Abstract
Pancreatic cancer is an aggressive cancer with low survival rates. Genetic and epigenetic dysregulation has been associated with the initiation and progression of pancreatic tumors. Multiple studies have pointed to the involvement of aberrant chromatin modifications in driving tumor behavior. ATP-dependent chromatin remodeling complexes regulate chromatin structure and have critical roles in stem cell maintenance, development, and cancer. Frequent mutations and chromosomal aberrations in the genes associated with subunits of the ATP-dependent chromatin remodeling complexes have been detected in different cancer types. In this review, we summarize the current literature on the genomic alterations and mechanistic studies of the ATP-dependent chromatin remodeling complexes in pancreatic cancer. Our review is focused on the four main subfamilies: SWItch/sucrose non-fermentable (SWI/SNF), imitation SWI (ISWI), chromodomain-helicase DNA-binding protein (CHD), and INOsitol-requiring mutant 80 (INO80). Finally, we discuss potential novel treatment options that use small molecules to target these complexes.
1. Introduction
Pancreatic cancer is an aggressive cancer with <10% survival at five years that is poised to become the second cause of cancer-related deaths by 2030 [1]. Currently, surgical resection is the only curative option, however >80% of the patients present with an unresectable tumor [2]. Absence of early diagnostic tools, chemoresistance, and lack of novel therapies contribute to the low survival rate. Although multiple studies have been done to characterize the disease, effective therapies that improve patient survival rate have not yet been developed.
Complex modifications are involved in pancreatic cancer initiation and progression. In addition to the mutations in the main oncogenes and tumor suppressors, the influence of the epigenetic dysregulation has been identified and is now increasingly being studied. Multiple studies highlighted the involvement of epigenetic dysregulation in cancer development, progression, and chemoresistance [3,4,5,6,7,8,9]. Epigenetics are changes that result in changes of gene expression without altering the DNA sequence and involve nucleosome remodeling, histone modifications, DNA methylation, and regulation through long noncoding RNAs (Figure 1). In eukaryotes, ~146 base pairs of genomic DNA is packed with an octamer of histone proteins to form the nucleosome, the basic unit of the chromatin [10]. Nucleosomes, linker histone protein, and non-histone proteins are further assembled into a highly organized chromatin structure that restricts access to the DNA [11]. Chromatin remodeling alters the chromatin structure and regulates accessibility of transcription factors and transcription machinery to the DNA, thus leading to dynamic regulation of gene expression.
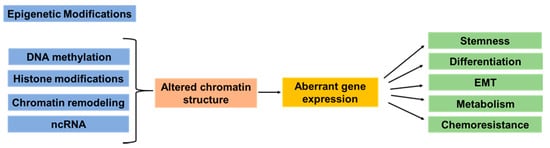
Figure 1.
Simplified overview of the epigenetic modifications involved in cancer initiation, progression, and metastasis. Epigenetic modifications include DNA methylation, histone modifications, chromatin remodeling, and noncoding RNA (ncRNA)s. Multiple studies have demonstrated that epigenetic dysregulation in cancer has been linked to altered chromatin structure and modulation of accessibility of transcription factors to the DNA. These alterations have been associated with aberrant expression of genes related to cancer cell stemness, cell differentiation, epithelial–mesenchymal transition (EMT), cell metabolism, and response to therapeutic drugs.
The two major enzyme groups involved in chromatin remodeling are ATP-dependent chromatin remodeling complexes that mobilize nucleosomes and histone-modifying enzymes that modify histones [12]. Histone-modifying enzymes covalently modify the N-terminal tails of the histones by using various post-translational modifications and alter the nucleosome structure and DNA-histone interactions [12,13,14]. Histone modifications are altered in cancer and contribute to cancer progression and metastasis [14,15]. Genome sequencing studies also reveal that chromatin regulatory proteins are highly mutated in cancer [16,17,18,19]. Specifically, in pancreatic cancer, chromosomal aberrations and/or mutations associated with ATP-dependent chromatin remodeling complexes have been detected in approximately one third of the samples [20,21], highlighting the involvement of aberrant chromatin remodeling in tumorigenesis. However, detailed reviews on the roles of the main subfamilies of the ATP-dependent chromatin-remodeling complexes in pancreatic cancer are limited. The scope of this review is to summarize the recent discoveries regarding the chromosomal alterations and mutations associated with subunits of the ATP-dependent chromatin remodeling complexes in pancreatic cancer and discuss their mechanistic roles and their targeting as a potential treatment strategy.
2. Epigenetic Dysregulation in Pancreatic Cancer Development and Heterogeneity
The majority (>90%) of the diagnosed pancreatic cancer cases are pancreatic ductal adenocarcinoma (PDAC), which develops from the exocrine ductal cells and is associated with mutations in several driver oncogenes and tumor-suppressor genes. The tumor-initiating oncogenic KRAS mutations, which are found in >90% of PDAC cases, initiate the process for noninvasive precursor lesions. Mutations in tumor-suppressor genes SMAD4, TP53, and p16/CDKN2A are detected in 50–70% of the PDAC cases [21,22]. In addition to the sporadic PDAC described above, it is estimated that 5–10% of pancreatic cancers occur due to inherited germline mutations [23], the most prominent ones being in the BRCA2 and CDKN2A genes.
PDAC development is associated with precursor lesions and the two major pathways that lead to PDAC include pancreatic intraepithelial neoplasms (PanIN) and intraductal papillary mucinous neoplasms (IPMN). Both pathways have distinct histological, genetic, and epigenetic changes associated with the multistep progression from low-grade precursor lesions to high-grade precursor lesions and invasive cancer [24,25]. The majority of the PDACs arise from PanINs, which are noninvasive microscopic flat or papillary intraepithelial lesions in the small intralobular pancreatic ducts [24]. In contrast, IPMNs are macroscopic cystic lesions that occur within the larger pancreatic ducts. IPMNs are heterogeneous and can be classified based on the site of origin and histological analysis. Some of the genetic changes in IPMNs are similar to the ones observed in PanINs and PDACs (KRAS, SMAD4, TP53, and p16/CDKN2A), whereas other mutations, such as activating GNAS and inactivating RNF43 mutations, are frequently observed only in IPMNs [26].
Multiple reports have highlighted that PDAC is associated with heterogeneity at both the genetic and epigenetic level, which might influence tumor progression. Several studies have classified the PDAC tumors based on transcriptional and genetic profiling [21,22,27,28,29,30,31]. The most widely used classification is by Moffitt et al., that defined two main tumor subtypes that are clinically and histopathologically different: the classical subtype and the basal subtype that is more aggressive with poorly differentiated tumors and poor outcome [29]. Multifactorial analysis and comparison of chromatin states and gene expression demonstrate that the two PDAC subtypes are associated with distinct chromatin states [32]. Another study by Hayashi and colleagues revealed that the basal subtype is associated with genetic alterations in the chromatin modifying genes, suggesting involvement of these genes in modulating tumor behavior [33]. Other studies have also highlighted that subtype development in PDAC is epigenetically driven and distinct epigenetic landscapes contribute to the PDAC heterogeneity [32,33,34,35]. Another interesting study compared the gene expression and DNA methylation by using PDAC patient-derived xenografts (PDXs) and demonstrated that the transcriptome and methylome have common patterns, highlighting that the main phenotypes in PDAC are established epigenetically [34]. Aberrant patterns of DNA methylation that can silence gene expression are commonly observed in PDAC, and they target tumor-suppressor genes involved in proliferation, apoptosis, cell adhesion, and major signaling pathways [7,31,32,36,37]. Supporting the fundamental role of epigenetic involvement in PDAC, another study concluded that epigenetic reprogramming involving DNA methylation and altered histone codes was associated with malignant gene expression and metastasis [35]. The role of epigenetic alterations in metastatic tumor progression was also confirmed by using PDAC mice models [38]. In addition, multiple sequencing studies have revealed chromosomal alterations and somatic non-silent mutations in components of the chromatin remodeling complexes in PDAC and other cancers [16,17,19,27,39,40,41,42]. Collectively, these reports suggest that epigenetic dysregulation and altered chromatin dynamics play an important role in PDAC. Comprehensive reviews of the roles of the four subfamilies of chromatin remodeling complexes in PDAC are lacking, presenting a knowledge gap, with the need for future studies.
3. ATP-Dependent Chromatin Remodeling Complexes
ATP-dependent chromatin remodeling complexes have essential functions during development; therefore, it is not surprising that genomic aberrations in genes encoding chromatin remodeling components contribute to different malignancies, such as cancer, including PDAC [6,32,33,35,43,44,45]. Epigenetic reprogramming has significant roles in lineage specification during pancreas development, and the development-specific subunit expression is important for altering the functional activity of the complexes [46,47,48,49]. The majority of the studies have focused on epigenetic regulation in endocrine β-cells [46,48], and only a few reports have analyzed the role of SWI/SNF complexes in acinar and ductal cells [48,50]. Understanding the function of these complexes in pancreas development can also aid in identification of pathways that can be targeted in PDAC.
ATP-dependent chromatin remodeling complexes bind to nucleosome cores and the surrounding DNA, and, using energy from ATP hydrolysis, they disrupt the DNA-histone interactions, slide or eject nucleosomes, alter nucleosome structures, and modulate the access of transcription factors to the DNA (Figure 2). In addition to modulating gene expression, some of the complexes are involved in nucleosome assembly and organization, following transcription at locations in which nucleosomes have been ejected, packing of DNA, following replication and DNA repair [45,51,52,53,54]. Based on the sequence homology of the catalytic ATPase and the accessory subunits, chromatin remodeling complexes are divided into four main subfamilies: SWItch/sucrose non-fermentable (SWI/SNF), imitation SWI (ISWI), chromodomain-helicase DNA-binding protein (CHD), and INOsitol-requiring mutant 80 (INO80) [52]. All of these complexes share a catalytic subunit containing a SWI2/SNF2-family ATPase domain that performs DNA translocation along the histone core of the nucleosome [52] and accessory subunits involved in target recognition, specificity, and modulation of the ATPase activity.
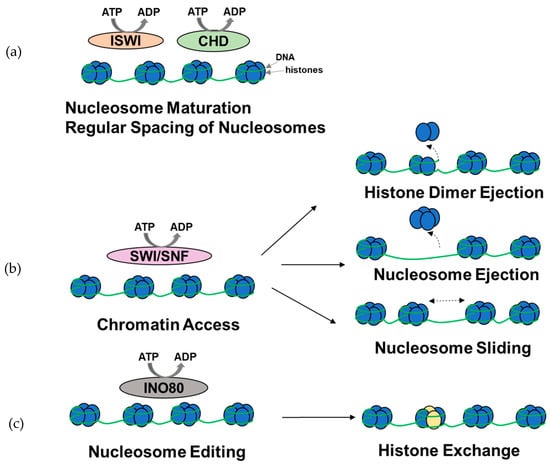
Figure 2.
Overview of the functions of ATP-dependent chromatin remodeling complexes. (a) A subset of ISWI and CHD complexes are involved in nucleosome assembly, maturation, and spacing. (b) SWI/SNF complexes are primarily involved in histone dimer ejection, nucleosome ejection, and nucleosome repositioning through sliding, thus modulating chromatin access. (c) INO80 complexes are involved in histone exchange. It should be noted that the complexes might be involved in other chromatin remodeling functions (figure adapted from [52]).
The differences in subunit compositions of each complex are associated with the cell-type, tissue-specific, or development-related roles of each complex [47,52]. SWI/SNF and INO80 subfamily complexes form large protein assemblies comprising up to 15 subunits, whereas most ISWI complexes and a subset of the CHD complexes are formed with <4 subunits.
3.1. SWI/SNF Subfamily
The SWI/SNF subfamily is involved in mobilizing the nucleosomes through repositioning, sliding, or ejection, and, typically, they facilitate chromatin access for transcription factors. The two main complexes are BAF and PBAF (Figure 3). Recently, a novel noncanonical complex, ncBAF, was identified [55]. The SWI/SNF complex is a multisubunit complex that includes a DNA-binding subunit (ARID1A, ARID1B, or PBRM1), an enzymatic ATPase subunit (BRM/SMARCA2 or BRG1/SMARCA4), three core subunits (SMARCB1, SMARCC1, and SMARCC2), accessory subunits, and BRM- or BRG1-associated factors (BAFs) that are essential for binding to DNA or proteins. The heterogeneity of the SWI/SNF complexes is associated with development and tissue-specific subtypes [44,56,57]. Multiple sequencing studies have identified the SWI/SNF complex as a major tumor suppressor in PDAC. Deletions or deleterious mutations in subunits of the SWI/SNF complexes were associated with 33–42% of the PDAC cases [20,21]. Genomic alterations were detected in multiple subunits of SWI/SNF complexes at varying frequencies [20,21,27] (Table 1). Additionally, SWI/SNF aberrations also modulate responsiveness to platinum-based treatment [58], indicating that detailed characterization of the human PDAC tumors can be used to identify biomarkers for improved treatment regimens.
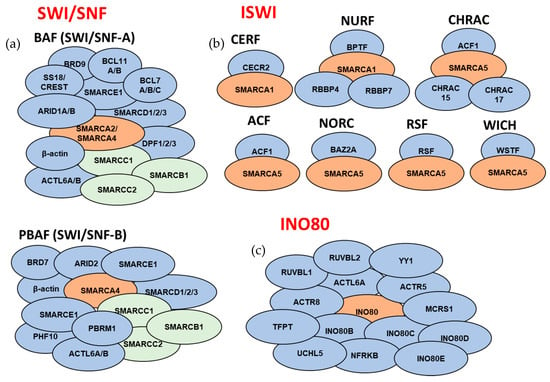
Figure 3.
Overview of the subunit compositions of the ATP-dependent chromatin remodeling complexes. Subunits that comprise the mammalian (a) SWI/SNF complexes, (b) ISWI complexes, and (c) INO80 complex (for SWI-SNF complexes: orange color corresponds to catalytic ATPase subunits, green color corresponds to core subunits, and blue color corresponds to accessory subunits; for ISWI and INO80 complexes: orange color corresponds to catalytic ATPase subunits). For subunits that are separated by dashes, only one of the subunits is present in a given complex. Subunit composition might be different based on tissue/cell types. SWI/SNF noncanonical complex ncBAF and INO80 subfamily complexes p400 and SRCAP are not included in the schematic.

Table 1.
Chromosomal copy number alteration (CNA) frequency (%) and mutational frequency (%) of genes encoding subunits of the ATP-dependent chromatin remodeling complexes.
3.2. ISWI Subfamily
ISWI complexes are involved in nucleosome organization following DNA replication and transcription. Specifically, they are involved in the maturation of DNA-histone complexes to nucleosomes, nucleosome sliding, and regular spacing of the nucleosomes [52]. Seven different mammalian ISWI complexes have been described so far: WICH, NoRC, RSF, ACF, CHRAC, NURF, and CERF (Figure 3). Each contains one of the two conserved ATPase subunits (SMARCA5 or SMARCA1) and accessory subunit(s) [53]. Different combinations of ATPases and accessory subunits might influence the chromatin remodeling reaction, such as the nucleosome spacing, and target the ISWI complex to different gene sets [53,59]. Most ISWI subfamily complexes are involved in repressing chromatin accessibility, whereas a subset such as the nucleosome remodeling factor (NURF) is involved in chromatin access and gene activation [53,60]. In addition, the ISWI complexes are involved in DNA damage response (DDR), which makes them a potential target in cancer [53].
