The Expression of Myeloproliferative Neoplasm-Associated Calreticulin Variants Depends on the Functionality of ER-Associated Degradation

 , , ,
, , ,  , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
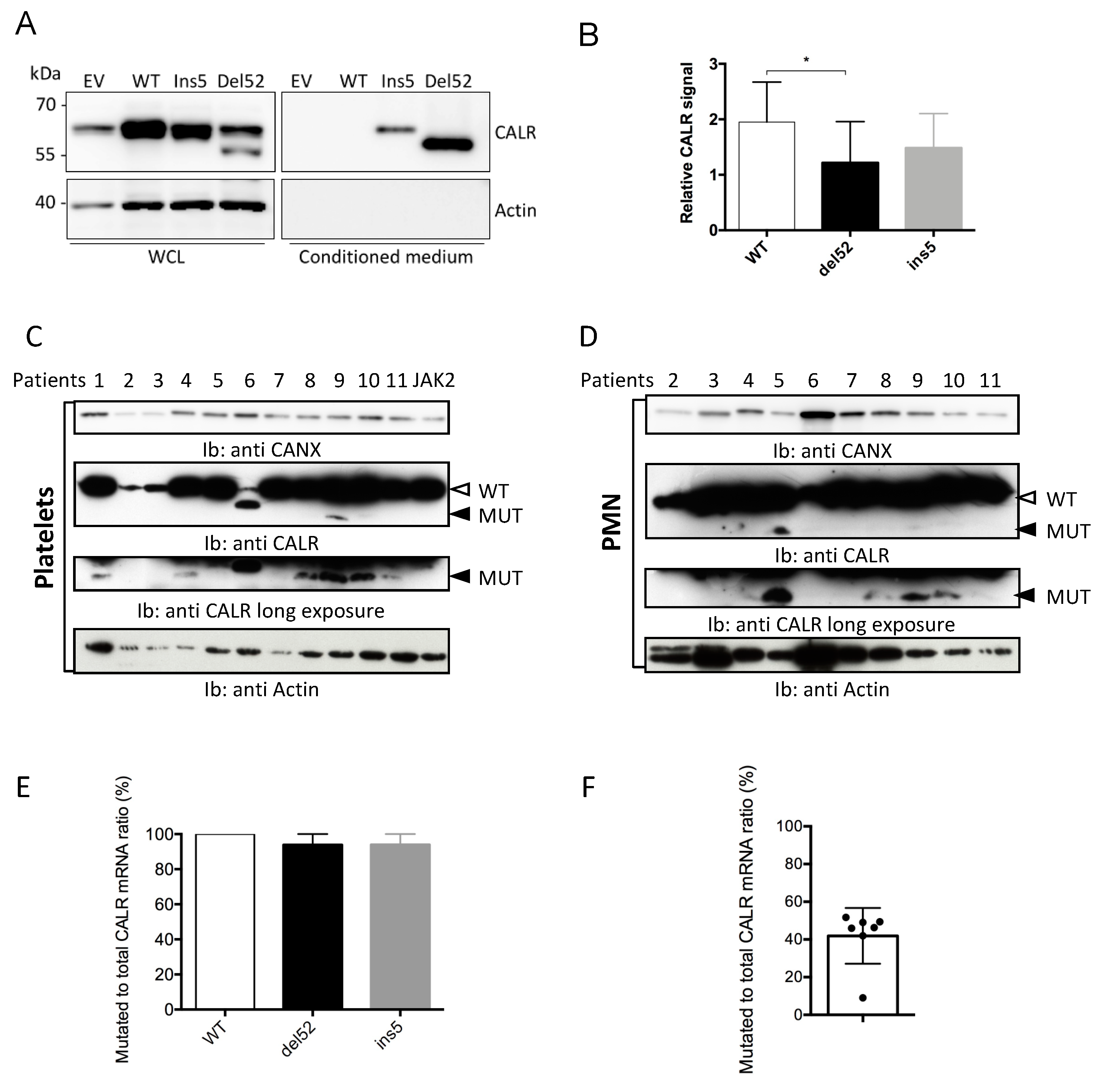
2.1. Mutant CALR are Faintly Expressed in Transfected and Primary Cells
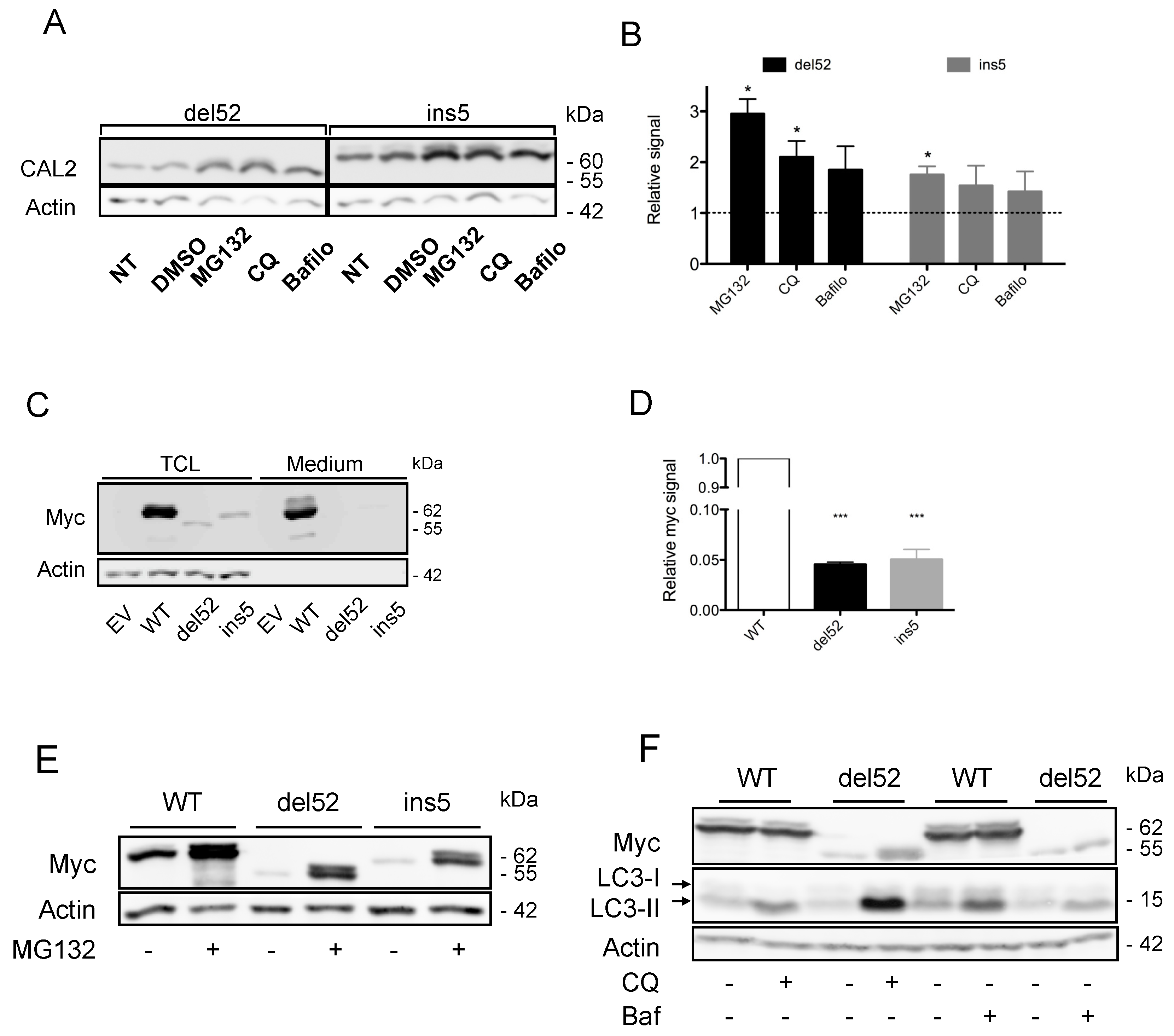
2.2. Proteasome-Mediated Degradation Participates in Low CALR Mutant Expression
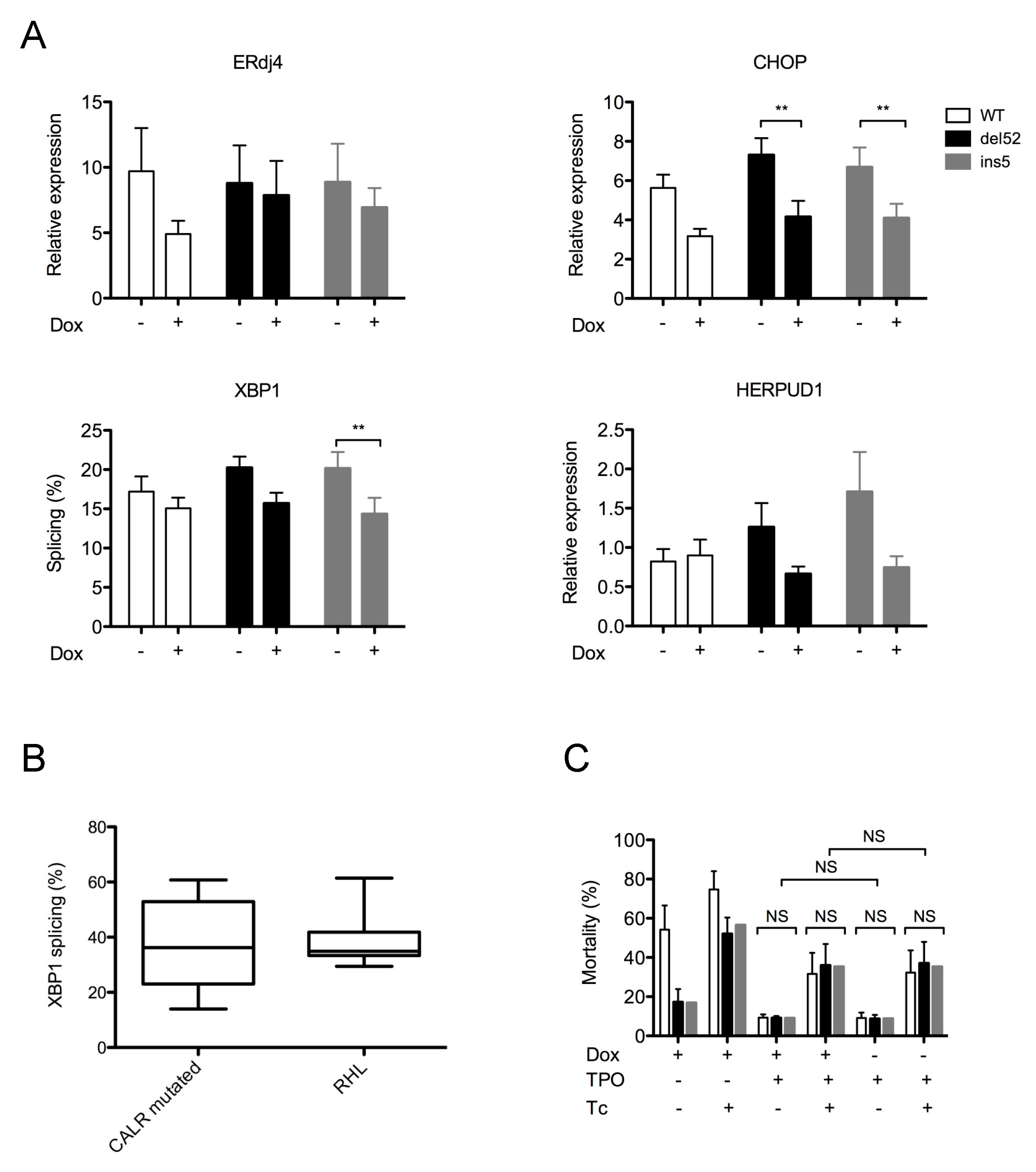
2.3. Mutant CALR Expression Does Not Alter ER Homeostasis
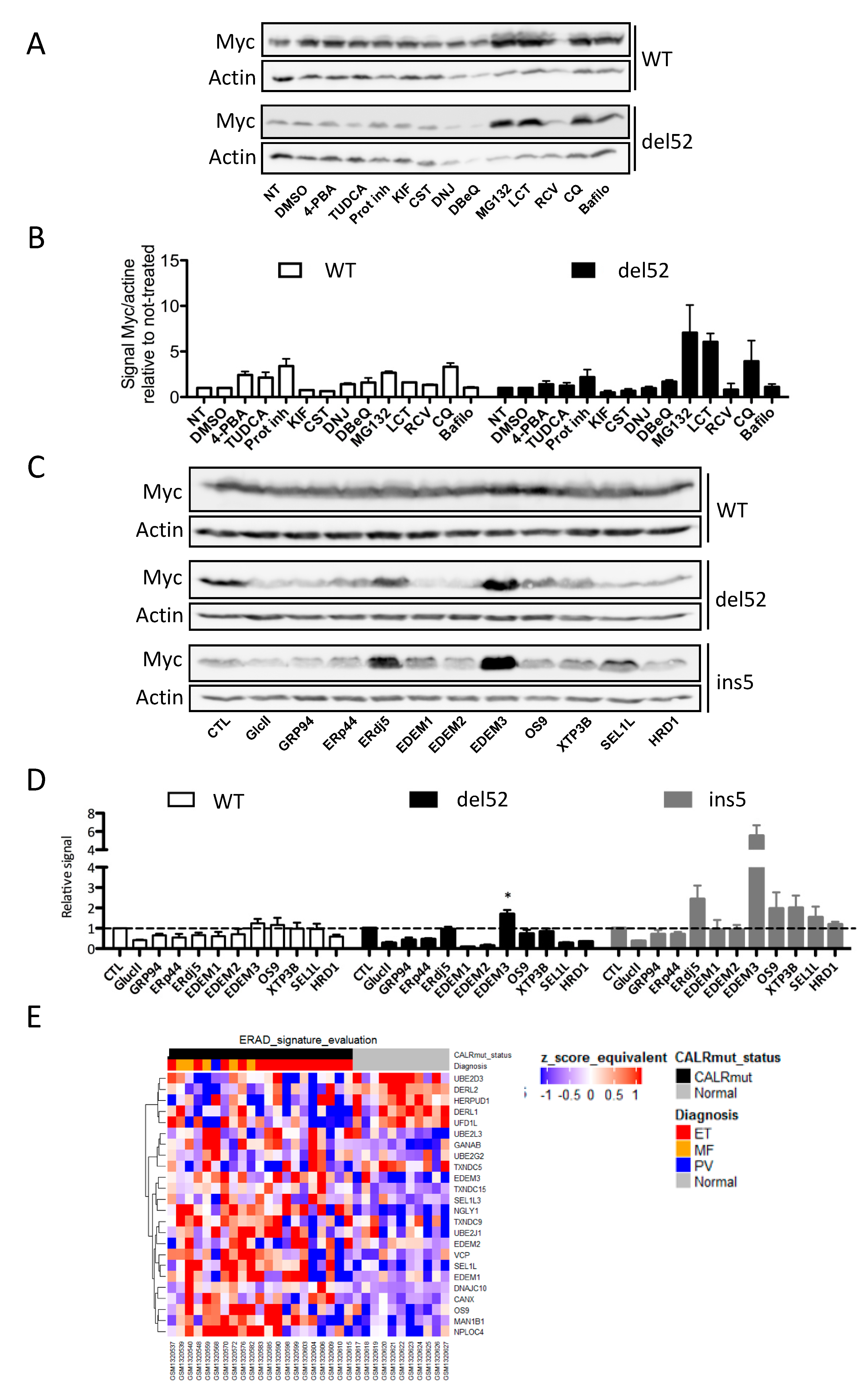
2.4. ERAD-Dependent Mechanisms of CALR Variants Degradation
2.5. Identification of a Network Involved in the ERAD-Dependent Degradation of CALR Variants
3. Discussion
4. Materials and Methods
4.1. Patients and DNA Constructs
4.2. Cell Culture and Lentiviral Transduction
4.3. Chemicals and Antibodies
4.4. RNA Interference
4.5. Coimmunoprecipitation and Western Blotting
4.6. RT-qPCR and ddPCR
4.7. Gene Expression Microarray Analysis and Statistical Selection
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651. [Google Scholar] [CrossRef]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344 Pt 2, 281–292. [Google Scholar] [CrossRef]
- Lamriben, L.; Graham, J.B.; Adams, B.M.; Hebert, D.N. N-Glycan-based ER molecular chaperone and protein quality control system: The Calnexin binding cycle. Traffic Cph. Den. 2016, 17, 308–326. [Google Scholar] [CrossRef] [PubMed]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.-M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef]
