The Protein Neddylation Inhibitor MLN4924 Suppresses Patient-Derived Glioblastoma Cells via Inhibition of ERK and AKT Signaling

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Neddylation Pathway is Overactivated in Patient-Derived Glioblastoma Specimens
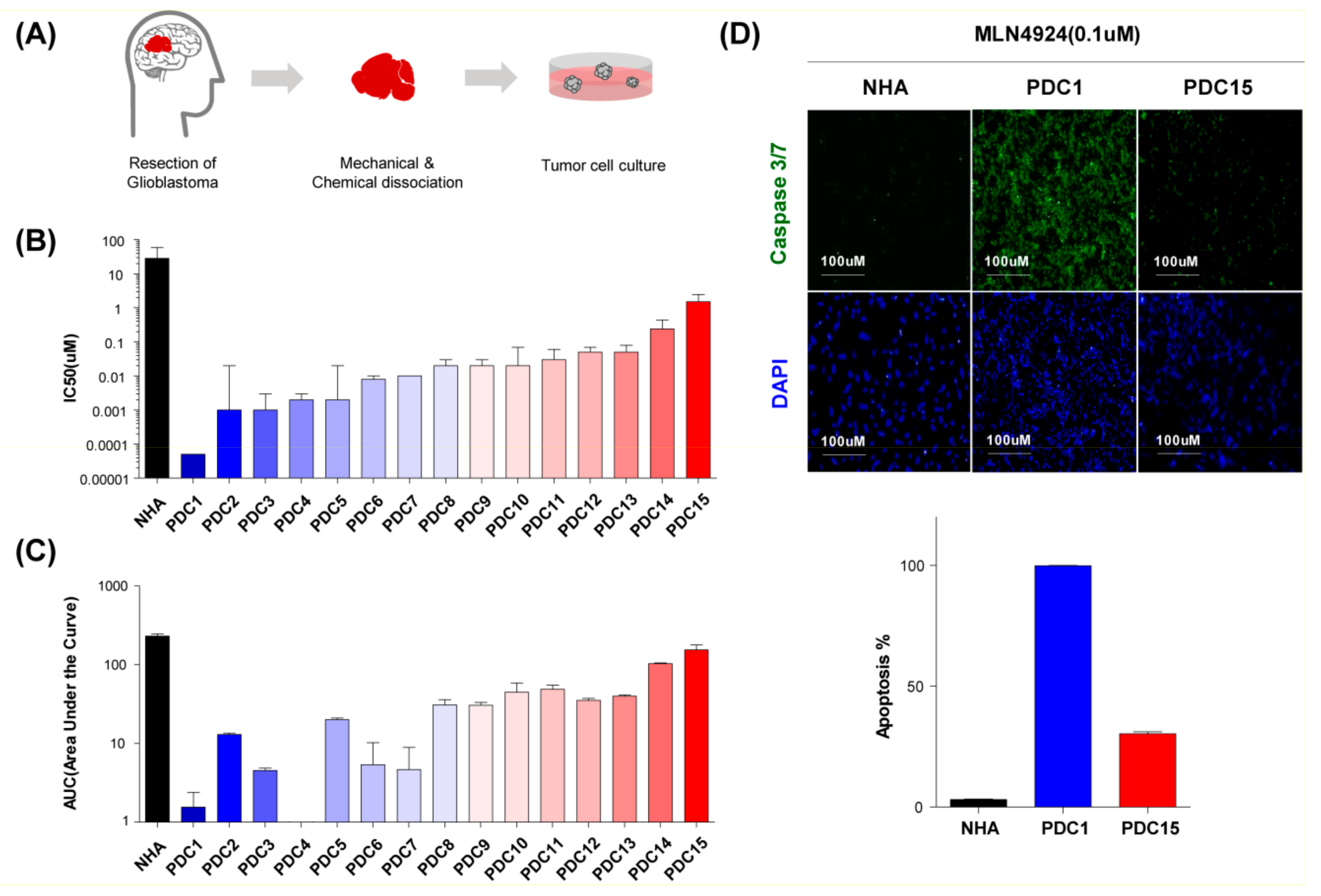
2.2. MLN4924 Inhibits Proliferation and Induces Apoptosis in Glioblastoma Cell Lines
2.3. MLN4924 Suppresses Cullin 1 Neddylation in Patient-Derived Glioblastoma Stem Cells
2.4. Response of MLN4924 is Related to ERK and AKT Signaling Pathway
2.5. Sensitivity of MLN4924 is Associated with Upregulation of ERK and AKT Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Patient-Derived Glioblastoma Specimens and Primary Cell Culture
4.3. Drugs
4.4. Immunofluorescence Staining and High Content Analysis
4.5. Western Blot Assay
4.6. Immunohistochemistry
4.7. Flow Cytometry-Based Apoptosis Detection
4.8. RNA Sequencing
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Khosla, D. Concurrent therapy to enhance radiotherapeutic outcomes in glioblastoma. Ann. Transl. Med. 2016, 4, 54. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.Y.; Staedtke, V.; Riggins, G.J. Molecular targeting of glioblastoma: Drug discovery and therapies. Trends Mol. Med. 2011, 17, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A.; Varshavsky, A. Basic Medical Research Award. The ubiquitin system. Nat. Med. 2000, 6, 1073–1081. [Google Scholar] [CrossRef]
- Pickart, C.M. Back to the future with ubiquitin. Cell 2004, 116, 181–190. [Google Scholar] [CrossRef]
- Shen, M.; Schmitt, S.; Buac, D.; Dou, Q.P. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin. Targets 2013, 17, 1091–1108. [Google Scholar] [CrossRef]
- Xirodimas, D.P. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochem. Soc. Trans. 2008, 36, 802–806. [Google Scholar] [CrossRef]
- Hua, W.; Li, C.; Yang, Z.; Li, L.; Jiang, Y.; Yu, G.; Zhu, W.; Liu, Z.; Duan, S.; Chu, Y.; et al. Suppression of glioblastoma by targeting the overactivated protein neddylation pathway. Neuro. Oncol. 2015, 17, 1333–1343. [Google Scholar] [CrossRef]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef]
- Zuo, W.; Huang, F.; Chiang, Y.J.; Li, M.; Du, J.; Ding, Y.; Zhang, T.; Lee, H.W.; Jeong, L.S.; Chen, Y.; et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-beta type II receptor. Mol. Cell 2013, 49, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Loftus, S.J.; Liu, G.; Carr, S.M.; Munro, S.; La Thangue, N.B. NEDDylation regulates E2F-1-dependent transcription. EMBO Rep. 2012, 13, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Sun, Y. SCF E3 ubiquitin ligases as anticancer targets. Curr. Cancer Drug Targets 2011, 11, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Zhang, M.; He, S.; Lu, K.; Chen, Y.; Xing, G.; Lu, Y.; Liu, P.; Li, Y.; Wang, S.; et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat. Commun. 2014, 5, 3733. [Google Scholar] [CrossRef]
- Li, L.; Wang, M.; Yu, G.; Chen, P.; Li, H.; Wei, D.; Zhu, J.; Xie, L.; Jia, H.; Shi, J.; et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J. Natl. Cancer Inst. 2014, 106, dju083. [Google Scholar] [CrossRef]
