln-doped upconversion nanocrystal | FL imaging | 2016 | [54] | |
| U87 | PLGA | Immunotherapy 4 | 2019 | [43] | |
| HCT 116 | Ln-doped upconversion nanocrystal | FL imaging | 2016 | [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harris, J.C.; Scully, M.A.; Day, E.S. Cancer Cell Membrane-Coated Nanoparticles for Cancer Management. Cancers 2019, 11, 1836. https://doi.org/10.3390/cancers11121836
Harris JC, Scully MA, Day ES. Cancer Cell Membrane-Coated Nanoparticles for Cancer Management. Cancers. 2019; 11(12):1836. https://doi.org/10.3390/cancers11121836
Chicago/Turabian StyleHarris, Jenna C., Mackenzie A. Scully, and Emily S. Day. 2019. "Cancer Cell Membrane-Coated Nanoparticles for Cancer Management" Cancers 11, no. 12: 1836. https://doi.org/10.3390/cancers11121836
APA StyleHarris, J. C., Scully, M. A., & Day, E. S. (2019). Cancer Cell Membrane-Coated Nanoparticles for Cancer Management. Cancers, 11(12), 1836. https://doi.org/10.3390/cancers11121836