3.3. CHD Subfamily
CHD subfamily comprises several complexes that have diverse functions, such as spacing of the nucleosomes, exposing the promoters, and editing the nucleosomes [52,61]. Most CHD members form multisubunit complexes and are involved in chromatin remodeling [61]. CHD3, CHD4, and CHD5 are components of the nucleosome remodeling deacetylase (NuRD) complex that is a transcriptional repressor and is the best characterized member of this subfamily. Multiple studies have investigated the role of NuRD in cancer [45,62,63,64]. The NurD complex and DNA methylation work cooperatively, demonstrating that both repressive histone marks and DNA hypermethylation are involved in the transcriptional silencing of tumor-suppressor genes [63,64], highlighting that multiple levels of epigenetic regulation are involved in cancer.
3.4. INO80 Subfamily
INO80 multisubunit complexes have diverse functions that include transcriptional regulation, DNA replication, and DNA repair. They are involved in shifting nucleosomes and histone dimer or histone variant exchange. The INO80 subfamily includes the INO80, p400, and SRCAP complexes. INO80 subfamily complexes form large multisubunit complexes that include a catalytic ATPase (INO80, p400, or SRCAP), helicases (RUVBL1, RUVBL2), actin related proteins (ACTL6A, ACTR5, and ACTR8), and other subunits [51] (Figure 3). INO80 has roles in development, but its role in pancreas development is unclear [65,66]. Although the alteration frequency of INO80 subunits is high in multiple cancers, including PDAC [67] (Table 1), only a few studies have been done. The findings pointed to the tumor-promoting role of the INO80 complex in several cancers [67,68,69]. It has been associated with opening the chromatin state in cancer cells and in embryonic stem cells, and enhancer- or superenhancer-mediated oncogenic transcription [67,68,70]. In addition, a high co-occurrence of alterations in subunits of INO80 and mTORC1 was observed in PDAC and other cancers, suggesting that disruption of these pathways might contribute to the metabolic dysregulation involved in tumorigenesis [71].
4. Mechanistic Studies of the ATP-Dependent Chromatin Remodeling Complexes in PDAC
Inactivating mutations in the SWI/SNF complexes are associated with various cancers, suggesting that they act as tumor repressors [16,72]. With slight exceptions, the involvement of the other three subfamilies in cancer has not been well characterized. Each complex is composed of multiple subunits that are associated with chromosomal alterations and/or mutations in PDAC (Table 1). The majority of the studies in PDAC have been focused on the role of few subunits of the SWI/SNF complex, whereas detailed mechanistic studies of the roles of the other subunits in PDAC are limited or missing. We summarize the current knowledge on the subunits of the ATP-dependent chromatin remodeling complexes in PDAC, and for cases that lack detailed mechanistic studies in PDAC, we include data from other cancers and/or stem cell studies, to provide evidence for their role.
4.1. SWI/SNF Subfamily
4.1.1. ARID1A
ARID1A encodes a DNA-binding subunit of the human SWI/SNF complex and is the most frequently mutated subunit of the SWI/SNF complex in PDAC [49] (Table 1). ARID1A expression is decreased in PDAC (Table 2) and is associated with survival outcomes [21]. Studies in mice and cell lines have demonstrated that ARID1A is a tumor suppressor that represses KRAS-induced precancerous lesion formation and suppresses ductal proliferation [49,82]. Pancreas specific Arid1a deletion in mice induced inflammation, formation of PanINs, and mucinous cysts [49]. ARID1A deletion in vitro resulted in global increase of active histone marks and increase in protein expression through induction of Myc, as well as acinar, to ductal metaplasia [49]. Similarly, Arid1a deletion in mice PDAC tumors (mutant Kras and hemizygous p53) led to decreased cancer-specific survival and poorly differentiated tumors [49]. Further characterization of the derivative Arid1a-deleted cells revealed a stem-cell-like and EMT profile resulting in a migratory and mesenchymal phenotype [49]. Furthermore, Arid1a deletion in mice with pancreatic expression of activated KRAS resulted in IPMN that progressed to PDAC [49,82].
Mechanistically, Arid1a deletion inhibited the mTOR pathway, suppressed SOX9 expression, and led to dedifferentiation of pancreatic ductal cells [82].
Table 2.
Summary of immunohistochemistry (IHC) analysis for subunits of ATP-dependent chromatin remodeling complexes in PDAC patient samples.
Another interesting study demonstrated that postnatal acute silencing of Arida1a in adult acinar cells harboring oncogenic Kras mutation accelerated acinar to ductal reprogramming leading to mucinous PDAC precursor lesions in mice. ATAC-seq analysis showed reduced chromatin accessibility, and further studies pointed that these sites correlate with access of transcription factors to enhancers related to acinar identity genes [94]. These observations support the tumor-suppressive role of ARID1A in pancreas.
4.1.2. ARID1B
ARID1B encodes an alternate DNA-binding subunit of the human SWI/SNF complex. The genomic alteration and mutation frequency of ARID1B is lower compared to ARID1A (Table 1). ARID1B expression is reduced in PDAC tumors (Table 2), and the gene is proposed to have a tumor-suppressive role. A limited number of studies in cell lines have been done to characterize the function of ARID1B. For instance, the pancreatic cancer cell line MIA PaCa-2 has a homozygous deletion of ARID1B and ectopic expression of ARID1B severely inhibited colony formation and anchorage independent growth of the cells [84]. Similarly, ARID1B knockdown promoted the growth-factor independent growth in normal human pancreatic duct epithelial (HPDE) cell line [20]. In addition, ARID1B transcription can also be epigenetically regulated through methylation [84].
ARID1A and ARID1B are mutually exclusive, and few studies have been done to characterize the functional dependency between ARID1A and ARID1B in cancer. ARID1A-deficient pancreatic cancer cells are selectively sensitive to ARID1B knockdown and have lower viability compared to ARID1A-expressing cells [21]. Similar findings were observed in a previous study which concluded that ARID1B is the preferential gene required for the survival of ARID1A-mutant cancer cell lines and loss of ARID1B in ARID1A-deficient background destabilized SWI/SNF and impaired proliferation, suggesting that ARID1B might be a potential target in ARID1A-mutant cancers [95].
4.1.3. SMARCA2
SMARCA2 is one of the mutually exclusive catalytic subunits of the SWI/SNF complex. It is generally accepted that loss of SMARCA2 expression is associated with formation of benign tumors [96]; however studies, of its role in PDAC mostly indicate an oncogenic function. Studies of patient samples have demonstrated a correlation between SMARCA2 expression, worse clinicopathological features, and worse survival [83,85,97] (Table 2). Limited mechanistic studies have been done to characterize the role of SMARCA2 in PDAC. In vivo studies using SMARCA2-silenced pancreatic cancer cells showed that mice had improved survival and decreased metastases [97]. Likewise, SMARCA2 knockdown in cell lines resulted in decreased proliferation and reduced invasion [85,97]. Mechanistically, SMARCA2 knockdown led to reduced activation of the JAK2/STAT3 pathway, inhibition of STAT3 phosphorylation and reduced transcription of STAT3 target genes [85]. Another study demonstrated the role of SMARCA2 in chemotherapy response. SMARCA2-downregulated pancreatic cancer cells had increased chemosensitivity to gemcitabine in vitro and in vivo [85]. Collectively, these studies suggest that further mechanistic studies are needed to delineate the role of SMARCA2 in PDAC.
4.1.4. SMARCA4
SMARCA4 is the other mutually exclusive catalytic subunit of the SWI/SNF complex that has significant roles in pancreas development. Early embryonic pancreas-specific removal of Smarca4 led to reduced multipotent pancreatic progenitor cell proliferation and resulted in pancreas hypoplasia [48], indicating its important role in modulating gene expression during development. SMARCA4 is the second most frequently mutated gene of the SWI/SNF subunits in PDAC and is one of the well-studied SWI/SNF subunits. In most cases, SMARCA4 acts as a tumor suppressor; however, it has context-specific oncogene roles [88]. Several studies indicated that SMARCA4 expression is increased in pancreatic cancer tissues [83,85,86] (Table 2). Further studies demonstrated that loss of SMARCA4 in pancreatic and other tumors is associated with E-cadherin loss, vimentin upregulation, and EMT [98].
Interestingly, SMARCA4 has stage-specific roles during PDAC progression, as demonstrated by the studies done in IPMNs, which are precursor lesions of PDAC. Contrary to the PDAC samples, SMARCA4 expression is reduced or lost in IPMNs. Analysis of normal pancreatic epithelium by IHC showed strong expression of SMARCA4, whereas reduced expression or loss of SMARCA4 was observed in surgically resected IPMNs [87]. Other studies also confirmed the differential expression of SMARCA4 in IPMNs compared to PDACs. For example, SMARCA4 expression is higher in human PDAC samples compared to the IPMN lesions [88,89]. Further characterization studies utilizing KrasG12D mouse models indicated the opposing roles of SMARCA4 in IPMN to PDAC progression. During early stages SMARCA4 acts as a tumor suppressor and inhibits dedifferentiation of ductal cells, whereas, at late stages, it induces EMT and promotes tumorigenesis [88]. Mechanistically, loss of Smarca4 promoted dedifferentiation of pancreatic ductal cells expressing oncogenic KrasG12D and led to development of IPMN lesions in vivo. Re-expressing SMARCA4 in a KrasG12D; Smarca4f/f IPMN-derived cell line resulted in enhanced tumorigenicity and EMT characteristics [88]. Similarly, other studies showed that SMARCA4 acts as a tumor suppressor during the oncogenic Kras-induced IPMN-PDAC formation in vivo. Pancreatic loss of Smarca4 and mutant Kras resulted in neoplastic cystic lesions that resembled human IPMNs and progressed to PDAC. Interestingly, opposing roles of SMARCA4 were detected during IPMN- and PanIN-PDAC progression, supporting the context-dependent and stage-specific roles of SMARCA4. Analysis of human samples revealed that reduction of SMARCA4 promoted PanIN-PDAC progression and resulted in poorer survival [89].
Several studies have been done to characterize the mechanistic role of SMARCA4. Characterization of SMARCA4-depleted IPMN-PDAC cells revealed the presence of repressive histone marks on the promoters of high-mobility group AT-hook 2 (Hmga2) gene, mediator of aggressive cancer phenotype, and other genes whose expression was reduced in IPMN-PDA [89]. Re-expressing SMARCA4 in a KrasG12D; Smarca4f/f IPMN-derived PDAC cell line upregulated Hmga2 expression through binding to its promoter and activating its transcription [88,89]. In addition, SMARCA4 binding to Sox9 regulatory elements was demonstrated [89]. Overexpression of Sox9 in KrasG12D; Smarca4f/f pancreatic ductal cells blocked duct dedifferentiation and inhibited upregulation of progenitor markers [88].
Further studies demonstrated the role of SMARCA4 in cell proliferation and chemoresistance. SMARCA4-deficient or SMARCA4-depleted pancreatic epithelial cells demonstrated increased sensitivity to the DNA-damaging agents cisplatin, oxaliplatin, irinotecan, and 5-fluorouracil [58]. Likewise, SMARCA4 knockdown led to reversal of chemoresistance to gemcitabine in MIA PaCa-2 cells [86]. Gemcitabine resistance has been linked to Akt signaling, and SMARCA4 knockdown led to reduced activation of Akt and increased sensitivity of cells to gemcitabine [86]. Furthermore, knockdown of SMARCA4 in pancreatic cancer cell lines PANC-1 and MIA PaCa-2 led to reduced growth in vitro and in vivo [86]. Conflicting results regarding the role of SMARCA4 in cell proliferation were obtained in another study. Re-expression of SMARCA4 in SMARCA4-deficient pancreatic cancer cell lines PANC-1 and Hs700T led to senescence and reduced cell growth. It is possible that the conflicting results are due to differences in the expression levels of the SWI/SNF subunits among different pancreatic cancer cell lines. MIA PaCa-2 cells express SMARCA4, whereas SMARCA4 protein levels are undetected in PANC-1 [20,98,99,100] and Hs700T cells [20].
Similar to the ARID1A/ARID1B functional dependency, SMARCA4 mutant cancer cells showed sensitivity to SMARCA2 depletion [101,102]. Likewise, SMARCA2 dependency was observed in SMARCA4-deficient cancer cells [103]. In addition to a panel of SMARCA2-deficient tested cells, SMARCA2-deficient pancreas carcinoma HuP-T4 cells were dependent on SMARCA4 [103]. These studies indicate the presence of SMARCA2/SMARCA4 paralog dependency for the maintenance of ATPase activity of the SWI/SNF complex and represent a novel treatment strategy of targeting SMARCA2 in SMARCA4-mutant cancers and vice versa.
4.1.5. SMARCC1
SMARCC1 is a core subunit of the SWI/SNF complex. Only one study has described the role of SMARCC1 in PDAC. Analysis of survival in recurrent PDAC pointed that SMARCC1 can be used as a predictor to gemcitabine therapy, as only SMARCC1-positive patients benefited from gemcitabine therapy [90]. Further studies in gemcitabine resistant clones of pancreatic cancer cell lines MIA PaCa-2 and PSN1 showed decreased expression of SMARCC1 [90]. IHC analysis demonstrated homogeneous nuclear staining of SMARCC1 in normal pancreatic ductal cells, whereas variable expression was observed in the pancreatic cancer lesions (Table 2). Mechanistically, SMARCC1 was identified as a tumor-suppressor gene in other cancer cell lines with roles in cell cycle and senescence [104]. SMARCC1 promoted breast cancer progression and metastasis through being recruited to unique chromatin regions, including the Myc target gene GADD45a [105]. Further studies are needed to characterize the role of SMARCC1 in PDAC.
4.1.6. ACTL6B
ACTL6B, a paralog of ACTL6A, has not been studied extensively in cancer. ACTL6B is amplified in PDAC (3–24%, Table 1), and detailed understanding of its role in tumor progression is needed. The role of ACTL6B in neuronal development and differentiation has been analyzed; however, studies in cancer are missing. Neuronal development involves ACTL6A to ACTL6B switch of the SWI/SNF complex subunits. Loss of ACTL6B resulted in impaired dendritic growth [56,106]. Expression of ACTL6B in ACTL6A-deficent mouse embryonic stem cells rescued the cells from cell death and maintained their undifferentiated state, indicating that ACTL6A and ACTL6B might have redundant functions depending on the cell type [107]. Given the amplification frequency of ACTL6B observed in PDAC, further studies are needed to understand its role in tumorigenicity.
Findings regarding the roles of the remaining subunits of the SWI/SNF subfamily complexes in PDAC are summarized in Table 3.
Table 3.
Functional studies of subunits of the SWI/SNF complexes in PDAC (or other cancers).
4.2. ISWI Subfamily
4.2.1. BPTF
BPTF is a component of the nucleosome remodeling factor (NURF) complex of the ISWI subfamily. BPTF expression has been shown to be increased in several cancer types and was associated with tumor progression and worse survival [147,148,149,150,151]. Although BPTF has been associated with deletions, amplifications, and mutations in PDAC (Table 1), functional studies on its role in PDAC are limited. Mechanistically, BPTF-activated human telomerase reverse-transcriptase (hTERT) expression and promoted stemness, proliferation, tumor growth, and metastasis associated with liver cancer [151]. Similarly, studies in other cell lines indicated that BPTF promoted proliferation and invasiveness in vitro [147,149,152]. Furthermore, other studies indicated that BPTF was associated with MYC signaling and promoted tumorigenesis [147,153]. In fibroblasts, BPTF knockdown led to changes in chromatin accessibility, reduced c-MYC recruitment to DNA, and decreased c-MYC-driven transcriptional signatures. BPTF knockdown suppressed the proliferation of pancreatic cancer cells and delayed the development of c-MYC-driven pancreatic tumors [153]. Taken together, these findings indicate that BPTF has an oncogenic role.