- Xu, J.; Li, L.; Yu, G.; Ying, W.; Gao, Q.; Zhang, W.; Li, X.; Ding, C.; Jiang, Y.; Wei, D.; et al. The neddylation-cullin 2-RBX1 E3 ligase axis targets tumor suppressor RhoB for degradation in liver cancer. Mol. Cell Proteom. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Barbier-Torres, L.; Delgado, T.C.; Garcia-Rodriguez, J.L.; Zubiete-Franco, I.; Fernandez-Ramos, D.; Buque, X.; Cano, A.; Gutierrez-de Juan, V.; Fernandez-Dominguez, I.; Lopitz-Otsoa, F.; et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget 2015, 6, 2509–2523. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Kane, R.C.; Dagher, R.; Farrell, A.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. Bortezomib for the treatment of mantle cell lymphoma. Clin. Cancer Res. 2007, 13, 5291–5294. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Petroski, M.D. Mechanism-based neddylation inhibitor. Chem. Biol. 2010, 17, 6–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nawrocki, S.T.; Griffin, P.; Kelly, K.R.; Carew, J.S. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin. Investig. Drugs 2012, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, H.; Sun, Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia 2011, 13, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O’Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef]
- Lin, J.J.; Milhollen, M.A.; Smith, P.G.; Narayanan, U.; Dutta, A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010, 70, 10310–10320. [Google Scholar] [CrossRef] [PubMed]
- Milhollen, M.A.; Narayanan, U.; Soucy, T.A.; Veiby, P.O.; Smith, P.G.; Amidon, B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011, 71, 3042–3051. [Google Scholar] [CrossRef]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef]
- Martens, T.; Laabs, Y.; Gunther, H.S.; Kemming, D.; Zhu, Z.; Witte, L.; Hagel, C.; Westphal, M.; Lamszus, K. Inhibition of glioblastoma growth in a highly invasive nude mouse model can be achieved by targeting epidermal growth factor receptor but not vascular endothelial growth factor receptor-2. Clin. Cancer Res. 2008, 14, 5447–5458. [Google Scholar] [CrossRef]
- Sausville, E.A.; Burger, A.M. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006, 66, 3351–3354. [Google Scholar] [CrossRef]
- Taillandier, L.; Antunes, L.; Angioi-Duprez, K.S. Models for neuro-oncological preclinical studies: solid orthotopic and heterotopic grafts of human gliomas into nude mice. J. Neurosci. Methods 2003, 125, 147–157. [Google Scholar] [CrossRef]
- Bonavia, R.; Inda, M.M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.M.; Kim, S.Y.; Jin, X.; Song, S.Y.; Kong, D.S.; Lee, J.I.; Jeon, J.W.; Kim, M.H.; Kang, B.G.; Jung, Y.; et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab. Invest. 2008, 88, 808–815. [Google Scholar] [CrossRef]
- Joo, K.M.; Jin, J.; Kim, E.; Ho Kim, K.; Kim, Y.; Gu Kang, B.; Kang, Y.J.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838. [Google Scholar] [CrossRef]
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y.L.; Park, W.Y.; Kong, D.S.; et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef]
