Omics-based Investigation of Diet-induced Obesity Synergized with HBx, Src, and p53 Mutation Accelerating Hepatocarcinogenesis in Zebrafish Model

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Overview of Omics-Based Investigation of Diet-Induced Obesity and Hepatocarcinogenesis in Zebrafish Model
2.2. Weight Changes in Zebrafish with Different Genetic Backgrounds by Three Feeding Methods
2.3. Impact of Genetic Factors on Steatosis and Cell Proliferation
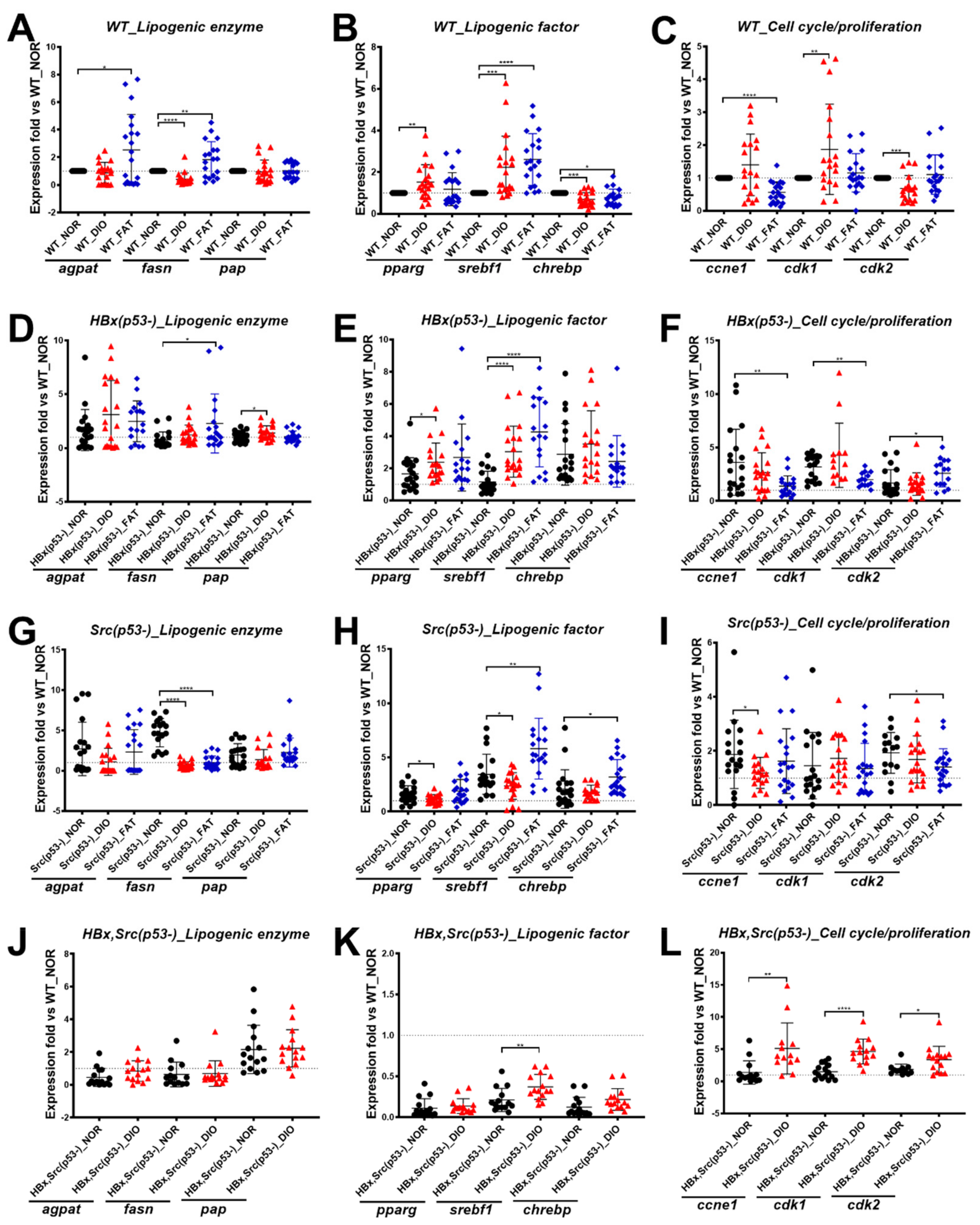
2.4. Validation of Lipogenic Enzymes/Factors and Cell Cycle–Related Genes in Wild-Type Fish after Overfeeding
2.5. Expression of Lipogenesis Factor, Lipogenesis Enzymes, and Cell Cycle–Related Genes in HBx(p53-), Src(p53-), and HBx,Src(p53-) Transgenic Fish after Overfeeding
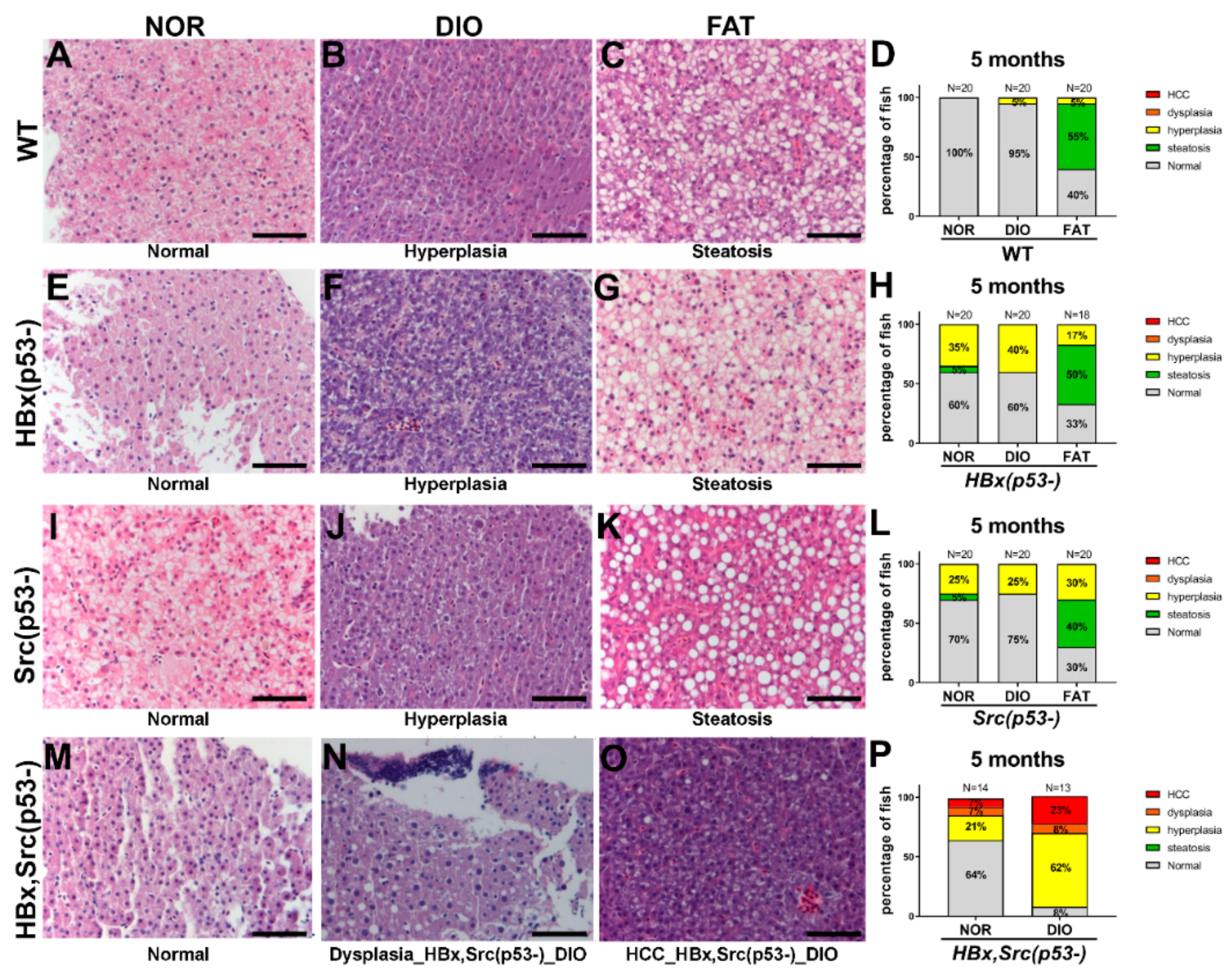
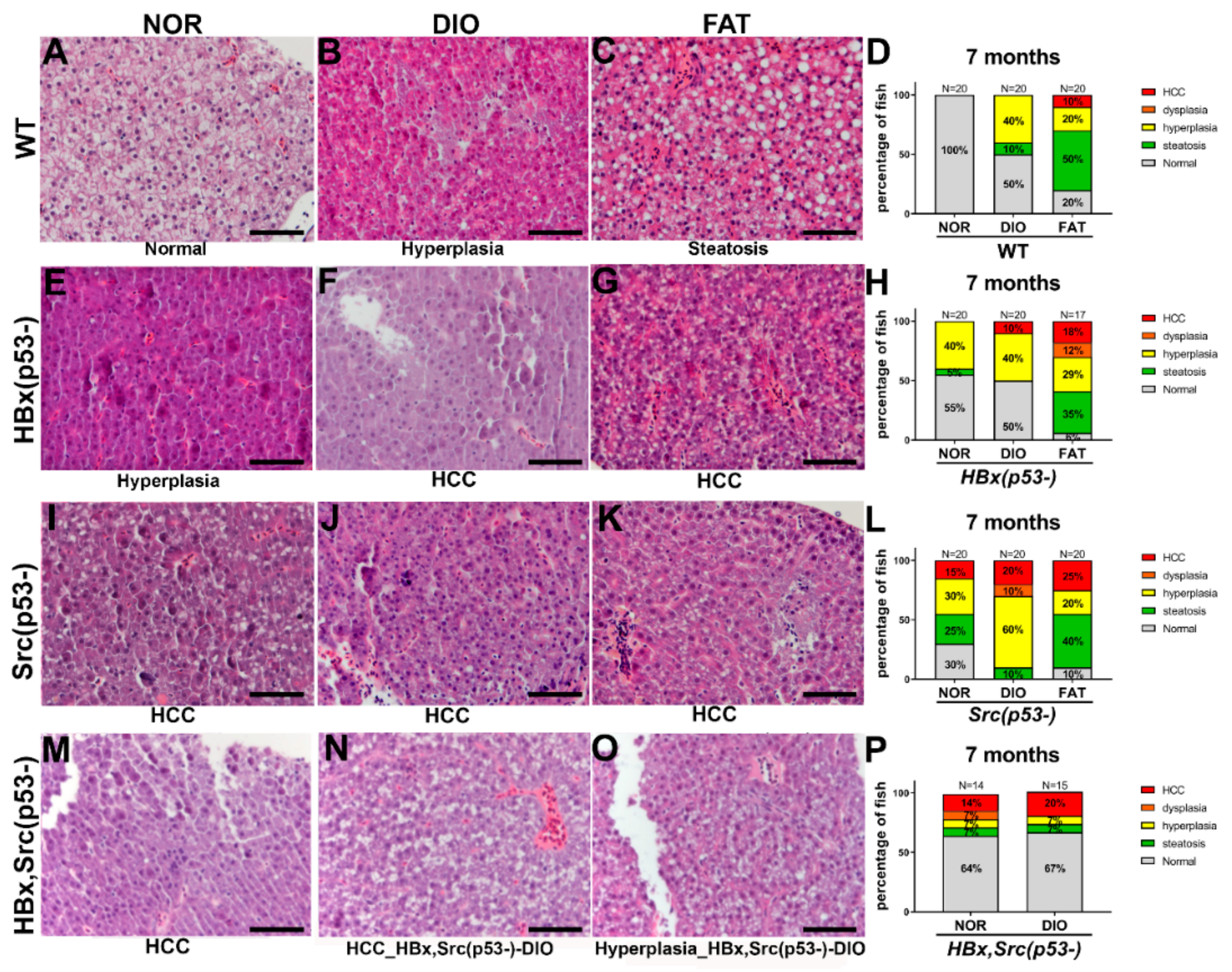
2.6. Liver Pathology after Diet-Induced Obesity
2.7. Global Omics Data Analysis
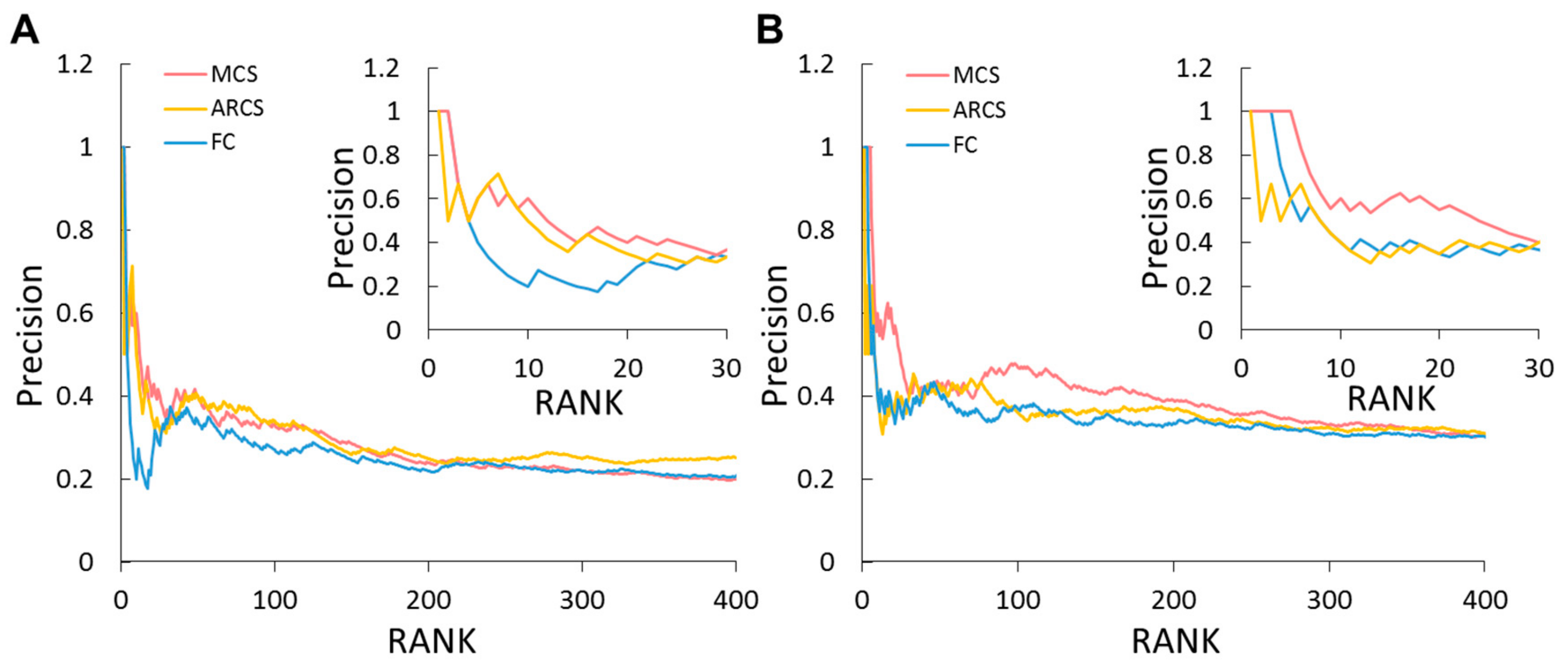
2.8. Similarity Estimation among Genes, Pathways, and System Levels of Global Omics Data Analysis
2.9. Identifying Potential Genes by Global Omics Data Analysis
3. Discussion
4. Materials and Methods
4.1. Zebrafish Maintenance and Transgenic Zebrafish Lines
4.2. Feeding Method
4.3. Body Weight Measurement and Liver Specimen Collection
4.4. RNA Analysis of Gene Expression
4.5. Quantitative PCR (qPCR)
- (1)
- 50 °C for 2 min, 95 °C for 5 min, 4 °C thereafter
- (2)
- 95 °C for 10 min
- (3)
- 95 °C for 15 sec, 60 °C for 1 min (40 cycles)
- (4)
- 95 °C for 15 sec, 60 °C for 15 sec, 95 °C for 15 sec
4.6. Histopathological Analysis
4.7. Statistical Analysis
4.8. GeneTitan™ Array and RNA-seq for Gene Expression Profiling
4.9. Global Omics Data Analysis
4.10. Scoring Function for Potential Gene Selection