In addition, BPTF expression was associated with chemoresistance. BPTF expression was associated with promoting resistance to BRAF inhibitors in melanoma [149], and its knockdown sensitized liver cancer cells to chemotherapeutic drugs [151].
Findings regarding the roles of the remaining subunits of the ISWI subfamily complexes in PDAC are summarized in Table 4.
Table 4.
Functional studies of subunits of the ISWI complexes in PDAC (or other cancers).
4.3. CHD Subfamily
4.3.1. CHD1
CHD1 is a component of the CHD chromatin remodeling subfamily. CHD1 binds to histone marks associated with active transcription [186], maintains an open chromatin state, and promotes pluripotency in mouse embryonic stem cells [187]. Studies of CHD1 in PDAC are limited. A single study in pancreatic cancer cells suggested that CHD1 might have a pro-oncogenic function. In pancreatic cancer, the hPaf1 subunit of the human RNA polymerase II-associated factor (PAF) complex is overexpressed [188], and it interacts with and regulates the expression of CHD1 and the nuclear import of CHD1, facilitating the nucleosomal remodeling in pancreatic cancer cells [189]. The pro-oncogenic function of CHD1 is supported by studies in other cell lines. Studies in colorectal adenocarcinoma cells demonstrated that KRAS mutation is associated with elevated SUMOylation of CHD1 and other proteins that supported the anchorage independent growth of the cells [190]. Furthermore, in prostate cancer, CHD1 loss sensitized cells to DNA damage, caused DNA repair defects, and enhanced therapy response to DNA-damaging therapy and PARP inhibitors [191,192].
4.3.2. CHD5
CHD5 is a component of the CHD chromatin remodeling subfamily and is a tumor suppressor [61,193]. Upstream factors, including the WNT/β-catenin pathway, are involved in the transcriptional regulation of CHD5 [61,194]. Limited studies have been performed to assess the function of CHD5 in PDAC. IHC analysis showed that low CHD5 expression correlated with worse patient outcomes (Table 2) in PDAC [93], and similar results were observed in other cancers [195,196]. Epigenetic silencing of CHD5 through methylation has been observed in multiple cancer types [194,195,196,197]. Low CHD5 expression and CHD5 depletion in several pancreatic cancer cell lines has been associated with DDR activation [93]. Furthermore, CHD5 is a component of the NuRD transcriptional repressor complex [195]. CHD5 has been linked to WEE1, which is a key regulator of cell-cycle progression that can act as an oncogene [198]. CHD5 represses WEE1 transcription in PANC-1 pancreatic cancer cells, thus acting as a tumor suppressor [199]. Similarly, WEE1 kinase inhibitor has recently shown promising results in combination therapy for PDAC [200]. Mechanistic studies in other cell types demonstrated that CHD5 expression suppressed expression of oncogenes, stem cell markers, and EMT markers in renal carcinoma cells [196]; and it resulted in reduced clonogenicity, cell proliferation, migration, and invasion in renal carcinoma cells and colorectal cancer cells [194,196].
4.3.3. CHD7
CHD7 is a component of the CHD chromatin remodeling subfamily. Mutations in the CHD7 gene cause a severe developmental human disorder, CHARGE syndrome [201], highlighting its role in neural stem cells and in development [202]. Mutations and/or altered gene expression of CHD7 are associated with various cancers, including breast cancer, gastric cancer, colon cancer [203,204,205], and PDAC (Table 1). CHD7 is also upregulated in gliomas, and mechanistic studies demonstrated that CHD7 overexpression enhanced cell migration and invasion in vitro and tumor growth in vivo [206]. Transcriptome analysis revealed that CHD7 altered the expression of adhesion molecules, stimulating cell motility and invasiveness [206].
A limited number of studies have focused on characterizing the role of CHD7 in PDAC. CHD7 is differentially methylated in PDAC [207]. CHD7 was dysregulated in over 90% of the analyzed PDAC samples, and low CHD7 expression was associated with increased survival in patients receiving adjuvant gemcitabine therapy [208]. Mechanistically, CHD7 depletion sensitized PDAC cells to gemcitabine by triggering DNA damage and delayed tumor xenograft growth [208]. CHD7 is amplified in PDAC (3.26–4.59%; Table 1), and further studies are needed to delineate its role.
Findings regarding the roles of the ATPase subunits of the CHD subfamily complexes in PDAC are summarized in Table 5.
Table 5.
Functional studies of the ATPase subunits of the CHD complexes in PDAC (or other cancers).
4.4. INO80 Subfamily
4.4.1. INO80
INO80 is the ATPase subunit of the INO80 complex. The majority of the studies focusing on the INO80 complex in cancer have been performed by using INO80 knockdowns. Several studies have been done to characterize its oncogenic role in cancer and maintenance of stem cells; however, studies in PDAC are missing. INO80 is upregulated in cancer cell lines and human cancer tissues, including lung cancer, colon cancer, and melanoma [67,68,69,218]. Functional studies demonstrated that INO80 is required for proliferation, viability, clonogenicity, and anchorage-independent growth of cancer cells in vitro and tumor formation in vivo [67,68,218]. Supporting these findings, INO80 knockdown led to smaller tumors in vivo and downregulation of stem-cell-specific factors, reduced proliferation, and reduced migration in vitro [68,69,218]. Mechanistically, INO80 occupies enhancers near cancer-associated genes and promotes their expression, thus enhancing tumorigenicity [67,68].
Similarly, INO80 is involved in the renewal of embryonic stem cells (ESCs) by maintaining open chromatin architecture and selectively activating pluripotency genes [70]. Further studies pointed that INO80 might promote nucleosome depletion, as ATAC-seq studies of INO-80 silenced cells showed a significant increase in nucleosome occupancy at INO-80 bound regions [68], thus supporting its role in promoting an open chromatin state.
4.4.2. INO80C
INO80C is a core subunit of the INO80 complex that is involved in nucleosome recognition [219]. A recent study pointed to its role as a novel potential tumor suppressor in KRASMUT PDAC and colorectal cancer (CRC) xenograft tumors. Analysis of TCGA data revealed frequent deep deletions of INO80C in PDAC, and association between INO80C deletion and worse prognosis of patients with KRASMUT PDAC and CRC was observed. Knockdown of INO80C in KRASMUT PDAC and CRC cell lines demonstrated enhanced growth of the xenografts in vivo [220]. Given the high frequency of deletions of INO80C in PDAC samples (2.17–18.35%, Table 1), further studies are needed to characterize its role.
Limited studies have been conducted in order to characterize the role of the other INO80 subfamily complexes in cancer [67,68,220]. Several reviews are focused on the roles of the INO80 subfamily complexes [51,219,221,222,223]. The remaining two complexes of the INO80 subfamily (Snf2-related CBP activator protein (SRCAP) and p400 [224,225] and their role in PDAC are not discussed in this review due to limited number of PDAC-specific studies.
Findings regarding the roles of the remaining subunits of the INO80 complex in PDAC are summarized in Table 6.
Table 6.
Functional studies of subunits of the INO80 complex in PDAC (or other cancers).
4.5. SWI/SNF and INO80 Subfamilies
4.5.1. ACTB
ACTB encodes β-actin, which is increased in PDAC and other cancers [267,268]. Studies in gastric cancer have indicated a higher level of β-actin in the primary tumor and a correlation between higher β-actin expression and lymph node metastasis [269]. Rearrangement of the actin cytoskeleton occurs during EMT [270] and, not surprisingly, downregulation of β-actin inhibited migration of gastric cancer cells [269]. As β-actin is implicated in cancer progression [267,271], further studies are needed to determine its role in PDAC.
It is important to distinguish the roles of cytosolic and nuclear β-actin in tumorigenesis. The nuclear isoform of β-actin is part of several chromatin remodeling complexes (SWI/SNF and INO80 p400). Nuclear β-actin was involved in the quiescence of breast epithelial cells, as growth factor removal induced downmodulation of nuclear β-actin, which led to growth arrest [272]. Signals from the extracellular matrix (ECM) decreased nuclear-actin export, resulting in accumulation of nuclear actin and activation of growth-related transcription and malignant progression of breast cancer [273]. Nuclear actin could be a potential therapeutic target, as doxorubicin treatment resulted in nuclear actin aggregates and affected the recruitment of nuclear DNA-damage repair factors [274].
Interesting findings have linked mechanotransduction to actin dynamics and modulating β-actin localization. High mechanical stress (stretched cells) led to nuclear β-actin/F-actin localization at the whole nucleoplasm compared to a perilaminar distribution of nuclear β-actin/F-actin in low-mechanical-stress cells. β-actin polymerizes to form filamentous (F) actin, which is an important component of the cytoskeleton and plays a role in motility [275]. These findings highlight the role of nuclear actins in linking extracellular mechanical signals to chromatin regulation.
ACTB is mostly regarded as a housekeeping gene and is widely used as an endogenous reference for quantification of protein/gene expression studies. Its differential increase in cancer samples suggests that it might not be an appropriate endogenous control. The comparison of four pancreatic ductal cell lines demonstrated that β-actin protein levels did not vary significantly across the cell lines. However, analysis of RNA seq data of 41 PDAC cell lines demonstrated that ACTB is one of the genes with the highest standard deviations [276,277]. Other studies have also demonstrated that β-actin might not be an appropriate control for real-time quantitative reverse-transcription PCR [278,279,280].
4.5.2. ACTL6A
ACTL6A is a component of the SWI/SNF complex and INO80 subfamily INO80 and P400 complexes. It encodes actin related proteins (ARPs) that resemble actin and have roles in chromatin modification and histone acetylation. Amplifications of ACTL6A are associated with PDAC (Table 1); however, mechanistic studies in PDAC are missing. Several studies demonstrated that ACTL6A is amplified and upregulated in different cancers [137,281,282,283,284,285]. ACTL6A has a protumorigenic function and its expression level correlated with worse clinicopathological features in liver cancer and in colon cancer [283,284]. Mechanistically, ACTL6A overexpression promoted migration and invasion and induced EMT in vitro [283,284] and promoted tumor growth and metastasis in a mouse liver cancer xenograft model [283]. Further studies demonstrated that ACTL6A targets SOX2 expression, which activates Notch1 signaling, leading to EMT [282,283].
Moreover, ACTL6A is associated with stem cell maintenance [107,281,282,286], including activation of the Hippo-YAP pathway [281], Nanog binding to pluripotency genes, and repression of differentiation genes [286], which might explain its role in cancer. As ACTL6A promotes a stem-cell-like state, it is not surprising that its levels are increased in cancer. Few studies have been done to delineate the chromatin-specific role of ACTL6A. ACTL6A binds to core histones and might modulate the interaction of the chromatin-modifying complexes with nucleosomes [287,288]. ACTL6A depletion accelerated the degradation of SMARCA4 and SMARCC2 and destabilized SMARCA4 chromatin remodeling complexes in several human cell lines [288]. Moreover, majority of the endogenous ACTL6A proteins are associated with SMARCA4 in the nucleus; thus, they are involved in chromatin modification [288].
5. Therapeutic Targeting of Chromatin Remodeling in Pancreatic Cancer
Chromatin remodeling complexes constitute only a portion of the epigenetic regulation mechanisms, and their roles in tumorigenesis have been highlighted in multiple studies. Genomic lesions are highly prevalent in ATP-dependent chromatin remodelers; however, specific small molecules that effectively target these complexes are limited. The complexity of the ATP-dependent chromatin remodelers poses a significant challenge for their pharmacological targeting. Several different approaches that involve siRNA libraries, inhibitor libraries, and computational modeling have been utilized to identify novel molecules. The majority of the studies have been focused on inhibitors that target the bromodomain domain or the ATPase domain of the subunits. Currently, there are very few potent and selective molecules targeting the subunits of the ATP-dependent chromatin remodeling complexes.
BPTF expression was associated with c-MYC signaling and tumorigenicity in multiple studies. A recent computational docking-based virtual screening identified C620-0696 as a potential inhibitor of BPTF. The addition of C620-0696 to BPTF overexpressing lung cancer cells resulted in cytotoxicity, suppression of c-Myc expression, and inhibition of migration and colony formation, indicating that targeting of BPTF can be further explored as a treatment strategy [289]. Another recent screening study demonstrated that GSK2801, an inhibitor of BAZ2A/B bromodomains of the ISWI complexes and BRD9 of the SWI/SNF complex, synergizes with bromodomain and extra-terminal motif (BET) inhibitors to induce apoptosis in triple-negative breast cancer in vitro [290]. GSK2801 did not result in a significant growth inhibition as a single agent, indicating the need for combinatorial treatment screens.
PFI-3 is a small molecule inhibitor that selectively targets the bromodomain domain of family VIII bromodomain proteins, which include SMARCA2, SMARCA4, and PBRM1 subunits of SWI/SNF complexes. Two studies showed that PFI-3 could not inhibit proliferation [291,292]. Further characterization studies demonstrated that PFI-3 cannot displace endogenous, full-length SMARCA2 from the chromatin, which raises the possibility that it cannot disrupt SMARCA2/SMARCA4-chromatin interaction. Further in vitro studies demonstrated that targeting the ATPase activity of SMARCA2 and SMARCA4 might be a more potent target in cancer [291,292,293].
Promising results have been observed with the active DNA-dependent ATPase A Domain inhibitor (ADAADi), which is the first-in-class inhibitor that inhibits the catalytic ATPase domain of the SWI2/SNF2 family members. ADAADi’s are natural products of aminoglycoside-resistant bacteria that compete with respect to the DNA effector needed for ATP hydrolysis of ATPases. Biochemical studies demonstrated that a subset of the ADAADi’s disrupted ATP-dependent nucleosome activity [294]. Studies in triple-negative breast cancer cell lines demonstrated that ADAADi’s decreased cell proliferation. However, it only targeted a subset of cells preferentially as treatment of cells with reduced SMARCA4 expression did not respond to the treatment [292]. Moreover, ADAADi treatment blocked drug efflux transporter gene expression; thus, it sensitized cells to chemotherapeutic drugs [292]. Studies in other cells lines demonstrated that ADAADi disrupted EMT, inhibited cell migration, and induced apoptosis. Treatment with ADAADi led to transcriptional changes which included repression of the tumor-promoting genes and upregulation of the pro-apoptotic and tumor-suppressors genes [293].
Recent screening study utilizing the proteolysis targeting chimera (PROTAC) technology has identified degraders of the SWI/SNF complex ATPase subunits SMARCA2/SMARCA4 and DNA binding subunit PBRM1 [295]. PROTACs degrade target proteins through recruitment of the ubiquitin proteasome system, which is achieved by using a target-binding ligand linked to a E3 ligase–binding ligand. In this case, the PROTAC ligand was targeted against the bromodomain motif of the proteins. The optimized PROTAC chemical probe ACBI1 resulted in complete degradation of SMARCA2/SMARCA4 and PBRM1. ACBI1 inhibited cell proliferation and induced apoptosis in leukemia cell lines with an intact BAF complex and SMARCA4-mutant cancer cells. These findings suggest that targeted degradation of BAF complex ATPases can be used as a potential treatment strategy.
Taken together, these studies suggest that targeting different domains of the ATPase subunits of the chromatin remodeling complexes can be used as a potential cancer treatment strategy. However, further studies are needed to determine their specificity and effect in normal cells and cancer cells. Multiple challenges are associated with identifying specific small compounds or probes against the subunits of the ATP-dependent chromatin modifying complexes. The compounds/probes have to be specific and target the critical domain of the subunits. In addition, as demonstrated by the PFI-3 study, targeting of the correct domain might not influence the activity of the complex within the cells. In addition, combinatorial treatment screening assays might need to be implemented to test the synergistic effect of drugs, as demonstrated by the GSK2801/BET inhibitor screening study.