- Han, L.; Schubert, C.; Köhler, J.; Schemionek, M.; Isfort, S.; Brümmendorf, T.H.; Koschmieder, S.; Chatain, N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 2016, 9, 45. [Google Scholar] [CrossRef]
- Kollmann, K.; Warsch, W.; Gonzalez-Arias, C.; Nice, F.L.; Avezov, E.; Milburn, J.; Li, J.; Dimitropoulou, D.; Biddie, S.; Wang, M.; et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia 2017, 31, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Garbati, M.R.; Welgan, C.A.; Landefeld, S.H.; Newell, L.F.; Agarwal, A.; Dunlap, J.B.; Chourasia, T.K.; Lee, H.; Elferich, J.; Traer, E.; et al. Mutant calreticulin-expressing cells induce monocyte hyperreactivity through a paracrine mechanism. Am. J. Hematol. 2016, 91, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Smirle, J.; Cameron, P.H.; Thomas, D.Y.; Bergeron, J.J.M. Calnexin phosphorylation: Linking cytoplasmic signalling to endoplasmic reticulum lumenal functions. Semin. Cell Dev. Biol. 2010, 21, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.H.; Chevet, E.; Pluquet, O.; Thomas, D.Y.; Bergeron, J.J.M. Calnexin phosphorylation attenuates the release of partially misfolded alpha1-antitrypsin to the secretory pathway. J. Biol. Chem. 2009, 284, 34570–34579. [Google Scholar] [CrossRef]
- Norez, C.; Vandebrouck, C.; Bertrand, J.; Noel, S.; Durieu, E.; Oumata, N.; Galons, H.; Antigny, F.; Chatelier, A.; Bois, P.; et al. Roscovitine is a proteostasis regulator that corrects the trafficking defect of F508del-CFTR by a CDK-independent mechanism. Br. J. Pharmacol. 2014, 171, 4831–4849. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.; Brunel, J.-P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef]