4.11. GeneTitan™ Array for Gene Expression Profiling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roberts, L.R.; Gores, G.J. Hepatocellular carcinoma: Molecular pathways and new therapeutic targets. Semin. Liver. Dis. 2005, 25, 212–225. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Samonakis, D.N.; Kouroumalis, E.A. Systemic treatment for hepatocellular carcinoma: Still unmet expectations. World J. Hepatol. 2017, 9, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Ayub, A.; Ashfaq, U.A.; Haque, A. HBV induced HCC: Major risk factors from genetic to molecular level. Biomed. Res. Int. 2013, 2013, 810461. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Yang, W.Y.; Tsai, S.M.; Lin, Y.M.; Chang, P.H.; Chen, J.R.; Wang, H.D.; Wu, J.L.; Jin, S.L.; Yuh, C.H. Liver-specific expressions of HBx and src in the p53 mutant trigger hepatocarcinogenesis in zebrafish. PLoS ONE 2013, 8, e76951. [Google Scholar] [CrossRef]
- Shi, D.R. The HBx protein expression in liver cancer. Zhonghua Bing Li Xue Za Zhi 1991, 20, 85–87. [Google Scholar] [CrossRef]
- Zhao, R.; Wu, Y.; Wang, T.; Zhang, Y.; Kong, D.; Zhang, L.; Li, X.; Wang, G.; Jin, Y.; Jin, X.; et al. Elevated Src expression associated with hepatocellular carcinoma metastasis in northern Chinese patients. Oncol. Lett. 2015, 10, 3026–3034. [Google Scholar] [CrossRef]
- Lu, J.W.; Hsia, Y.; Yang, W.Y.; Lin, Y.I.; Li, C.C.; Tsai, T.F.; Chang, K.W.; Shieh, G.S.; Tsai, S.F.; Wang, H.D.; et al. Identification of the common regulators for hepatocellular carcinoma induced by hepatitis B virus X antigen in a mouse model. Carcinogenesis 2012, 33, 209–219. [Google Scholar] [CrossRef]
- Qi, L.N.; Bai, T.; Chen, Z.S.; Wu, F.X.; Chen, Y.Y.; De Xiang, B.; Peng, T.; Han, Z.G.; Li, L.Q. The p53 mutation spectrum in hepatocellular carcinoma from Guangxi, China: Role of chronic hepatitis B virus infection and aflatoxin B1 exposure. Liver Int. 2015, 35, 999–1009. [Google Scholar] [CrossRef]