6. Conclusions
ATP-dependent chromatin remodeling complexes are involved in the dynamic regulation of gene transcription. Perturbation of the ATP-dependent chromatin remodeling complexes has been associated with cancer, including PDAC. Although the expression of these genes appears to have an impact on PDAC progression and chemoresistance, functional data regarding the role of majority of the individual subunits in PDAC is missing. Detailed understanding of the effect of chromosomal aberrations and mutations associated with components of the ATP-dependent chromatin remodeling complexes in oncogenesis might lead to the discovery of downstream therapeutic targets.
Currently, the ATP-dependent chromatin remodeling complexes are divided into four subfamilies. We noticed that the mechanistic studies have focused on a limited number of complexes, particularly the SWI/SNF subfamily complexes. The remaining subfamilies (ISWI, CHD, and INO80) have not been studied extensively in PDAC, and detailed studies to understand their involvement in PDAC are urgently needed. A noncanonical BAF complex and several subunits of the SWI/SNF complexes (BCL7, BRD7, and BRD9) have been recently identified and detailed studies regarding their function are missing. Multiple subunits, including ACTL6B, SMARCD3, DPF1, DPF2, BCL7B, BCL7C, BRD9, BICRA, BICRAL, SS18, SS18L, CHRAC1, INO80C, RUVBL2, UCHL5, and TFPT, display a high percentage of chromosomal aberrations and/or mutations in PDAC; therefore, mechanistic studies are needed to delineate their role in transcriptional regulation and oncogenesis.
Expression of several of the subunits (SMARCA4, BCL11B, BPTF, SMARCA2, CHD1, CHD4, CHD7, SMARCD1, and SMARCE1) also correlated with chemoresistance and chemosensitivity. Therefore, further mechanistic understanding of their function might be important to identify pathways that can increase sensitivity to current drug regimens.
Targeting the ATP-dependent chromatin remodeling complexes has demonstrated promising results in decreasing cancer cell proliferation in vitro. Recent studies, using either ADAADi’s or PROTACs, have focused on targeting the bromodomain domains and the ATPase domains of the SWI/SNF complex ATPase subunits SMARCA2 and SMARCA4. Another study identified a bromodomain inhibitor, GSK2801, that targets BAZ2A/B and BRD9 and has shown successful results in a combinatorial treatment. It would be beneficial to test these compounds in pancreatic cancer cell lines.
In conclusion, ATP-dependent chromatin remodeling complexes modulate gene expression, and, with few exceptions, detailed studies regarding their role in PDAC are lacking. Studies exploring their mechanistic roles in PDAC are needed for our understanding of PDAC chromatin biology, identification of novel therapeutic targets, and development of specific cancer therapeutics. Furthermore, the expression of individual subunits or complexes can be used as prognostic markers to predict response to therapy.
Author Contributions
Conceptualization, N.H. and N.A.; literature review and data curation, N.H. and N.A.; writing—original draft preparation, review, and editing, N.H. and N.A.
Funding
This research was funded by National Institute of Health, grant number R01CA185357.
Acknowledgments
We thank Anup Sharma, John Kunstman, and Paulomi Aldo for discussions and suggestions.
Conflicts of Interest
N.A. has received grant funding from Cepheid and Astex and has served as consultant to Ethicon. She has licensed methylation biomarkers to Cepheid.
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Lomberk, G.; Dusetti, N.; Iovanna, J.; Urrutia, R. Emerging epigenomic landscapes of pancreatic cancer in the era of precision medicine. Nat. Commun. 2019, 10, 3875. [Google Scholar] [CrossRef]
- Yi, J.M.; Guzzetta, A.A.; Bailey, V.J.; Downing, S.R.; Van Neste, L.; Chiappinelli, K.B.; Keeley, B.P.; Stark, A.; Herrera, A.; Wolfgang, C.; et al. Novel methylation biomarker panel for the early detection of pancreatic cancer. Clin. Cancer Res. 2013, 19, 6544–6555. [Google Scholar] [CrossRef]
- Morel, D.; Almouzni, G.; Soria, J.C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2017, 28, 254–269. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A. Epigenetic changes in cancer. Annu. Rev. Pathol. 2009, 4, 229–249. [Google Scholar] [CrossRef]
- Luger, K.; Rechsteiner, T.J.; Flaus, A.J.; Waye, M.M.; Richmond, T.J. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 1997, 272, 301–311. [Google Scholar] [CrossRef]
- Luo, R.X.; Dean, D.C. Chromatin remodeling and transcriptional regulation. J. Natl. Cancer Inst. 1999, 91, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Torres, K.; Liu, X.; Liu, C.G.; Pollock, R.E. An overview of chromatin-regulating proteins in cells. Curr. Protein Pept. Sci. 2016, 17, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Leroy, G.; Dimaggio, P.A.; Chan, E.Y.; Zee, B.M.; Blanco, M.A.; Bryant, B.; Flaniken, I.Z.; Liu, S.; Kang, Y.; Trojer, P.; et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 2013, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Crabtree, G.R. Mammalian swi/snf chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian swi/snf complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and functions of atp-dependent chromatin-remodeling enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef]
- Valencia, A.M.; Kadoch, C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol. 2019, 21, 152–161. [Google Scholar] [CrossRef]
- Shain, A.H.; Giacomini, C.P.; Matsukuma, K.; Karikari, C.A.; Bashyam, M.D.; Hidalgo, M.; Maitra, A.; Pollack, J.R. Convergent structural alterations define switch/sucrose nonfermentable (swi/snf) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E252–E259. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef] [PubMed]
- Brosens, L.A.; Hackeng, W.M.; Offerhaus, G.J.; Hruban, R.H.; Wood, L.D. Pancreatic adenocarcinoma pathology: Changing “landscape”. J. Gastrointest. Oncol. 2015, 6, 358–374. [Google Scholar]
- Patra, K.C.; Bardeesy, N.; Mizukami, Y. Diversity of precursor lesions for pancreatic cancer: The genetics and biology of intraductal papillary mucinous neoplasm. Clin. Transl. Gastroenterol. 2017, 8, e86. [Google Scholar] [CrossRef]
- Wu, J.; Jiao, Y.; Dal Molin, M.; Maitra, A.; de Wilde, R.F.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Wolfgang, C.L.; Canto, M.I.; et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 21188–21193. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 2018, 155, 1999–2013. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Hayashi, A.; Fan, J.; Chen, R.; Ho, Y.; Makohon-Moore, A.P.; Zhong, Y.; Hong, J.; Sakamoto, H.; Attiyeh, M.A.; Kohutek, Z.A.; et al. The genetic basis of transcriptional heterogeneity for basal-like features in pancreatic ductal adenocarcinoma. bioRxiv 2019. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Natale, F.; Vivo, M.; Falco, G.; Angrisano, T. Deciphering DNA methylation signatures of pancreatic cancer and pancreatitis. Clin. Epigenetics 2019, 11, 132. [Google Scholar] [CrossRef]
- Thompson, M.J.; Rubbi, L.; Dawson, D.W.; Donahue, T.R.; Pellegrini, M. Pancreatic cancer patient survival correlates with DNA methylation of pancreas development genes. PLoS ONE 2015, 10, e0128814. [Google Scholar] [CrossRef]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef]
- Kumar, R.; Li, D.Q.; Muller, S.; Knapp, S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016, 35, 4423–4436. [Google Scholar] [CrossRef]
- Hohmann, A.F.; Vakoc, C.R. A rationale to target the swi/snf complex for cancer therapy. Trends Genet. 2014, 30, 356–363. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Nunez, F.J.; Mendez, F.; Brosnan-Cashman, J.A.; Meeker, A.K.; Lowenstein, P.R.; Castro, M.G. Mutated chromatin regulatory factors as tumor drivers in cancer. Cancer Res. 2017, 77, 227–233. [Google Scholar] [CrossRef]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Brien, G.L.; Verrijzer, C.P. Dangerous liaisons: Interplay between swi/snf, nurd, and polycomb in chromatin regulation and cancer. Genes Dev. 2019, 33, 936–959. [Google Scholar] [CrossRef]
- McKenna, B.; Guo, M.; Reynolds, A.; Hara, M.; Stein, R. Dynamic recruitment of functionally distinct swi/snf chromatin remodeling complexes modulates pdx1 activity in islet beta cells. Cell Rep. 2015, 10, 2032–2042. [Google Scholar] [CrossRef]
- Campbell, S.A.; Hoffman, B.G. Chromatin regulators in pancreas development and diabetes. Trends Endocrinol. Metab. 2016, 27, 142–152. [Google Scholar] [CrossRef]
- Spaeth, J.M.; Liu, J.H.; Peters, D.; Guo, M.; Osipovich, A.B.; Mohammadi, F.; Roy, N.; Bhushan, A.; Magnuson, M.A.; Hebrok, M.; et al. The pdx1-bound swi/snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet beta-cell function. Diabetes 2019, 68, 1806–1818. [Google Scholar] [CrossRef]
- Wang, W.; Friedland, S.C.; Guo, B.; O’Dell, M.R.; Alexander, W.B.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; Myers, J.R.; Ashton, J.M.; et al. Arid1a, a swi/snf subunit, is critical to acinar cell homeostasis and regeneration and is a barrier to transformation and epithelial-mesenchymal transition in the pancreas. Gut 2019, 68, 1245–1258. [Google Scholar] [CrossRef]
- Wang, S.C.; Nassour, I.; Xiao, S.; Zhang, S.; Luo, X.; Lee, J.; Li, L.; Sun, X.; Nguyen, L.H.; Chuang, J.C.; et al. Swi/snf component arid1a restrains pancreatic neoplasia formation. Gut 2019, 68, 1259–1270. [Google Scholar] [CrossRef]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The ino80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160290. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of atp-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Aydin, O.Z.; Vermeulen, W.; Lans, H. Iswi chromatin remodeling complexes in the DNA damage response. Cell Cycle 2014, 13, 3016–3025. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.C.; Crabtree, G.R. Atp-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [CrossRef]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical swi/snf complex is a synthetic lethal target in cancers driven by baf complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef]
- Lessard, J.; Wu, J.I.; Ranish, J.A.; Wan, M.; Winslow, M.M.; Staahl, B.T.; Wu, H.; Aebersold, R.; Graef, I.A.; Crabtree, G.R. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 2007, 55, 201–215. [Google Scholar] [CrossRef]
- Lessard, J.A.; Crabtree, G.R. Chromatin regulatory mechanisms in pluripotency. Annu. Rev. Cell Dev. Biol. 2010, 26, 503–532. [Google Scholar] [CrossRef]
- Davidson, J.; Shen, Z.; Gong, X.; Pollack, J.R. Swi/snf aberrations sensitize pancreatic cancer cells to DNA crosslinking agents. Oncotarget 2018, 9, 9608–9617. [Google Scholar] [CrossRef][Green Version]
- Oppikofer, M.; Bai, T.; Gan, Y.; Haley, B.; Liu, P.; Sandoval, W.; Ciferri, C.; Cochran, A.G. Expansion of the iswi chromatin remodeler family with new active complexes. EMBO Rep. 2017, 18, 1697–1706. [Google Scholar] [CrossRef]
- Xiao, H.; Sandaltzopoulos, R.; Wang, H.M.; Hamiche, A.; Ranallo, R.; Lee, K.M.; Fu, D.; Wu, C. Dual functions of largest nurf subunit nurf301 in nucleosome sliding and transcription factor interactions. Mol. Cell 2001, 8, 531–543. [Google Scholar] [CrossRef]
- Mills, A.A. The chromodomain helicase DNA-binding chromatin remodelers: Family traits that protect from and promote cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026450. [Google Scholar] [CrossRef] [PubMed]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. 2015, 165, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Geutjes, E.J.; de Lint, K.; Roepman, P.; Bruurs, L.; Yu, L.R.; Wang, W.; van Blijswijk, J.; Mohammad, H.; de Rink, I.; et al. The nurd complex cooperates with dnmts to maintain silencing of key colorectal tumor suppressor genes. Oncogene 2014, 33, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Huang, W.; Bellani, M.; Seidman, M.M.; Wu, K.; Fan, D.; Nie, Y.; Cai, Y.; Zhang, Y.W.; Yu, L.R.; et al. Chd4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 2017, 31, 653–668 e657. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Elsayed, Z.; Peterkin, V.; Alkatib, S.; Bennett, D.; Landry, J.W. Ino80 is essential for proximal-distal axis asymmetry in part by regulating bmp4 expression. BMC Biol. 2016, 14, 18. [Google Scholar] [CrossRef]
- Rhee, S.; Chung, J.I.; King, D.A.; D’Amato, G.; Paik, D.T.; Duan, A.; Chang, A.; Nagelberg, D.; Sharma, B.; Jeong, Y.; et al. Endothelial deletion of ino80 disrupts coronary angiogenesis and causes congenital heart disease. Nat. Commun. 2018, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, B.; Wang, L.; Li, P.; Bennett, B.D.; Snyder, R.; Garantziotis, S.; Fargo, D.C.; Cox, A.D.; Chen, L.; et al. Ino80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene 2017, 36, 1430–1439. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, L.; Zhang, S.; Bennett, B.D.; He, F.; Zhang, Y.; Xiong, C.; Han, L.; Diao, L.; Li, P.; et al. Ino80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016, 30, 1440–1453. [Google Scholar] [CrossRef]
- Lee, S.A.; Lee, H.S.; Hur, S.K.; Kang, S.W.; Oh, G.T.; Lee, D.; Kwon, J. Ino80 haploinsufficiency inhibits colon cancer tumorigenesis via replication stress-induced apoptosis. Oncotarget 2017, 8, 115041–115053. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Du, Y.; Ward, J.M.; Shimbo, T.; Lackford, B.; Zheng, X.; Miao, Y.L.; Zhou, B.; Han, L.; Fargo, D.C.; et al. Ino80 facilitates pluripotency gene activation in embryonic stem cell self-renewal, reprogramming, and blastocyst development. Cell Stem Cell 2014, 14, 575–591. [Google Scholar] [CrossRef]