- Xylinas, E.; Hassler, M.R.; Zhuang, D.; Krzywinski, M.; Erdem, Z.; Robinson, B.D.; Elemento, O.; Clozel, T.; Shariat, S.F. An Epigenomic Approach to Improving Response to Neoadjuvant Cisplatin Chemotherapy in Bladder Cancer. Biomolecules 2016, 6. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, N.; Li, C.; Wang, H.; Fang, Y.; Wang, J.; Zheng, X. Anticancer drug sensitivity prediction in cell lines from baseline gene expression through recursive feature selection. BMC Cancer 2015, 15, 489. [Google Scholar] [CrossRef]
- Staunton, J.E.; Slonim, D.K.; Coller, H.A.; Tamayo, P.; Angelo, M.J.; Park, J.; Scherf, U.; Lee, J.K.; Reinhold, W.O.; Weinstein, J.N.; et al. Chemosensitivity prediction by transcriptional profiling. Proc. Natl. Acad. Sci. USA 2001, 98, 10787–10792. [Google Scholar] [CrossRef]
- Hausser, H.J.; Brenner, R.E. Phenotypic instability of Saos-2 cells in long-term culture. Biochem. Biophys. Res. Commun. 2005, 333, 216–222. [Google Scholar] [CrossRef]
- Haney, S.A.; LaPan, P.; Pan, J.; Zhang, J. High-content screening moves to the front of the line. Drug Discov. Today 2006, 11, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Lefloch, R.; Pouyssegur, J.; Lenormand, P. Total ERK1/2 activity regulates cell proliferation. Cell Cycle 2009, 8, 705–711. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Bertrand, F.E.; Ludwig, D.E.; Basecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008, 22, 708–722. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Chappell, W.H.; Russo, S.; Ove, R.; Milella, M.; Tafuri, A.; Lunghi, P.; Bonati, A.; et al. Emerging Raf inhibitors. Expert Opin. Emerg. Drugs 2009, 14, 633–648. [Google Scholar] [CrossRef]
- Xing, J.; Ginty, D.D.; Greenberg, M.E. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 1996, 273, 959–963. [Google Scholar] [CrossRef]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, R.C.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011, 3, 192–222. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Shin, H.; Oh, J.-W.; Oh, Y.J.; Her, N.-G.; Nam, D.-H. The Protein Neddylation Inhibitor MLN4924 Suppresses Patient-Derived Glioblastoma Cells via Inhibition of ERK and AKT Signaling. Cancers 2019, 11, 1849. https://doi.org/10.3390/cancers11121849
Han S, Shin H, Oh J-W, Oh YJ, Her N-G, Nam D-H. The Protein Neddylation Inhibitor MLN4924 Suppresses Patient-Derived Glioblastoma Cells via Inhibition of ERK and AKT Signaling. Cancers. 2019; 11(12):1849. https://doi.org/10.3390/cancers11121849
Chicago/Turabian StyleHan, Suji, Hyemi Shin, Jeong-Woo Oh, Yun Jeong Oh, Nam-Gu Her, and Do-Hyun Nam. 2019. "The Protein Neddylation Inhibitor MLN4924 Suppresses Patient-Derived Glioblastoma Cells via Inhibition of ERK and AKT Signaling" Cancers 11, no. 12: 1849. https://doi.org/10.3390/cancers11121849
APA StyleHan, S., Shin, H., Oh, J.-W., Oh, Y. J., Her, N.-G., & Nam, D.-H. (2019). The Protein Neddylation Inhibitor MLN4924 Suppresses Patient-Derived Glioblastoma Cells via Inhibition of ERK and AKT Signaling. Cancers, 11(12), 1849. https://doi.org/10.3390/cancers11121849