- Gouas, D.A.; Villar, S.; Ortiz-Cuaran, S.; Legros, P.; Ferro, G.; Kirk, G.D.; Lesi, O.A.; Mendy, M.; Bah, E.; Friesen, M.D.; et al. TP53 R249S mutation, genetic variations in HBX and risk of hepatocellular carcinoma in The Gambia. Carcinogenesis 2012, 33, 1219–1224. [Google Scholar] [CrossRef]
- Guo, X.; Jin, M.; Yang, M.; Liu, K.; Li, J.W. Type 2 diabetes mellitus and the risk of hepatitis C virus infection: A systematic review. Sci. Rep. 2013, 3, 2981. [Google Scholar] [CrossRef] [PubMed]
- Arase, Y.; Kobayashi, M.; Suzuki, F.; Suzuki, Y.; Kawamura, Y.; Akuta, N.; Kobayashi, M.; Sezaki, H.; Saito, S.; Hosaka, T.; et al. Effect of type 2 diabetes on risk for malignancies includes hepatocellular carcinoma in chronic hepatitis C. Hepatology 2013, 57, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Hohensee, T.; Haybaeck, J. Steatosis and steatohepatitis: Complex disorders. Int. J. Mol. Sci. 2014, 15, 9924–9944. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Freedman, N.D.; Demuth, J.; Yang, B.; Van Den Eeden, S.K.; Engel, L.S.; McGlynn, K.A. Obesity, diabetes, serum glucose, and risk of primary liver cancer by birth cohort, race/ethnicity, and sex: Multiphasic health checkup study. Cancer Epidemiol. 2016, 42, 140–146. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef]
- Ray, K. Gut microbiota: Obesity-induced microbial metabolite promotes HCC. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 442. [Google Scholar] [CrossRef]
- Gan, L.; Liu, Z.; Sun, C. Obesity linking to hepatocellular carcinoma: A global view. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 97–102. [Google Scholar] [CrossRef]