- Beckwith, S.L.; Schwartz, E.K.; Garcia-Nieto, P.E.; King, D.A.; Gowans, G.J.; Wong, K.M.; Eckley, T.L.; Paraschuk, A.P.; Peltan, E.L.; Lee, L.R.; et al. The ino80 chromatin remodeler sustains metabolic stability by promoting tor signaling and regulating histone acetylation. PLoS Genet. 2018, 14, e1007216. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant swi/snf complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018, 6, 271–281. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 2018, 33, 676–689. [Google Scholar] [CrossRef]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018, 23, 227–238. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated tcga pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Fukuda, A.; Ogawa, S.; Maruno, T.; Takada, Y.; Tsuda, M.; Hiramatsu, Y.; Araki, O.; Nagao, M.; Yoshikawa, T.; et al. Arid1a maintains differentiation of pancreatic ductal cells and inhibits development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2018, 155, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Numata, M.; Morinaga, S.; Watanabe, T.; Tamagawa, H.; Yamamoto, N.; Shiozawa, M.; Nakamura, Y.; Kameda, Y.; Okawa, S.; Rino, Y.; et al. The clinical significance of swi/snf complex in pancreatic cancer. Int. J. Oncol. 2013, 42, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Khursheed, M.; Kolla, J.N.; Kotapalli, V.; Gupta, N.; Gowrishankar, S.; Uppin, S.G.; Sastry, R.A.; Koganti, S.; Sundaram, C.; Pollack, J.R.; et al. Arid1b, a member of the human swi/snf chromatin remodeling complex, exhibits tumour-suppressor activities in pancreatic cancer cell lines. Br. J. Cancer 2013, 108, 2056–2062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, F.; Du, C.; Guo, H.; Ma, L.; Liu, X.; Kornmann, M.; Tian, X.; Yang, Y. Brm/smarca2 promotes the proliferation and chemoresistance of pancreatic cancer cells by targeting jak2/stat3 signaling. Cancer Lett. 2017, 402, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tian, X.; Wang, F.; Ma, Y.; Kornmann, M.; Yang, Y. Brg1 promotes chemoresistance of pancreatic cancer cells through crosstalking with akt signalling. Eur. J. Cancer 2014, 50, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, M.; Hong, S.M.; Hebbar, S.; Sharma, R.; Scrimieri, F.; de Wilde, R.F.; Mayo, S.C.; Goggins, M.; Wolfgang, C.L.; Schulick, R.D.; et al. Loss of expression of the swi/snf chromatin remodeling subunit brg1/smarca4 is frequently observed in intraductal papillary mucinous neoplasms of the pancreas. Hum. Pathol. 2012, 43, 585–591. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef]
- Von Figura, G.; Fukuda, A.; Roy, N.; Liku, M.E.; Morris Iv, J.P.; Kim, G.E.; Russ, H.A.; Firpo, M.A.; Mulvihill, S.J.; Dawson, D.W.; et al. The chromatin regulator brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2014, 16, 255–267. [Google Scholar] [CrossRef]
- Iwagami, Y.; Eguchi, H.; Nagano, H.; Akita, H.; Hama, N.; Wada, H.; Kawamoto, K.; Kobayashi, S.; Tomokuni, A.; Tomimaru, Y.; et al. Mir-320c regulates gemcitabine-resistance in pancreatic cancer via smarcc1. Br. J. Cancer 2013, 109, 502–511. [Google Scholar] [CrossRef]
- Taniuchi, K.; Furihata, M.; Naganuma, S.; Dabanaka, K.; Hanazaki, K.; Saibara, T. Bcl7b, a predictor of poor prognosis of pancreatic cancers, promotes cell motility and invasion by influencing creb signaling. Am. J. Cancer Res. 2018, 8, 387–404. [Google Scholar] [PubMed]
- Arpalahti, L.; Saukkonen, K.; Hagstrom, J.; Mustonen, H.; Seppanen, H.; Haglund, C.; Holmberg, C.I. Nuclear ubiquitin c-terminal hydrolase l5 expression associates with increased patient survival in pancreatic ductal adenocarcinoma. Tumour Biol. 2017, 39, 1010428317710411. [Google Scholar] [CrossRef] [PubMed]
- Hall, W.A.; Petrova, A.V.; Colbert, L.E.; Hardy, C.W.; Fisher, S.B.; Saka, B.; Shelton, J.W.; Warren, M.D.; Pantazides, B.G.; Gandhi, K.; et al. Low chd5 expression activates the DNA damage response and predicts poor outcome in patients undergoing adjuvant therapy for resected pancreatic cancer. Oncogene 2014, 33, 5450–5456. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Alonso-Curbelo, D.; Morris, J.P.t.; Koche, R.; Saborowski, M.; Wilkinson, J.E.; Lowe, S.W. Arid1a restrains kras-dependent changes in acinar cell identity. Elife 2018, 7, e35216. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. Arid1b is a specific vulnerability in arid1a-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef]
- Guerrero-Martinez, J.A.; Reyes, J.C. High expression of smarca4 or smarca2 is frequently associated with an opposite prognosis in cancer. Sci. Rep. 2018, 8, 2043. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Guo, H.; Wang, F.; Ma, L.; Du, C.; Wang, Y.; Wang, Q.; Kornmann, M.; Tian, X.; et al. Brm transcriptionally regulates mir-302a-3p to target socs5/stat3 signaling axis to potentiate pancreatic cancer metastasis. Cancer Lett. 2019, 449, 215–225. [Google Scholar] [CrossRef]
- Marquez-Vilendrer, S.B.; Thompson, K.; Lu, L.; Reisman, D. Mechanism of brg1 silencing in primary cancers. Oncotarget 2016, 7, 56153–56169. [Google Scholar] [CrossRef]
- Strobeck, M.W.; Knudsen, K.E.; Fribourg, A.F.; DeCristofaro, M.F.; Weissman, B.E.; Imbalzano, A.N.; Knudsen, E.S. Brg-1 is required for rb-mediated cell cycle arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 7748–7753. [Google Scholar] [CrossRef]
- Reisman, D.N.; Strobeck, M.W.; Betz, B.L.; Sciariotta, J.; Funkhouser, W., Jr.; Murchardt, C.; Yaniv, M.; Sherman, L.S.; Knudsen, E.S.; Weissman, B.E. Concomitant down-regulation of brm and brg1 in human tumor cell lines: Differential effects on rb-mediated growth arrest vs cd44 expression. Oncogene 2002, 21, 1196–1207. [Google Scholar] [CrossRef][Green Version]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.; et al. Functional epigenetics approach identifies brm/smarca2 as a critical synthetic lethal target in brg1-deficient cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 3128–3133. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Helming, K.C.; Wang, X.; Kim, Y.; Vazquez, F.; Jagani, Z.; Hahn, W.C.; Roberts, C.W. Residual complexes containing smarca2 (brm) underlie the oncogenic drive of smarca4 (brg1) mutation. Mol. Cell. Biol. 2014, 34, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Ehrenhofer-Wolfer, K.; Puchner, T.; Schwarz, C.; Rippka, J.; Blaha-Ostermann, S.; Strobl, U.; Hormann, A.; Bader, G.; Kornigg, S.; Zahn, S.; et al. Smarca2-deficiency confers sensitivity to targeted inhibition of smarca4 in esophageal squamous cell carcinoma cell lines. Sci. Rep. 2019, 9, 11661. [Google Scholar] [CrossRef]
- DelBove, J.; Rosson, G.; Strobeck, M.; Chen, J.; Archer, T.K.; Wang, W.; Knudsen, E.S.; Weissman, B.E. Identification of a core member of the swi/snf complex, baf155/smarcc1, as a human tumor suppressor gene. Epigenetics 2011, 6, 1444–1453. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Z.; Meyer, M.B.; Saha, S.; Yu, M.; Guo, A.; Wisinski, K.B.; Huang, W.; Cai, W.; Pike, J.W.; et al. Carm1 methylates chromatin remodeling factor baf155 to enhance tumor progression and metastasis. Cancer Cell 2014, 25, 21–36. [Google Scholar] [CrossRef]
- Bell, S.; Rousseau, J.; Peng, H.; Aouabed, Z.; Priam, P.; Theroux, J.F.; Jefri, M.; Tanti, A.; Wu, H.; Kolobova, I.; et al. Mutations in actl6b cause neurodevelopmental deficits and epilepsy and lead to loss of dendrites in human neurons. Am. J. Hum. Genet. 2019, 104, 815–834. [Google Scholar] [CrossRef]
- Zhu, B.; Ueda, A.; Song, X.; Horike, S.I.; Yokota, T.; Akagi, T. Baf53a is involved in survival of mouse es cells, which can be compensated by baf53b. Sci. Rep. 2017, 7, 14059. [Google Scholar] [CrossRef]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the swi/snf complex gene pbrm1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Porter, E.G.; Dhiman, A.; Chowdhury, B.; Carter, B.C.; Lin, H.; Stewart, J.C.; Kazemian, M.; Wendt, M.K.; Dykhuizen, E.C. Pbrm1 regulates stress response in epithelial cells. iScience 2019, 15, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Kohashi, K.; Oda, Y. Oncogenic roles of smarcb1/ini1 and its deficient tumors. Cancer Sci. 2017, 108, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations of a chromatin-remodeling gene smarcc2 in gastric and colorectal cancers with microsatellite instability. APMIS 2013, 121, 168–169. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef]
- Nakazato, H.; Takeshima, H.; Kishino, T.; Kubo, E.; Hattori, N.; Nakajima, T.; Yamashita, S.; Igaki, H.; Tachimori, Y.; Kuniyoshi, Y.; et al. Early-stage induction of swi/snf mutations during esophageal squamous cell carcinogenesis. PLoS ONE 2016, 11, e0147372. [Google Scholar] [CrossRef]
- Revill, K.; Wang, T.; Lachenmayer, A.; Kojima, K.; Harrington, A.; Li, J.; Hoshida, Y.; Llovet, J.M.; Powers, S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 2013, 145, 1424–1435. [Google Scholar] [CrossRef]
- Hong, C.F.; Lin, S.Y.; Chou, Y.T.; Wu, C.W. Microrna-7 compromises p53 protein-dependent apoptosis by controlling the expression of the chromatin remodeling factor smarcd1. J. Biol. Chem. 2016, 291, 1877–1889. [Google Scholar] [CrossRef]
- Oh, J.; Sohn, D.H.; Ko, M.; Chung, H.; Jeon, S.H.; Seong, R.H. Baf60a interacts with p53 to recruit the swi/snf complex. J. Biol. Chem. 2008, 283, 11924–11934. [Google Scholar] [CrossRef]
- Arts, F.A.; Keogh, L.; Smyth, P.; O’Toole, S.; Ta, R.; Gleeson, N.; O’Leary, J.J.; Flavin, R.; Sheils, O. Mir-223 potentially targets swi/snf complex protein smarcd1 in atypical proliferative serous tumor and high-grade ovarian serous carcinoma. Hum. Pathol. 2017, 70, 98–104. [Google Scholar] [CrossRef]
- Inoue, C.; Zhao, C.; Tsuduki, Y.; Udono, M.; Wang, L.; Nomura, M.; Katakura, Y. Smarcd1 regulates senescence-associated lipid accumulation in hepatocytes. NPJ Aging Mech. Dis. 2017, 3, 11. [Google Scholar] [CrossRef]
- Shen, J.; Xiao, Z.; Wu, W.K.; Wang, M.H.; To, K.F.; Chen, Y.; Yang, W.; Li, M.S.; Shin, V.Y.; Tong, J.H.; et al. Epigenetic silencing of mir-490-3p reactivates the chromatin remodeler smarcd1 to promote helicobacter pylori-induced gastric carcinogenesis. Cancer Res. 2015, 75, 754–765. [Google Scholar] [CrossRef]
- Van de Wijngaart, D.J.; Dubbink, H.J.; Molier, M.; de Vos, C.; Trapman, J.; Jenster, G. Functional screening of fxxlf-like peptide motifs identifies smarcd1/baf60a as an androgen receptor cofactor that modulates tmprss2 expression. Mol. Endocrinol. 2009, 23, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Witzel, M.; Petersheim, D.; Fan, Y.; Bahrami, E.; Racek, T.; Rohlfs, M.; Puchalka, J.; Mertes, C.; Gagneur, J.; Ziegenhain, C.; et al. Chromatin-remodeling factor smarcd2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat. Genet. 2017, 49, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Zhu, L.; Gao, Y.; Zhang, X.; Yan, Y.; Cen, J.; Li, R.; Zeng, R.; Liao, L.; Hou, C.; et al. Baf60b-mediated atm-p53 activation blocks cell identity conversion by sensing chromatin opening. Cell Res. 2017, 27, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.V.; Prat, A.; Abell, A.N.; Zawistowski, J.S.; Sciaky, N.; Karginova, O.A.; Zhou, B.; Golitz, B.T.; Perou, C.M.; Johnson, G.L. Swi/snf chromatin-remodeling factor smarcd3/baf60c controls epithelial-mesenchymal transition by inducing wnt5a signaling. Mol. Cell. Biol. 2013, 33, 3011–3025. [Google Scholar] [CrossRef]
- Sokol, E.S.; Feng, Y.X.; Jin, D.X.; Tizabi, M.D.; Miller, D.H.; Cohen, M.A.; Sanduja, S.; Reinhardt, F.; Pandey, J.; Superville, D.A.; et al. Smarce1 is required for the invasive progression of in situ cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 4153–4158. [Google Scholar] [CrossRef]
- Link, K.A.; Balasubramaniam, S.; Sharma, A.; Comstock, C.E.; Godoy-Tundidor, S.; Powers, N.; Cao, K.H.; Haelens, A.; Claessens, F.; Revelo, M.P.; et al. Targeting the baf57 swi/snf subunit in prostate cancer: A novel platform to control androgen receptor activity. Cancer Res. 2008, 68, 4551–4558. [Google Scholar] [CrossRef]
- Papadakis, A.I.; Sun, C.; Knijnenburg, T.A.; Xue, Y.; Grernrum, W.; Holzel, M.; Nijkamp, W.; Wessels, L.F.; Beijersbergen, R.L.; Bernards, R.; et al. Smarce1 suppresses egfr expression and controls responses to met and alk inhibitors in lung cancer. Cell Res. 2015, 25, 445–458. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kurita, T.; Nishio, K.; Tsukada, J.; Hachisuga, T.; Morimoto, Y.; Iwai, Y.; Izumi, H. Expression of baf57 in ovarian cancer cells and drug sensitivity. Cancer Sci. 2015, 106, 359–366. [Google Scholar] [CrossRef]
- Banga, S.S.; Peng, L.; Dasgupta, T.; Palejwala, V.; Ozer, H.L. Phf10 is required for cell proliferation in normal and sv40-immortalized human fibroblast cells. Cytogenet. Genome Res. 2009, 126, 227–242. [Google Scholar] [CrossRef]
- Anbunathan, H.; Verstraten, R.; Singh, A.D.; Harbour, J.W.; Bowcock, A.M. Integrative copy number analysis of uveal melanoma reveals novel candidate genes involved in tumorigenesis including a tumor suppressor role for phf10/baf45a. Clin. Cancer Res. 2019, 25, 5156–5166. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Herlong, F.H.; Stroehlein, J.R.; Mishra, L. Mutations of chromatin structure regulating genes in human malignancies. Curr. Protein Pept. Sci. 2016, 17, 411–437. [Google Scholar] [CrossRef]