- Timms, R.T.; Menzies, S.A.; Tchasovnikarova, I.A.; Christensen, L.C.; Williamson, J.C.; Antrobus, R.; Dougan, G.; Ellgaard, L.; Lehner, P.J. Genetic dissection of mammalian ERAD through comparative haploid and CRISPR forward genetic screens. Nat. Commun. 2016, 7, 11786. [Google Scholar] [CrossRef]
- Lau, W.W.Y.; Hannah, R.; Green, A.R.; Göttgens, B. The JAK-STAT signaling pathway is differentially activated in CALR-positive compared with JAK2V617F-positive ET patients. Blood 2015, 125, 1679–1681. [Google Scholar] [CrossRef]
- Nam, A.S.; Kim, K.-T.; Chaligne, R.; Izzo, F.; Ang, C.; Abu-Zeinah, G.; Omans, N.D.; Pastore, A.; Taylor, J.; Alonso, A.; et al. High throughput droplet single-cell Genotyping of Transcriptomes (GoT) reveals the cell identity dependency of the impact of somatic mutations. bioRxiv 2018, 444687. [Google Scholar] [CrossRef]
- Salati, S.; Genovese, E.; Carretta, C.; Zini, R.; Bartalucci, N.; Prudente, Z.; Pennucci, V.; Ruberti, S.; Rossi, C.; Rontauroli, S.; et al. Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Barosi, G.; Gattoni, E.; Guglielmelli, P.; Campanelli, R.; Facchetti, F.; Fisogni, S.; Goldberg, J.; Marchioli, R.; Hoffman, R.; Vannucchi, A.M.; et al. Phase I/II study of single-agent bortezomib for the treatment of patients with myelofibrosis. Clinical and biological effects of proteasome inhibition. Am. J. Hematol. 