- Abarca-Gómez, L.; Abdeen, Z.A.; Hamid, Z.A.; Abu-Rmeileh, N.M.; Acosta-Cazares, B.; Acuin, C.; Adams, R.J.; Aekplakorn, W.; Afsana, K.; Aguilar-Salinas, C.A.; et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Farrell, G.C.; Wong, V.W.; Chitturi, S. NAFLD in Asia—As common and important as in the West. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 307–318. [Google Scholar] [CrossRef]
- Huang, S.F.; Chang, I.C.; Hong, C.C.; Yen, T.C.; Chen, C.L.; Wu, C.C.; Tsai, C.C.; Ho, M.C.; Lee, W.C.; Yu, H.C.; et al. Metabolic risk factors are associated with non-hepatitis B non-hepatitis C hepatocellular carcinoma in Taiwan, an endemic area of chronic hepatitis B. Hepatol. Commun. 2018, 2, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Mejia-Guevara, I.; Estrada, K.; Liu, S.Y.; Glymour, M.M. Association of a Genetic Risk Score with Body Mass Index Across Different Birth Cohorts. JAMA 2016, 316, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Spitsbergen, J.M.; Kent, M.L. The state of the art of the zebrafish model for toxicology and toxicologic pathology research—Advantages and current limitations. Toxicol. Pathol. 2003, 31, 62–87. [Google Scholar]
- Zon, L.I.; Peterson, R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug. Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Huiting, L.N.; Laroche, F.; Feng, H. The Zebrafish as a Tool to Cancer Drug Discovery. Austin J. Pharmacol. Ther. 2015, 3, 1069. [Google Scholar]
- Kirchberger, S.; Sturtzel, C.; Pascoal, S.; Distel, M. Quo natas, Danio?-Recent Progress in Modeling Cancer in Zebrafish. Front. Oncol. 2017, 7, 186. [Google Scholar] [CrossRef]
- Schimmenti, L.A. Zebrafish and the Cancer Moonshot. Zebrafish 2017, 14, 390–392. [Google Scholar] [CrossRef]
- Lu, J.W.; Liao, C.Y.; Yang, W.Y.; Lin, Y.M.; Jin, S.L.; Wang, H.D.; Yuh, C.H. Overexpression of endothelin 1 triggers hepatocarcinogenesis in zebrafish and promotes cell proliferation and migration through the AKT pathway. PLoS ONE 2014, 9, e85318. [Google Scholar] [CrossRef]