- Duan, Y.; Tian, L.; Gao, Q.; Liang, L.; Zhang, W.; Yang, Y.; Zheng, Y.; Pan, E.; Li, S.; Tang, N. Chromatin remodeling gene arid2 targets cyclin d1 and cyclin e1 to suppress hepatoma cell progression. Oncotarget 2016, 7, 45863–45875. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, J.; Han, Y.; Huang, Z.; Ying, J.; Bi, X.; Zhao, J.; Fang, Y.; Zhou, H.; Zhou, J.; et al. Arid2: A new tumor suppressor gene in hepatocellular carcinoma. Oncotarget 2011, 2, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Z.; Shen, J. Brd7: A novel tumor suppressor gene in different cancers. Am. J. Transl. Res. 2016, 8, 742–748. [Google Scholar]
- Gatchalian, J.; Malik, S.; Ho, J.; Lee, D.S.; Kelso, T.W.R.; Shokhirev, M.N.; Dixon, J.R.; Hargreaves, D.C. A non-canonical brd9-containing baf chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 2018, 9, 5139. [Google Scholar] [CrossRef]
- Sima, X.; He, J.; Peng, J.; Xu, Y.; Zhang, F.; Deng, L. The genetic alteration spectrum of the swi/snf complex: The oncogenic roles of brd9 and actl6a. PLoS ONE 2019, 14, e0222305. [Google Scholar] [CrossRef]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of brd9 reverses oncogenic gene expression in synovial sarcoma. Elife 2018, 7, e41305. [Google Scholar] [CrossRef]
- Uehara, T.; Kage-Nakadai, E.; Yoshina, S.; Imae, R.; Mitani, S. The tumor suppressor bcl7b functions in the wnt signaling pathway. PLoS Genet. 2015, 11, e1004921. [Google Scholar] [CrossRef]
- Lazarus, K.A.; Hadi, F.; Zambon, E.; Bach, K.; Santolla, M.F.; Watson, J.K.; Correia, L.L.; Das, M.; Ugur, R.; Pensa, S.; et al. Bcl11a interacts with sox2 to control the expression of epigenetic regulators in lung squamous carcinoma. Nat. Commun. 2018, 9, 3327. [Google Scholar] [CrossRef]
- Khaled, W.T.; Choon Lee, S.; Stingl, J.; Chen, X.; Raza Ali, H.; Rueda, O.M.; Hadi, F.; Wang, J.; Yu, Y.; Chin, S.F.; et al. Bcl11a is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat. Commun. 2015, 6, 5987. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Luo, N.; Hu, Y.; Li, X.; Zhang, K. Mir-137 suppresses triple-negative breast cancer stemness and tumorigenesis by perturbing bcl11a-dnmt1 interaction. Cell. Physiol. Biochem. 2018, 47, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Grabarczyk, P.; Nahse, V.; Delin, M.; Przybylski, G.; Depke, M.; Hildebrandt, P.; Volker, U.; Schmidt, C.A. Increased expression of bcl11b leads to chemoresistance accompanied by g1 accumulation. PLoS ONE 2010, 5, e12532. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Kentsis, A.; Sanda, T.; Holmfeldt, L.; Chen, S.C.; Zhang, J.; Protopopov, A.; Chin, L.; Dahlberg, S.E.; Neuberg, D.S.; et al. The bcl11b tumor suppressor is mutated across the major molecular subtypes of t-cell acute lymphoblastic leukemia. Blood 2011, 118, 4169–4173. [Google Scholar] [CrossRef]
- Sakamaki, A.; Katsuragi, Y.; Otsuka, K.; Tomita, M.; Obata, M.; Iwasaki, T.; Abe, M.; Sato, T.; Ochiai, M.; Sakuraba, Y.; et al. Bcl11b swi/snf-complex subunit modulates intestinal adenoma and regeneration after gamma-irradiation through wnt/beta-catenin pathway. Carcinogenesis 2015, 36, 622–631. [Google Scholar] [CrossRef]
- Qiu, Z.; Ghosh, A. A calcium-dependent switch in a crest-brg1 complex regulates activity-dependent gene expression. Neuron 2008, 60, 775–787. [Google Scholar] [CrossRef]
- Pan, Y.; Yuan, F.; Li, Y.; Wang, G.; Lin, Z.; Chen, L. Bromodomain phdfinger transcription factor promotes glioma progression and indicates poor prognosis. Oncol. Rep. 2019, 41, 246–256. [Google Scholar]
- Dar, A.A.; Majid, S.; Bezrookove, V.; Phan, B.; Ursu, S.; Nosrati, M.; De Semir, D.; Sagebiel, R.W.; Miller, J.R., III; Debs, R.; et al. Bptf transduces mitf-driven prosurvival signals in melanoma cells. Proc. Natl. Acad. Sci. USA 2016, 113, 6254–6258. [Google Scholar] [CrossRef]
- Dar, A.A.; Nosrati, M.; Bezrookove, V.; de Semir, D.; Majid, S.; Thummala, S.; Sun, V.; Tong, S.; Leong, S.P.; Minor, D.; et al. The role of bptf in melanoma progression and in response to braf-targeted therapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Xiao, S.; Liu, L.; Lu, X.; Long, J.; Zhou, X.; Fang, M. The prognostic significance of bromodomain phd-finger transcription factor in colorectal carcinoma and association with vimentin and e-cadherin. J. Cancer Res. Clin. Oncol. 2015, 141, 1465–1474. [Google Scholar] [CrossRef]
- Zhao, X.; Zheng, F.; Li, Y.; Hao, J.; Tang, Z.; Tian, C.; Yang, Q.; Zhu, T.; Diao, C.; Zhang, C.; et al. Bptf promotes hepatocellular carcinoma growth by modulating htert signaling and cancer stem cell traits. Redox Biol. 2019, 20, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Balbas-Martinez, C.; Sagrera, A.; Carrillo-de-Santa-Pau, E.; Earl, J.; Marquez, M.; Vazquez, M.; Lapi, E.; Castro-Giner, F.; Beltran, S.; Bayes, M.; et al. Recurrent inactivation of stag2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 2013, 45, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Richart, L.; Carrillo-de Santa Pau, E.; Rio-Machin, A.; de Andres, M.P.; Cigudosa, J.C.; Lobo, V.J.S.; Real, F.X. Bptf is required for c-myc transcriptional activity and in vivo tumorigenesis. Nat. Commun. 2016, 7, 10153. [Google Scholar] [CrossRef]
- Eckey, M.; Kuphal, S.; Straub, T.; Rummele, P.; Kremmer, E.; Bosserhoff, A.K.; Becker, P.B. Nucleosome remodeler snf2l suppresses cell proliferation and migration and attenuates wnt signaling. Mol. Cell. Biol. 2012, 32, 2359–2371. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ye, Y.; Xiao, Y.; Wang, W.; Wang, Q.; Yearsley, K.; Wani, A.A.; Yan, Q.; Gao, J.X.; Shetuni, B.S.; Barsky, S.H. Inhibition of expression of the chromatin remodeling gene, snf2l, selectively leads to DNA damage, growth inhibition, and cancer cell death. Mol. Cancer Res. 2009, 7, 1984–1999. [Google Scholar] [CrossRef] [PubMed]
- Gigek, C.O.; Lisboa, L.C.; Leal, M.F.; Silva, P.N.; Lima, E.M.; Khayat, A.S.; Assumpcao, P.P.; Burbano, R.R.; Smith Mde, A. Smarca5 methylation and expression in gastric cancer. Cancer Investig. 2011, 29, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Mao, X.; Li, B.; Guan, S.; Yao, F.; Jin, F. Overexpression of smarca5 correlates with cell proliferation and migration in breast cancer. Tumour Biol. 2015, 36, 1895–1902. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, J.; Liu, Q.; Hong, X.; Li, T.; Zhu, Y.; He, L.; Zheng, B.; Li, M. Snf2h promotes hepatocellular carcinoma proliferation by activating the wnt/beta-catenin signaling pathway. Oncol. Lett. 2016, 12, 1329–1336. [Google Scholar] [CrossRef]
- Dluhosova, M.; Curik, N.; Vargova, J.; Jonasova, A.; Zikmund, T.; Stopka, T. Epigenetic control of spi1 gene by ctcf and iswi atpase smarca5. PLoS ONE 2014, 9, e87448. [Google Scholar] [CrossRef]
- Klement, K.; Luijsterburg, M.S.; Pinder, J.B.; Cena, C.S.; Del Nero, V.; Wintersinger, C.M.; Dellaire, G.; van Attikum, H.; Goodarzi, A.A. Opposing iswi- and chd-class chromatin remodeling activities orchestrate heterochromatic DNA repair. J. Cell Biol. 2014, 207, 717–733. [Google Scholar] [CrossRef]
- Toiber, D.; Erdel, F.; Bouazoune, K.; Silberman, D.M.; Zhong, L.; Mulligan, P.; Sebastian, C.; Cosentino, C.; Martinez-Pastor, B.; Giacosa, S.; et al. Sirt6 recruits snf2h to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 2013, 51, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Kokavec, J.; Zikmund, T.; Savvulidi, F.; Kulvait, V.; Edelmann, W.; Skoultchi, A.I.; Stopka, T. The ISWI atpase smarca5 (snf2h) is required for proliferation and differentiation of hematopoietic stem and progenitor cells. Stem Cells 2017, 35, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Bossi, D.; Cicalese, A.; Dellino, G.I.; Luzi, L.; Riva, L.; D’Alesio, C.; Diaferia, G.R.; Carugo, A.; Cavallaro, E.; Piccioni, R.; et al. In vivo genetic screens of patient-derived tumors revealed unexpected frailty of the transformed phenotype. Cancer Discov. 2016, 6, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Zhang, X.T.; Liu, X.L.; Fan, L.; Li, C.; Sun, Y.; Liang, X.H.; Wang, J.B.; Mei, Q.B.; Zhang, F.; et al. Wstf promotes proliferation and invasion of lung cancer cells by inducing emt via pi3k/akt and il-6/stat3 signaling pathways. Cell. Signal. 2016, 28, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. Wstf regulates the h2a.X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar] [CrossRef]
- Ansari, D.; Andersson, R.; Bauden, M.P.; Andersson, B.; Connolly, J.B.; Welinder, C.; Sasor, A.; Marko-Varga, G. Protein deep sequencing applied to biobank samples from patients with pancreatic cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 369–380. [Google Scholar] [CrossRef]
- Gu, L.; Frommel, S.C.; Oakes, C.C.; Simon, R.; Grupp, K.; Gerig, C.Y.; Bar, D.; Robinson, M.D.; Baer, C.; Weiss, M.; et al. Baz2a (tip5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat. Genet. 2015, 47, 22–30. [Google Scholar] [CrossRef]
- Postepska-Igielska, A.; Krunic, D.; Schmitt, N.; Greulich-Bode, K.M.; Boukamp, P.; Grummt, I. The chromatin remodelling complex norc safeguards genome stability by heterochromatin formation at telomeres and centromeres. EMBO Rep. 2013, 14, 704–710. [Google Scholar] [CrossRef]
- Sheu, J.J.; Choi, J.H.; Guan, B.; Tsai, F.J.; Hua, C.H.; Lai, M.T.; Wang, T.L.; Shih Ie, M. Rsf-1, a chromatin remodelling protein, interacts with cyclin e1 and promotes tumour development. J. Pathol. 2013, 229, 559–568. [Google Scholar] [CrossRef]
- Sheu, J.J.; Guan, B.; Choi, J.H.; Lin, A.; Lee, C.H.; Hsiao, Y.T.; Wang, T.L.; Tsai, F.J.; Shih Ie, M. Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J. Biol. Chem. 2010, 285, 38260–38269. [Google Scholar] [CrossRef]
- Wang, X.; Sheu, J.J.; Lai, M.T.; Yin-Yi Chang, C.; Sheng, X.; Wei, L.; Gao, Y.; Wang, X.; Liu, N.; Xie, W.; et al. Rsf-1 overexpression determines cancer progression and drug resistance in cervical cancer. Biomedicine 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.I.; Ahn, J.H.; Lee, K.T.; Shih Ie, M.; Choi, J.H. Rsf1 is a positive regulator of nf-kappab-induced gene expression required for ovarian cancer chemoresistance. Cancer Res. 2014, 74, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Sheu, J.J.; Guan, B.; Jinawath, N.; Markowski, P.; Wang, T.L.; Shih Ie, M. Functional analysis of 11q13.5 amplicon identifies rsf-1 (hbxap) as a gene involved in paclitaxel resistance in ovarian cancer. Cancer Res. 2009, 69, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, F.; Villagrasa, P.; Matsui, J.; Kotliar, D.; Castro, V.; Akavia, U.D.; Chen, B.J.; Saucedo-Cuevas, L.; Rodriguez Barrueco, R.; Llobet-Navas, D.; et al. Integration of genomic data enables selective discovery of breast cancer drivers. Cell 2014, 159, 1461–1475. [Google Scholar] [CrossRef]
- Min, S.; Kim, K.; Kim, S.G.; Cho, H.; Lee, Y. Chromatin-remodeling factor, rsf1, controls p53-mediated transcription in apoptosis upon DNA strand breaks. Cell Death Dis. 2018, 9, 1079. [Google Scholar] [CrossRef]
- Li, X.; Ding, D.; Yao, J.; Zhou, B.; Shen, T.; Qi, Y.; Ni, T.; Wei, G. Chromatin remodeling factor baz1a regulates cellular senescence in both cancer and normal cells. Life Sci. 2019, 229, 225–232. [Google Scholar] [CrossRef]
- Kostrhon, S.; Kontaxis, G.; Kaufmann, T.; Schirghuber, E.; Kubicek, S.; Konrat, R.; Slade, D. A histone-mimicking interdomain linker in a multidomain protein modulates multivalent histone binding. J. Biol. Chem. 2017, 292, 17643–17657. [Google Scholar] [CrossRef]
- Mahmood, S.F.; Gruel, N.; Chapeaublanc, E.; Lescure, A.; Jones, T.; Reyal, F.; Vincent-Salomon, A.; Raynal, V.; Pierron, G.; Perez, F.; et al. A sirna screen identifies rad21, eif3h, chrac1 and tanc2 as driver genes within the 8q23, 8q24.3 and 17q23 amplicons in breast cancer with effects on cell growth, survival and transformation. Carcinogenesis 2014, 35, 670–682. [Google Scholar] [CrossRef]
- Wang, Y.L.; Faiola, F.; Xu, M.; Pan, S.; Martinez, E. Human atac is a gcn5/pcaf-containing acetylase complex with a novel nc2-like histone fold module that interacts with the tata-binding protein. J. Biol. Chem. 2008, 283, 33808–33815. [Google Scholar] [CrossRef]
- Bolognese, F.; Forni, C.; Caretti, G.; Frontini, M.; Minuzzo, M.; Mantovani, R. The pole3 bidirectional unit is regulated by myc and e2fs. Gene 2006, 366, 109–116. [Google Scholar] [CrossRef]
- Thornley, J.A.; Trask, H.W.; Ringelberg, C.S.; Ridley, C.J.; Wang, S.; Sal-Lari, R.C.; Moore, J.H.; Korc, M.; Tomlinson, C.R. Smad4-dependent polysome rna recruitment in human pancreatic cancer cells. Mol. Carcinog. 2012, 51, 771–782. [Google Scholar] [CrossRef]
- Van Gool, I.C.; Eggink, F.A.; Freeman-Mills, L.; Stelloo, E.; Marchi, E.; de Bruyn, M.; Palles, C.; Nout, R.A.; de Kroon, C.D.; Osse, E.M.; et al. Pole proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin. Cancer Res. 2015, 21, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, J.; Jiang, J.; Yuan, J.; Zhang, Y.; Wei, X.; Loo, N.; Wang, Y.; Pan, Y.; Zhang, T.; et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun. 2019, 10, 733. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Park, E.J.; Lee, H.S.; Lee, Y.S.; Kwon, J. Genome-wide screen of human bromodomain-containing proteins identifies cecr2 as a novel DNA damage response protein. Mol. Cells 2012, 34, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Dawe, C.E.; Kooistra, M.K.; Fairbridge, N.A.; Pisio, A.C.; McDermid, H.E. Role of chromatin remodeling gene cecr2 in neurulation and inner ear development. Dev. Dyn. 2011, 240, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Marfella, C.G.; Imbalzano, A.N. The chd family of chromatin remodelers. Mutat. Res. 2007, 618, 30–40. [Google Scholar] [CrossRef]