2010, 85, 616–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesa, R.A.; Verstovsek, S.; Rivera, C.; Pardanani, A.; Hussein, K.; Lasho, T.; Wu, W.; Tefferi, A. Bortezomib therapy in myelofibrosis: A phase II clinical trial. Leukemia 2008, 22, 1636–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.J.; Palazzo, A.; Chaabane, C.; Albarran, L.; Polidano, E.; Lebozec, K.; Dally, S.; Nurden, P.; Enouf, J.; Debili, N.; et al. Crucial role for endoplasmic reticulum stress during megakaryocyte maturation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2750–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshad, N.; Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecquet, C.; Chachoua, I.; Roy, A.; Balligand, T.; Vertenoeil, G.; Leroy, E.; Albu, R.-I.; Defour, J.-P.; Nivarthi, H.; Hug, E.; et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood 2019, 133, 2669–2681. [Google Scholar] [CrossRef]
- Sollazzo, D.; Forte, D.; Polverelli, N.; Perricone, M.; Romano, M.; Luatti, S.; Vianelli, N.; Cavo, M.; Palandri, F.; Catani, L. Circulating calreticulin is increased in myelofibrosis: Correlation with interleukin-6 plasma levels, bone marrow fibrosis, and splenomegaly. Mediators Inflamm. 2016, 2016, 5860657. [Google Scholar] [CrossRef]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef]
- Nivarthi, H.; Chen, D.; Cleary, C.; Kubesova, B.; Jäger, R.; Bogner, E.; Marty, C.; Pecquet, C.; Vainchenker, W.; Constantinescu, S.N.; et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia 2016, 30, 1759–1763. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.M.; Mancino, L.; Thammavongsa, V.; Cantley, R.L.; Raghavan, M. A polypeptide binding conformation of calreticulin is induced by heat shock, calcium depletion, or by deletion of the C-terminal acidic region. Mol. Cell 2004, 15, 913–923. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Baral, A.J.; Beeson, D.; Rivera, J.F.; Ko, A.; Florescu, N.; Birrane, G.; Chen, E.; Mullally, A. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 2018, 131, 782–786. [Google Scholar] [CrossRef]
- Bernasconi, R.; Galli, C.; Calanca, V.; Nakajima, T.; Molinari, M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol. 2010, 188, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T.; et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.-Y.; Huang, C.-H.; Zhuang, Y.-H.; Christianson, J.C.; Chen, X. EDEM2 and OS-9 are required for ER-associated degradation of non-glycosylated sonic hedgehog. PLoS ONE 2014, 9, e92164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda-Shimizu, Y.; Hendershot, L.M. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell 2007, 28, 544–554. [Google Scholar] [CrossRef] [Green Version]
- Pronier, E.; Cifani, P.; Merlinsky, T.R.; Berman, K.B.; Somasundara, A.V.H.; Rampal, R.K.; LaCava, J.; Wei, K.E.; Pastore, F.; Maag, J.L.V.; et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome activation by small molecules. Cell Chem. Biol. 2017, 24, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, A.; Higa, A.; Jégou, G.; Jouan, F.; Pineau, R.; Saas, L.; Avril, T.; Pluquet, O.; Chevet, E. Alterations of EDEM1 functions enhance ATF6 pro-survival signaling. FEBS J. 2018, 285, 4146–4164. [Google Scholar] [CrossRef]