- Lu, J.W.; Yang, W.Y.; Lin, Y.M.; Jin, S.L.; Yuh, C.H. Hepatitis B virus X antigen and aflatoxin B1 synergistically cause hepatitis, steatosis and liver hyperplasia in transgenic zebrafish. Acta Histochem. 2013, 115, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.C.; Hsiao, Y.C.; Yang, W.Y.; Tsai, S.L.; Lin, H.K.; Liao, C.Y.; Lu, J.W.; Chou, Y.T.; Wang, H.D.; Yuh, C.H. Up-Regulation of Golgi α-Mannosidase IA and Down-Regulation of Golgi α-Mannosidase IC Activates Unfolded Protein Response During Hepatocarcinogenesis. Hepatol. Commun. 2017, 1, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.T.; Chen, L.Y.; Tsai, S.L.; Tu, H.C.; Lu, J.W.; Ciou, S.C.; Wang, H.D.; Yuh, C.H. Ribose-5-Phosphate Isomerase a Overexpression Promotes Liver Cancer Development in Transgenic Zebrafish via Activation of ERK and beta-catenin Pathways. Carcinogenesis 2019, 40, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Maddison, L.A.; Chen, W. Zebrafish as a Model for Obesity and Diabetes. Front. Cell Dev. Biol. 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minchin, J.E.N.; Scahill, C.M.; Staudt, N.; Busch-Nentwich, E.M.; Rawls, J.F. Deep phenotyping in zebrafish reveals genetic and diet-induced adiposity changes that may inform disease risk. J. Lipid Res. 2018, 59, 1536–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Nishimura, Y.; Zang, L.; Hirano, M.; Shimada, Y.; Wang, Z.; Umemoto, N.; Kuroyanagi, J.; Nishimura, N.; Tanaka, T. Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol. 2010, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Weber, G. Enzymology of cancer cells (first of two parts). N. Engl. J. Med. 1977, 296, 486–492. [Google Scholar] [CrossRef]
- Weber, G. Enzymology of cancer cells (second of two parts). N. Engl. J. Med. 1977, 296, 541–551. [Google Scholar] [CrossRef]