- Gaspar-Maia, A.; Alajem, A.; Polesso, F.; Sridharan, R.; Mason, M.J.; Heidersbach, A.; Ramalho-Santos, J.; McManus, M.T.; Plath, K.; Meshorer, E.; et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 2009, 460, 863–868. [Google Scholar] [CrossRef]
- Chaudhary, K.; Deb, S.; Moniaux, N.; Ponnusamy, M.P.; Batra, S.K. Human rna polymerase ii-associated factor complex: Dysregulation in cancer. Oncogene 2007, 26, 7499–7507. [Google Scholar] [CrossRef]
- Dey, P.; Ponnusamy, M.P.; Deb, S.; Batra, S.K. Human rna polymerase ii-association factor 1 (hpaf1/pd2) regulates histone methylation and chromatin remodeling in pancreatic cancer. PLoS ONE 2011, 6, e26926. [Google Scholar] [CrossRef]
- Yu, B.; Swatkoski, S.; Holly, A.; Lee, L.C.; Giroux, V.; Lee, C.S.; Hsu, D.; Smith, J.L.; Yuen, G.; Yue, J.; et al. Oncogenesis driven by the ras/raf pathway requires the sumo e2 ligase ubc9. Proc. Natl. Acad. Sci. USA 2015, 112, E1724–E1733. [Google Scholar] [CrossRef]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of chd1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016, 17, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. Chd1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.; Papazoglu, C.; Wu, Y.; Capurso, D.; Brodt, M.; Francis, D.; Bredel, M.; Vogel, H.; Mills, A.A. Chd5 is a tumor suppressor at human 1p36. Cell 2007, 128, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, M.; Paul, T.A.; Brodeur, G.M.; Shokrani, B.; Brim, H.; Ashktorab, H. Epigenetic silencing of chd5, a novel tumor-suppressor gene, occurs in early colorectal cancer stages. Cancer 2014, 120, 172–180. [Google Scholar] [CrossRef]
- Kolla, V.; Naraparaju, K.; Zhuang, T.; Higashi, M.; Kolla, S.; Blobel, G.A.; Brodeur, G.M. The tumour suppressor chd5 forms a nurd-type chromatin remodelling complex. Biochem. J. 2015, 468, 345–352. [Google Scholar] [CrossRef][Green Version]
- Du, Z.; Li, L.; Huang, X.; Jin, J.; Huang, S.; Zhang, Q.; Tao, Q. The epigenetic modifier chd5 functions as a novel tumor suppressor for renal cell carcinoma and is predominantly inactivated by promoter cpg methylation. Oncotarget 2016, 7, 21618–21630. [Google Scholar] [CrossRef]
- Mulero-Navarro, S.; Esteller, M. Chromatin remodeling factor chd5 is silenced by promoter cpg island hypermethylation in human cancer. Epigenetics 2008, 3, 210–215. [Google Scholar] [CrossRef]
- Wang, F.; Zhu, Y.; Huang, Y.; McAvoy, S.; Johnson, W.B.; Cheung, T.H.; Chung, T.K.; Lo, K.W.; Yim, S.F.; Yu, M.M.; et al. Transcriptional repression of wee1 by kruppel-like factor 2 is involved in DNA damage-induced apoptosis. Oncogene 2005, 24, 3875–3885. [Google Scholar] [CrossRef]
- Quan, J.; Adelmant, G.; Marto, J.A.; Look, A.T.; Yusufzai, T. The chromatin remodeling factor chd5 is a transcriptional repressor of wee1. PLoS ONE 2014, 9, e108066. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose escalation trial of the wee1 inhibitor adavosertib (azd1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Bouazoune, K.; Kingston, R.E. Chromatin remodeling by the chd7 protein is impaired by mutations that cause human developmental disorders. Proc. Natl. Acad. Sci. USA 2012, 109, 19238–19243. [Google Scholar] [CrossRef] [PubMed]
- Engelen, E.; Akinci, U.; Bryne, J.C.; Hou, J.; Gontan, C.; Moen, M.; Szumska, D.; Kockx, C.; van Ijcken, W.; Dekkers, D.H.; et al. Sox2 cooperates with chd7 to regulate genes that are mutated in human syndromes. Nat. Genet. 2011, 43, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Chung, N.G.; Kang, M.R.; Yoo, N.J.; Lee, S.H. Genetic and expressional alterations of chd genes in gastric and colorectal cancers. Histopathology 2011, 58, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Yamamoto, E.; Madireddi, P.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.O.; Sugai, T.; et al. Colorectal carcinomas with cpg island methylator phenotype 1 frequently contain mutations in chromatin regulators. Gastroenterology 2014, 146, 530–538 e535. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Guo, X.; Jiang, Y.; Yu, H.; Liu, L.; Shan, W.; Yang, Z.Q. Genotranscriptomic meta-analysis of the chd family chromatin remodelers in human cancers-initial evidence of an oncogenic role for chd7. Mol. Oncol. 2017, 11, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.A.C.; Schneider, H.; DeOcesano-Pereira, C.; Lichtenstein, F.; Andrade, F.; Fujita, A.; Trombetta-Lima, M.; Weller, M.; Bowman-Colin, C.; Sogayar, M.C. Chd7 promotes glioblastoma cell motility and invasiveness through transcriptional modulation of an invasion signature. Sci. Rep. 2019, 9, 3952. [Google Scholar] [CrossRef]
- Mishra, N.K.; Guda, C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 2017, 8, 28990–29012. [Google Scholar] [CrossRef]
- Colbert, L.E.; Petrova, A.V.; Fisher, S.B.; Pantazides, B.G.; Madden, M.Z.; Hardy, C.W.; Warren, M.D.; Pan, Y.; Nagaraju, G.P.; Liu, E.A.; et al. Chd7 expression predicts survival outcomes in patients with resected pancreatic cancer. Cancer Res. 2014, 74, 2677–2687. [Google Scholar] [CrossRef]
- Rodriguez, D.; Bretones, G.; Quesada, V.; Villamor, N.; Arango, J.R.; Lopez-Guillermo, A.; Ramsay, A.J.; Baumann, T.; Quiros, P.M.; Navarro, A.; et al. Mutations in chd2 cause defective association with active chromatin in chronic lymphocytic leukemia. Blood 2015, 126, 195–202. [Google Scholar] [CrossRef]
- Semba, Y.; Harada, A.; Maehara, K.; Oki, S.; Meno, C.; Ueda, J.; Yamagata, K.; Suzuki, A.; Onimaru, M.; Nogami, J.; et al. Chd2 regulates chromatin for proper gene expression toward differentiation in mouse embryonic stem cells. Nucleic Acids Res. 2017, 45, 8758–8772. [Google Scholar] [CrossRef]
- Moore, S.; Berger, N.D.; Luijsterburg, M.S.; Piett, C.G.; Stanley, F.K.T.; Schrader, C.U.; Fang, S.; Chan, J.A.; Schriemer, D.C.; Nagel, Z.D.; et al. The chd6 chromatin remodeler is an oxidative DNA damage response factor. Nat. Commun. 2019, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Sawada, G.; Ueo, H.; Matsumura, T.; Uchi, R.; Ishibashi, M.; Mima, K.; Kurashige, J.; Takahashi, Y.; Akiyoshi, S.; Sudo, T.; et al. Chd8 is an independent prognostic indicator that regulates wnt/beta-catenin signaling and the cell cycle in gastric cancer. Oncol. Rep. 2013, 30, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.A.; Tremblay, V.; Lin, G.; Bochar, D.A. Chd8 is an atp-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell. Biol. 2008, 28, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Peng, H.; Huang, X.X.; Xia, Y.B.; Hu, K.F.; Zhang, Z.M. Decreased expression of chromodomain helicase DNA-binding protein 9 is a novel independent prognostic biomarker for colorectal cancer. Braz. J. Med. Biol. Res. 2018, 51, e7588. [Google Scholar] [CrossRef]
- Hibi, K.; Kitamura, Y.H.; Mizukami, H.; Goto, T.; Sakuraba, K.; Sakata, M.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; et al. Frequent cdh3 demethylation in advanced gastric carcinoma. Anticancer Res. 2009, 29, 3945–3947. [Google Scholar]
- Hibi, K.; Goto, T.; Mizukami, H.; Kitamura, Y.H.; Sakuraba, K.; Sakata, M.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; et al. Demethylation of the cdh3 gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2009, 29, 2215–2217. [Google Scholar]
- Wang, H.C.; Chou, C.L.; Yang, C.C.; Huang, W.L.; Hsu, Y.C.; Luo, C.W.; Chen, T.J.; Li, C.F.; Pan, M.R. Over-expression of chd4 is an independent biomarker of poor prognosis in patients with rectal cancers receiving concurrent chemoradiotherapy. Int. J. Mol. Sci. 2019, 20, 4087. [Google Scholar] [CrossRef]
- Sheng, W.; Chen, Y.; Gong, Y.; Dong, T.; Zhang, B.; Gao, W. Mir-148a inhibits self-renewal of thyroid cancer stem cells via repressing ino80 expression. Oncol. Rep. 2016, 36, 3387–3396. [Google Scholar] [CrossRef][Green Version]
- Chen, L.; Conaway, R.C.; Conaway, J.W. Multiple modes of regulation of the human ino80 snf2 atpase by subunits of the ino80 chromatin-remodeling complex. Proc. Natl. Acad. Sci. USA 2013, 110, 20497–20502. [Google Scholar] [CrossRef]
- Yau, E.H.; Kummetha, I.R.; Lichinchi, G.; Tang, R.; Zhang, Y.; Rana, T.M. Genome-wide crispr screen for essential cell growth mediators in mutant kras colorectal cancers. Cancer Res. 2017, 77, 6330–6339. [Google Scholar] [CrossRef]
- Runge, J.S.; Raab, J.R.; Magnuson, T. Identification of two distinct classes of the human ino80 complex genome-wide. G3 2018, 8, 1095–1102. [Google Scholar] [CrossRef]
- Su, J.; Sui, Y.; Ding, J.; Li, F.; Shen, S.; Yang, Y.; Lu, Z.; Wang, F.; Cao, L.; Liu, X.; et al. Human ino80/yy1 chromatin remodeling complex transcriptionally regulates the brca2- and cdkn1a-interacting protein (bccip) in cells. Protein Cell 2016, 7, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Ayala, R.; Willhoft, O.; Aramayo, R.J.; Wilkinson, M.; McCormack, E.A.; Ocloo, L.; Wigley, D.B.; Zhang, X. Structure and regulation of the human ino80-nucleosome complex. Nature 2018, 556, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Han, J.; Chen, H.; Liu, T.; Huen, M.S.Y.; Yang, Y.; Guo, C.; Huang, J. The human srcap chromatin remodeling complex promotes DNA-end resection. Curr. Biol. 2014, 24, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.J.; Shen, X. Chromatin remodelling beyond transcription: The ino80 and swr1 complexes. Nat. Rev. Mol. Cell Biol. 2009, 10, 373–384. [Google Scholar] [CrossRef]
- Fang, L.T.; Lee, S.; Choi, H.; Kim, H.K.; Jew, G.; Kang, H.C.; Chen, L.; Jablons, D.; Kim, I.J. Comprehensive genomic analyses of a metastatic colon cancer to the lung by whole exome sequencing and gene expression analysis. Int. J. Oncol. 2014, 44, 211–221. [Google Scholar] [CrossRef]
- Thakur, A.; Bollig, A.; Wu, J.; Liao, D.J. Gene expression profiles in primary pancreatic tumors and metastatic lesions of ela-c-myc transgenic mice. Mol. Cancer 2008, 7, 11. [Google Scholar] [CrossRef]
- Shen, X.; Ranallo, R.; Choi, E.; Wu, C. Involvement of actin-related proteins in atp-dependent chromatin remodeling. Mol. Cell 2003, 12, 147–155. [Google Scholar] [CrossRef]
- Willhoft, O.; Bythell-Douglas, R.; McCormack, E.A.; Wigley, D.B. Synergy and antagonism in regulation of recombinant human ino80 chromatin remodeling complex. Nucleic Acids Res. 2016, 44, 8179–8188. [Google Scholar] [CrossRef]
- Gentili, C.; Castor, D.; Kaden, S.; Lauterbach, D.; Gysi, M.; Steigemann, P.; Gerlich, D.W.; Jiricny, J.; Ferrari, S. Chromosome missegregation associated with ruvbl1 deficiency. PLoS ONE 2015, 10, e0133576. [Google Scholar] [CrossRef]
- Tarangelo, A.; Lo, N.; Teng, R.; Kim, E.; Le, L.; Watson, D.; Furth, E.E.; Raman, P.; Ehmer, U.; Viatour, P. Recruitment of pontin/reptin by e2f1 amplifies e2f transcriptional response during cancer progression. Nat. Commun. 2015, 6, 10028. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Chauvet, S.; Huber, O.; Usseglio, F.; Rothbacher, U.; Aragnol, D.; Kemler, R.; Pradel, J. Pontin52 and reptin52 function as antagonistic regulators of beta-catenin signalling activity. EMBO J. 2000, 19, 6121–6130. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.A.; McMahon, S.B.; Cole, M.D. An atpase/helicase complex is an essential cofactor for oncogenic transformation by c-myc. Mol. Cell 2000, 5, 321–330. [Google Scholar] [CrossRef]
- Haurie, V.; Menard, L.; Nicou, A.; Touriol, C.; Metzler, P.; Fernandez, J.; Taras, D.; Lestienne, P.; Balabaud, C.; Bioulac-Sage, P.; et al. Adenosine triphosphatase pontin is overexpressed in hepatocellular carcinoma and coregulated with reptin through a new posttranslational mechanism. Hepatology 2009, 50, 1871–1883. [Google Scholar] [CrossRef]
- Grigoletto, A.; Lestienne, P.; Rosenbaum, J. The multifaceted proteins reptin and pontin as major players in cancer. Biochim. Biophys. Acta 2011, 1815, 147–157. [Google Scholar] [CrossRef]
- Lauscher, J.C.; Elezkurtaj, S.; Dullat, S.; Lipka, S.; Grone, J.; Buhr, H.J.; Huber, O.; Kruschewski, M. Increased pontin expression is a potential predictor for outcome in sporadic colorectal carcinoma. Oncol. Rep. 2012, 28, 1619–1624. [Google Scholar] [CrossRef][Green Version]
- Sun, Q.; Li, F.; Yu, S.; Zhang, X.; Shi, F.; She, J. Pontin acts as a potential biomarker for poor clinical outcome and promotes tumor invasion in hilar cholangiocarcinoma. Biomed. Res. Int. 2018, 2018, 6135016. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, J.; Yan, L.; Tang, Y.; Zhang, W.; Li, D.; Zang, Y.; Kong, F.; Xu, Z. Cytoplasmic expression of pontin in renal cell carcinoma correlates with tumor invasion, metastasis and patients’ survival. PLoS ONE 2015, 10, e0118659. [Google Scholar] [CrossRef]
- Taniuchi, K.; Furihata, M.; Iwasaki, S.; Tanaka, K.; Shimizu, T.; Saito, M.; Saibara, T. Ruvbl1 directly binds actin filaments and induces formation of cell protrusions to promote pancreatic cancer cell invasion. Int. J. Oncol. 2014, 44, 1945–1954. [Google Scholar] [CrossRef]
- Mao, Y.Q.; Houry, W.A. The role of pontin and reptin in cellular physiology and cancer etiology. Front. Mol. Biosci. 2017, 4, 58. [Google Scholar] [CrossRef]