- Mansier, O.; Migeon, M.; Saint-Lézer, A.; James, C.; Verger, E.; Robin, M.; Socié, G.; Bidet, A.; Mahon, F.-X.; Cassinat, B.; et al. Quantification of the mutant CALR allelic burden by digital PCR: Application to minimal residual disease evaluation after bone marrow transplantation. J. Mol. Diagn. 2016, 18, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Fessart, D.; Domblides, C.; Avril, T.; Eriksson, L.A.; Begueret, H.; Pineau, R.; Malrieux, C.; Dugot-Senant, N.; Lucchesi, C.; Chevet, E.; et al. Secretion of protein disulphide isomerase AGR2 confers tumorigenic properties. eLife 2016, 5, e13887. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansier, O.; Prouzet-Mauléon, V.; Jégou, G.; Barroso, K.; Raymundo, D.P.; Chauveau, A.; Dumas, P.-Y.; Lagarde, V.; Turcq, B.; Pasquet, J.-M.; et al. The Expression of Myeloproliferative Neoplasm-Associated Calreticulin Variants Depends on the Functionality of ER-Associated Degradation. Cancers 2019, 11, 1921. https://doi.org/10.3390/cancers11121921
Mansier O, Prouzet-Mauléon V, Jégou G, Barroso K, Raymundo DP, Chauveau A, Dumas P-Y, Lagarde V, Turcq B, Pasquet J-M, et al. The Expression of Myeloproliferative Neoplasm-Associated Calreticulin Variants Depends on the Functionality of ER-Associated Degradation. Cancers. 2019; 11(12):1921. https://doi.org/10.3390/cancers11121921
Chicago/Turabian StyleMansier, Olivier, Valérie Prouzet-Mauléon, Gwénaële Jégou, Kim Barroso, Diana Pelizzari Raymundo, Aurélie Chauveau, Pierre-Yves Dumas, Valérie Lagarde, Béatrice Turcq, Jean-Max Pasquet, and et al. 2019. "The Expression of Myeloproliferative Neoplasm-Associated Calreticulin Variants Depends on the Functionality of ER-Associated Degradation" Cancers 11, no. 12: 1921. https://doi.org/10.3390/cancers11121921