- Weber, G. Enzymes of purine metabolism in cancer. Clin. Biochem. 1983, 16, 57–63. [Google Scholar] [CrossRef]
- Djiogue, S.; Nwabo Kamdje, A.H.; Vecchio, L.; Kipanyula, M.J.; Farahna, M.; Aldebasi, Y.; Seke Etet, P.F. Insulin resistance and cancer: The role of insulin and IGFs. Endocr. Relat. Cancer 2013, 20, R1–R17. [Google Scholar] [CrossRef] [Green Version]
- Pinero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutierrez-Sacristan, A.; Deu-Pons, J.; Centeno, E.; Garcia-Garcia, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Bjermo, H.; Riserus, U. Role of hepatic desaturases in obesity-related metabolic disorders. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.; Stefan, N.; Cegan, A.; Walenta, M.; Wagner, S.; Konigsrainer, A.; Konigsrainer, I.; Machicao, F.; Schick, F.; Haring, H.U.; et al. Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J. Clin. Endocrinol. Metab. 2011, 96, E1126–E1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, O.; Pramfalk, C.; Pedrelli, M.; Olin, M.; Steffensen, K.R.; Eriksson, M.; Parini, P. Genetic depletion of Soat2 diminishes hepatic steatosis via genes regulating de novo lipogenesis and by GLUT2 protein in female mice. Dig. Liver Dis. 2019, 51, 1016–1022. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Pirola, C.J. Repurposing drugs to target nonalcoholic steatohepatitis. World J. Gastroenterol. 2019, 25, 1783–1796. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, Q.L.; Shen, L.; Tao, Y.Y.; Liu, C.H. MicroRNA-101 suppresses liver fibrosis by downregulating PI3K/Akt/mTOR signaling pathway. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 575–584. [Google Scholar] [CrossRef]