- Grigoletto, A.; Neaud, V.; Allain-Courtois, N.; Lestienne, P.; Rosenbaum, J. The atpase activity of reptin is required for its effects on tumor cell growth and viability in hepatocellular carcinoma. Mol. Cancer Res. 2013, 11, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.; Taras, D.; Grigoletto, A.; Haurie, V.; Nicou, A.; Dugot-Senant, N.; Costet, P.; Rousseau, B.; Rosenbaum, J. In vivo silencing of reptin blocks the progression of human hepatocellular carcinoma in xenografts and is associated with replicative senescence. J. Hepatol. 2010, 52, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Menard, L.; Haurie, V.; Taras, D.; Blanc, J.F.; Moreau-Gaudry, F.; Metzler, P.; Hugues, M.; Boyault, S.; Lemiere, S.; et al. Overexpression and role of the atpase and putative DNA helicase ruvb-like 2 in human hepatocellular carcinoma. Hepatology 2007, 46, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, C.; Yue, X.; Li, X.; Liu, J.; Yu, H.; Belyi, V.A.; Yang, Q.; Feng, Z.; Hu, W. Pontin, a new mutant p53-binding protein, promotes gain-of-function of mutant p53. Cell Death Differ. 2015, 22, 1824–1836. [Google Scholar] [CrossRef]
- Yuan, P.; He, X.H.; Rong, Y.F.; Cao, J.; Li, Y.; Hu, Y.P.; Liu, Y.; Li, D.; Lou, W.; Liu, M.F. Kras/nf-kappab/yy1/mir-489 signaling axis controls pancreatic cancer metastasis. Cancer Res. 2017, 77, 100–111. [Google Scholar] [CrossRef]
- Jiang, W.; Zhao, S.; Shen, J.; Guo, L.; Sun, Y.; Zhu, Y.; Ma, Z.; Zhang, X.; Hu, Y.; Xiao, W.; et al. The mir-135b-bmal1-yy1 loop disturbs pancreatic clockwork to promote tumourigenesis and chemoresistance. Cell Death Dis. 2018, 9, 149. [Google Scholar] [CrossRef]
- De Nigris, F.; Crudele, V.; Giovane, A.; Casamassimi, A.; Giordano, A.; Garban, H.J.; Cacciatore, F.; Pentimalli, F.; Marquez-Garban, D.C.; Petrillo, A.; et al. Cxcr4/yy1 inhibition impairs vegf network and angiogenesis during malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 14484–14489. [Google Scholar] [CrossRef]
- Palmer, M.B.; Majumder, P.; Cooper, J.C.; Yoon, H.; Wade, P.A.; Boss, J.M. Yin yang 1 regulates the expression of snail through a distal enhancer. Mol. Cancer Res. 2009, 7, 221–229. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, J.; Wu, Y.; Shi, G.; Yuan, H.; Lu, Z.; Zhu, Q.; Wu, P.; Lu, C.; Guo, F.; et al. Yy1 suppresses proliferation and migration of pancreatic ductal adenocarcinoma by regulating the cdkn3/mdm2/p53/p21 signaling pathway. Int. J. Cancer 2018, 142, 1392–1404. [Google Scholar] [CrossRef]
- Zhang, J.J.; Zhu, Y.; Zhang, X.F.; Liu, D.F.; Wang, Y.; Yang, C.; Shi, G.D.; Peng, Y.P.; Zhang, K.; Tian, L.; et al. Yin yang-1 suppresses pancreatic ductal adenocarcinoma cell proliferation and tumor growth by regulating sox2ot-sox2 axis. Cancer Lett. 2017, 408, 144–154. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, J.J.; Ge, W.L.; Chen, L.; Yuan, H.; Meng, L.D.; Huang, X.M.; Shen, P.; Miao, Y.; Jiang, K.R. Yy1 inhibits the migration and invasion of pancreatic ductal adenocarcinoma by downregulating the fer/stat3/mmp2 signaling pathway. Cancer Lett. 2019, 463, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.A.; Bonavida, B. Targeting the overexpressed yy1 in cancer inhibits emt and metastasis. Crit. Rev. Oncog. 2017, 22, 49–61. [Google Scholar] [CrossRef]
- Khachigian, L.M. The yin and yang of yy1 in tumor growth and suppression. Int. J. Cancer 2018, 143, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Huang, W.; Kute, T.E.; Miller, L.D.; Zhang, Q.; Hatcher, H.; Wang, J.; Stovall, D.B.; Russell, G.B.; Cao, P.D.; et al. Yin yang 1 plays an essential role in breast cancer and negatively regulates p27. Am. J. Pathol. 2012, 180, 2120–2133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Stovall, D.B.; Inoue, K.; Sui, G. The oncogenic role of yin yang 1. Crit. Rev. Oncog. 2011, 16, 163–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.X.; Zhou, K.C.; Cao, Y. Mcrs1 overexpression, which is specifically inhibited by mir-129*, promotes the epithelial-mesenchymal transition and metastasis in non-small cell lung cancer. Mol. Cancer 2014, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhou, K.; Huang, Y.; Cao, Y. The candidate oncogene (mcrs1) promotes the growth of human lung cancer cells via the mir-155-rb1 pathway. J. Exp. Clin. Cancer Res. 2015, 34, 121. [Google Scholar] [CrossRef]
- Shi, H.; Chen, S.; Jin, H.; Xu, C.; Dong, G.; Zhao, Q.; Wang, W.; Zhang, H.; Lin, W.; Zhang, J.; et al. Downregulation of msp58 inhibits growth of human colorectal cancer cells via regulation of the cyclin d1-cyclin-dependent kinase 4-p21 pathway. Cancer Sci. 2009, 100, 1585–1590. [Google Scholar] [CrossRef]
- Fawal, M.A.; Brandt, M.; Djouder, N. Mcrs1 binds and couples rheb to amino acid-dependent mtorc1 activation. Dev. Cell 2015, 33, 67–81. [Google Scholar] [CrossRef]
- Li, C.; Chen, M.; Zhao, P.; Ayana, D.A.; Wang, L.; Jiang, Y. Expression of mcrs1 and mcrs2 and their correlation with serum carcinoembryonic antigen in colorectal cancer. Exp. Ther. Med. 2016, 12, 589–596. [Google Scholar] [CrossRef][Green Version]
- Vander Linden, R.T.; Hemmis, C.W.; Schmitt, B.; Ndoja, A.; Whitby, F.G.; Robinson, H.; Cohen, R.E.; Yao, T.; Hill, C.P. Structural basis for the activation and inhibition of the uch37 deubiquitylase. Mol. Cell 2015, 57, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Fu, D.; Tang, W.; Cai, Y.; Ma, D.; Wang, H.; Xue, R.; Liu, T.; Huang, X.; Dong, L.; et al. Ubiquitin c-terminal hydrolase 37, a novel predictor for hepatocellular carcinoma recurrence, promotes cell migration and invasion via interacting and deubiquitinating prp19. Biochim. Biophys. Acta 2013, 1833, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, D.; Xi, J.; Ji, Z.; Liu, T.; Ma, Y.; Zhao, Y.; Dong, L.; Wang, Q.; Shen, X. Expression and clinical significance of uch37 in human esophageal squamous cell carcinoma. Dig. Dis. Sci. 2012, 57, 2310–2317. [Google Scholar] [CrossRef] [PubMed]
- Nan, L.; Jacko, A.M.; Tan, J.; Wang, D.; Zhao, J.; Kass, D.J.; Ma, H.; Zhao, Y. Ubiquitin carboxyl-terminal hydrolase-l5 promotes tgfbeta-1 signaling by de-ubiquitinating and stabilizing smad2/smad3 in pulmonary fibrosis. Sci. Rep. 2016, 6, 33116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yao, X.; Pang, S.; Chen, P.; Jiang, W.; Shan, Z.; Zhang, Q. The deubiquitinase uchl5/uch37 positively regulates hedgehog signaling by deubiquitinating smoothened. J. Mol. Cell Biol. 2018, 10, 243–257. [Google Scholar] [CrossRef]
- Barber, K.E.; Harrison, C.J.; Broadfield, Z.J.; Stewart, A.R.; Wright, S.L.; Martineau, M.; Strefford, J.C.; Moorman, A.V. Molecular cytogenetic characterization of tcf3 (e2a)/19p13.3 rearrangements in b-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer 2007, 46, 478–486. [Google Scholar] [CrossRef]
- Guo, C.; Liu, S.; Wang, J.; Sun, M.Z.; Greenaway, F.T. Actb in cancer. Clin. Chim. Acta 2013, 417, 39–44. [Google Scholar] [CrossRef]
- Hu, X.; Du, S.; Yu, J.; Yang, X.; Yang, C.; Zhou, D.; Wang, Q.; Qin, S.; Yan, X.; He, L.; et al. Common housekeeping proteins are upregulated in colorectal adenocarcinoma and hepatocellular carcinoma, making the total protein a better “housekeeper”. Oncotarget 2016, 7, 66679–66688. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Z.; Chen, J.; Liu, F.; Bai, L. Overexpression of beta-actin is closely associated with metastasis of gastric cancer. Hepatogastroenterology 2013, 60, 620–623. [Google Scholar]
- Morris, H.T.; Machesky, L.M. Actin cytoskeletal control during epithelial to mesenchymal transition: Focus on the pancreas and intestinal tract. Br. J. Cancer 2015, 112, 613–620. [Google Scholar] [CrossRef]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Spencer, V.A.; Costes, S.; Inman, J.L.; Xu, R.; Chen, J.; Hendzel, M.J.; Bissell, M.J. Depletion of nuclear actin is a key mediator of quiescence in epithelial cells. J. Cell Sci. 2011, 124, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Fiore, A.; Spencer, V.A.; Mori, H.; Carvalho, H.F.; Bissell, M.J.; Bruni-Cardoso, A. Laminin-111 and the level of nuclear actin regulate epithelial quiescence via exportin-6. Cell Rep. 2017, 19, 2102–2115. [Google Scholar] [CrossRef] [PubMed]
- Pfitzer, L.; Moser, C.; Gegenfurtner, F.; Arner, A.; Foerster, F.; Atzberger, C.; Zisis, T.; Kubisch-Dohmen, R.; Busse, J.; Smith, R.; et al. Targeting actin inhibits repair of doxorubicin-induced DNA damage: A novel therapeutic approach for combination therapy. Cell Death Dis. 2019, 10, 302. [Google Scholar] [CrossRef]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The swi/snf complex is a mechanoregulated inhibitor of yap and taz. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Nyberg, K.D.; Scott, M.B.; Welsh, A.M.; Nguyen, A.H.; Wu, N.; Hohlbauch, S.V.; Geisse, N.A.; Gibb, E.A.; Robertson, A.G.; et al. Stiffness of pancreatic cancer cells is associated with increased invasive potential. Integr. Biol. 2016, 8, 1232–1245. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Lemma, S.; Avnet, S.; Salerno, M.; Chano, T.; Baldini, N. Identification and validation of housekeeping genes for gene expression analysis of cancer stem cells. PLoS ONE 2016, 11, e0149481. [Google Scholar] [CrossRef]
- Deindl, E.; Boengler, K.; van Royen, N.; Schaper, W. Differential expression of gapdh and beta3-actin in growing collateral arteries. Mol. Cell. Biochem. 2002, 236, 139–146. [Google Scholar] [CrossRef]
- Selvey, S.; Thompson, E.W.; Matthaei, K.; Lea, R.A.; Irving, M.G.; Griffiths, L.R. Beta-actin--an unsuitable internal control for rt-pcr. Mol. Cell. Probes 2001, 15, 307–311. [Google Scholar] [CrossRef]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. Actl6a is co-amplified with p63 in squamous cell carcinoma to drive yap activation, regenerative proliferation, and poor prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.Q.; Zhong, L.; Yao, J.J.; Liu, D.D.; Yuan, Z.; Liu, J.M.; Chen, M.; Yao, S.F.; Zhao, Y.; Liu, L.; et al. Actl6a interacts with p53 in acute promyelocytic leukemia cell lines to affect differentiation via the sox2/notch1 signaling pathway. Cell. Signal. 2019, 53, 390–399. [Google Scholar] [CrossRef]
- Xiao, S.; Chang, R.M.; Yang, M.Y.; Lei, X.; Liu, X.; Gao, W.B.; Xiao, J.L.; Yang, L.Y. Actin-like 6a predicts poor prognosis of hepatocellular carcinoma and promotes metastasis and epithelial-mesenchymal transition. Hepatology 2016, 63, 1256–1271. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Yang, H.; Xiao, S. Actl6a expression promotes invasion, metastasis and epithelial mesenchymal transition of colon cancer. BMC Cancer 2018, 18, 1020. [Google Scholar] [CrossRef]
- Wee, Y.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. Identification of novel prognosis-related genes associated with cancer using integrative network analysis. Sci. Rep. 2018, 8, 3233. [Google Scholar] [CrossRef]
- Lu, W.; Fang, L.; Ouyang, B.; Zhang, X.; Zhan, S.; Feng, X.; Bai, Y.; Han, X.; Kim, H.; He, Q.; et al. Actl6a protects embryonic stem cells from differentiating into primitive endoderm. Stem Cells 2015, 33, 1782–1793. [Google Scholar] [CrossRef]
- Gerhold, C.B.; Winkler, D.D.; Lakomek, K.; Seifert, F.U.; Fenn, S.; Kessler, B.; Witte, G.; Luger, K.; Hopfner, K.P. Structure of actin-related protein 8 and its contribution to nucleosome binding. Nucleic Acids Res. 2012, 40, 11036–11046. [Google Scholar] [CrossRef]
- Nishimoto, N.; Watanabe, M.; Watanabe, S.; Sugimoto, N.; Yugawa, T.; Ikura, T.; Koiwai, O.; Kiyono, T.; Fujita, M. Heterocomplex formation by arp4 and beta-actin is involved in the integrity of the brg1 chromatin remodeling complex. J. Cell Sci. 2012, 125, 3870–3882. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Q.; Leung, E.L.H.; Li, Y.; Fan, X.; Wu, Q.; Yao, X.; Liu, L. Compound c620-0696, a new potent inhibitor targeting bptf, the chromatin-remodeling factor in non-small-cell lung cancer. Front. Med. 2019. [Google Scholar] [CrossRef]
- Bevill, S.M.; Olivares-Quintero, J.F.; Sciaky, N.; Golitz, B.T.; Singh, D.; Beltran, A.S.; Rashid, N.U.; Stuhlmiller, T.J.; Hale, A.; Moorman, N.J.; et al. Gsk2801, a baz2/brd9 bromodomain inhibitor, synergizes with bet inhibitors to induce apoptosis in triple-negative breast cancer. Mol. Cancer Res. 2019, 17, 1503–1518. [Google Scholar] [CrossRef]
- Vangamudi, B.; Paul, T.A.; Shah, P.K.; Kost-Alimova, M.; Nottebaum, L.; Shi, X.; Zhan, Y.; Leo, E.; Mahadeshwar, H.S.; Protopopov, A.; et al. The smarca2/4 atpase domain surpasses the bromodomain as a drug target in swi/snf-mutant cancers: Insights from cdna rescue and pfi-3 inhibitor studies. Cancer Res. 2015, 75, 3865–3878. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sharma, S.; Cui, H.; LeBlanc, S.E.; Zhang, H.; Muthuswami, R.; Nickerson, J.A.; Imbalzano, A.N. Targeting the chromatin remodeling enzyme brg1 increases the efficacy of chemotherapy drugs in breast cancer cells. Oncotarget 2016, 7, 27158–27175. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, R.; Hussain, S.; Goel, K.; Sharma, S.; Bisht, D.; Chanana, U.B.; Hockensmith, J.W.; Muthuswami, R. Altering mammalian transcription networking with adaadi: An inhibitor of atp-dependent chromatin remodeling. BioRxiv 2019. [Google Scholar] [CrossRef]
- Muthuswami, R.; Mesner, L.D.; Wang, D.; Hill, D.A.; Imbalzano, A.N.; Hockensmith, J.W. Phosphoaminoglycosides inhibit swi2/snf2 family DNA-dependent molecular motor domains. Biochemistry 2000, 39, 4358–4365. [Google Scholar] [CrossRef]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. Baf complex vulnerabilities in cancer demonstrated via structure-based protac design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).