- Su, Z.L.; Su, C.W.; Huang, Y.L.; Yang, W.Y.; Sampurna, B.P.; Ouchi, T.; Lee, K.L.; Wu, C.S.; Wang, H.D.; Yuh, C.H. A Novel AURKA Mutant-Induced Early-Onset Severe Hepatocarcinogenesis Greater than Wild-Type via Activating Different Pathways in Zebrafish. Cancers 2019, 11, 927. [Google Scholar] [CrossRef] [Green Version]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two hits? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Oikawa, T.; Nakano, M.; Saeki, C.; Torisu, Y.; Kajihara, M.; Saruta, M. Recent Insights into the Multiple Pathways Driving Non-alcoholic Steatohepatitis-Derived Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis-(NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Jaramillo, M.; Spooner, M.H.; Lohr, C.V.; Wong, C.P.; Zhang, W.; Jump, D.B. Lipidomic and transcriptomic analysis of western diet-induced nonalcoholic steatohepatitis (NASH) in female Ldlr−/− mice. PLoS ONE 2019, 14, e0214387. [Google Scholar] [CrossRef] [Green Version]
- Dalmas, E. Innate immune priming of insulin secretion. Curr. Opin. Immunol. 2019, 56, 44–49. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, F.; Feng, L.; Shan, Q.; Zheng, G.H.; Wang, Y.J.; Lu, J.; Fan, S.H.; Sun, C.H.; Wu, D.M.; et al. FBXW7 suppresses HMGB1-mediated innate immune signaling to attenuate hepatic inflammation and insulin resistance in a mouse model of nonalcoholic fatty liver disease. Mol. Med. 2019, 25, 29. [Google Scholar] [CrossRef] [Green Version]
- Kammerl, I.E.; Meiners, S. Proteasome function shapes innate and adaptive immune responses. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L328–L336. [Google Scholar] [CrossRef] [Green Version]
- Yaqoob, P. Fatty acids and the immune system: From basic science to clinical applications. Proc. Nutr. Soc. 2004, 63, 89–104. [Google Scholar] [CrossRef] [Green Version]
- You, M.S.; Jiang, Y.J.; Yuh, C.H.; Wang, C.M.; Tang, C.H.; Chuang, Y.J.; Lin, B.H.; Wu, J.L.; Hwang, S.P. A sketch of the Taiwan Zebrafish Core Facility. Zebrafish 2016, 13 (Suppl. 1), 24–29. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Knuth, D.E.; Patashnik, O. Concrete Mathematics: A Foundation for Computer Science; Addison-Wesley: Boston, MA, USA, 1989; p. 625. [Google Scholar]
- Lin, C.Y.; Lin, Y.W.; Yu, S.W.; Lo, Y.S.; Yang, J.M. MoNetFamily: A web server to infer homologous modules and module-module interaction networks in vertebrates. Nucleic Acids Res. 2012, 40, W263–W270. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, 3. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Normal Diet (NOR) | Diet-Induced Obesity (DIO) | High-Fat Diet (FAT) | |
|---|---|---|---|---|
| Time | ||||
| 09:00 | Powdered feed (0.22g) | powdered feed (0.22 g) | powdered feed (0.22 g) | |
| 11:00 | None | brine shrimp 2 mL | high-fat fish food (0.22 g) | |
| 15:00 | Artemia 0.5mL | brine shrimp 2 mL | high-fat fish food (0.22 g) brine shrimp 0.5 mL | |
| 17:30 | None | brine shrimp 2 mL | high-fat fish food (0.22 g) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, W.-Y.; Rao, P.-S.; Luo, Y.-C.; Lin, H.-K.; Huang, S.-H.; Yang, J.-M.; Yuh, C.-H. Omics-based Investigation of Diet-induced Obesity Synergized with HBx, Src, and p53 Mutation Accelerating Hepatocarcinogenesis in Zebrafish Model. Cancers 2019, 11, 1899. https://doi.org/10.3390/cancers11121899
Yang W-Y, Rao P-S, Luo Y-C, Lin H-K, Huang S-H, Yang J-M, Yuh C-H. Omics-based Investigation of Diet-induced Obesity Synergized with HBx, Src, and p53 Mutation Accelerating Hepatocarcinogenesis in Zebrafish Model. Cancers. 2019; 11(12):1899. https://doi.org/10.3390/cancers11121899
Chicago/Turabian StyleYang, Wan-Yu, Pei-Shu Rao, Yong-Chun Luo, Hua-Kuo Lin, Sing-Han Huang, Jinn-Moon Yang, and Chiou-Hwa Yuh. 2019. "Omics-based Investigation of Diet-induced Obesity Synergized with HBx, Src, and p53 Mutation Accelerating Hepatocarcinogenesis in Zebrafish Model" Cancers 11, no. 12: 1899. https://doi.org/10.3390/cancers11121899